

Abstract
Mutations in the Aristaless-related homeobox (ARX) gene are found in a spectrum of epilepsy and X-linked intellectual disability disorders. During development Arx is expressed in pallial ventricular zone (VZ) progenitor cells where the excitatory projection neurons of the cortex are born. Arx−/Y mice were shown to have decreased proliferation in the cortical VZ resulting in smaller brains; however, the basis for this reduced proliferation was not established. To determine the role of ARX on cell cycle dynamics in cortical progenitor cells, we generated cerebral cortex-specific Arx mouse mutants (cKO). The loss of pallial Arx resulted in the reduction of cortical progenitor cells, particularly the proliferation of intermediate progenitor cells (IPCs) was affected. Later in development and postnatally cKO brains showed a reduction of upper layer but not deeper layer neurons consistent with the IPC defect. Transcriptional profile analysis of E14.5 Arx-ablated cortices compared with control revealed that CDKN1C, an inhibitor of cell cycle progression, is overexpressed in the cortical VZ and SVZ of Arx KOs throughout corticogenesis. We also identified ARX as a direct regulator of Cdkn1c transcription. Together these data support a model where ARX regulates the expansion of cortical progenitor cells through repression of Cdkn1c.
Keywords: cell cycle, forebrain development, neurogenesis, neurodevelopmental disorders, TBR2
Introduction
Mutations in the Aristaless-related homeobox gene (ARX) are a common cause of X-linked intellectual disability. Over 40 different mutations have been identified in ARX resulting in a spectrum of disorders that vary from mild intellectual disability with no brain malformations to severe brain malformations such as lissencephaly and hydranencephaly (Gecz et al. 2006). Currently the pathogenesis of these ARX-related disorders are poorly defined.
During mouse cerebral cortical development Arx is expressed in the ventricular zone (VZ) (Colasante et al. 2008; Colombo et al. 2004; Friocourt et al. 2006). Arx deficient mice die shortly after birth with a thin and disorganized neocortex in addition to other brain abnormalities (Kitamura et al. 2002). The neocortical alterations appear to be the result of decreased pallial progenitor cell proliferation (Friocourt et al. 2008; Kitamura et al. 2002). However, it remains unclear how Arx regulates cortical progenitor proliferation, cell specification, and layer formation in the neocortex.
To define the endogenous function of Arx in the cortical VZ, Arx-floxed mice were mated to Emx1-Cre mice, removing Arx specifically in the dorsal telencephalon. The number and proliferation rate of progenitor cells, their cell cycle length, and final cortical laminar fate were then assayed. Our data show that Arx regulates the expansion of both radial glial cells (RGC) and intermediate progenitor cells (IPC), with a more pronounced effect on the latter. The decrease in proliferation in the Arx cKO mice resulted in a loss of neurons specifically in the upper layers of the neocortex. This is consistent with the hypothesis that IPC derived neurons contribute to all cortical layers, but predominately upper layers (Nieto et al. 2004; Sessa et al. 2008; Tarabykin et al. 2001; Zimmer et al. 2004).
We also identified a cohort of genes whose expression is consistently altered in Arx KO mice cortices when compared with wild-type mice. Among them, Cdkn1c, encoding for CDKN1C, was found to be a direct target and its role in the identified IPC phenotype was analyzed. As a member of the Cip/Kip family of cyclin-dependent kinase inhibitors (CDKi), CDKN1C antagonizes cell cycle progression by inhibiting G1/S transition (Sherr and Roberts 1999). Interestingly, CDKN1C, whose expression is normally detected in the cortical SVZ, was overexpressed in both the VZ and SVZ of Arx KO cortices. Thus, ARX appears to regulate cortical progenitor pool expansion by repressing the premature expression of Cdkn1c in the cerebral cortex.
Methods
Mice
Arx conditional knock out mice (Arxfl/fl) (Fulp et al. 2008) and Arx knock out mice (Arx−/y) (Collombat et al. 2003) were maintained on a C57Bl/6 background. Timed-pregnant mice were considered embryonic day 0.5 (E0.5) on the morning of the confirmation of the vaginal plug. To inactivate Arx in the dorsal telencephalon during development Arxfl/fl mice were crossed with Emx1-Cre animals (Jin et al. 2000). Genotyping of Arx conditional knock out and germline knock out mice was performed as described (Collombat et al. 2003; Fulp et al. 2008; Jin et al. 2000). Sex assessment for control embryos was performed by Sry PCR (SryFw 5′-CAGAAATGAACTACTGCATCCC-3′ and SryRev 5′-AACTTGTGCCTCTCACCACG-3′).
Tbr2::GFP transgenic mice (Kwon and Hadjantonakis 2007) were crossed with Arxfl/y;Emx1-Cre animals. Mice were maintained at the Children's Hospital of Philadelphia and at the San Raffaele Scientific Institute lab animal facilities. All experiments were approved by the Institutional Animal Care and Use Committees of the Children's Hospital of Philadelphia and of San Raffaele Scientific Institute.
BrdU Administration for Cell Cycle Dynamic Analysis
BrdU was administered (50 mg/kg, i.p.) to pregnant mice 1 h before embryo harvesting to assess the S-phase labeling index (LI). A cumulative BrdU-labeling protocol was followed to determine the growth fraction (GF) (fraction of the cell population that is proliferative), to determine the time to reach the GF (Tc−Ts), and, subsequently, to estimate the length of the cell cycle (Tc) and the duration of S-phase (Ts) for both radial glia and intermediate progenitors (Takahashi et al. 1995). Pregnant mice were injected with BrdU (50 mg/kg, i.p.) starting at E13.5, with injections every 3 h and with survival times of 0.5 h. Thus, a total of 6 time points were examined: 0.5, 3.5, 6.5, 9.5, 12.5, and 15.5 h. For cell cycle exit experiment, BrdU (50 mg/kg, i.p.) was injected in a pregnant female at E13.5 and 24 h later embryos were harvested and processed for analysis.
Histological Techniques
Nissl Staining
Sections were stained in a 0.1% cresyl violet solution for 5–10 min and then, rinsed quickly in distilled water. Thereafter, sections were dehydrated in 100% ethanol (2 × 5 min), dried and coverslipped with Eukitt (Electron Microscopy Science).
RNA In Situ Hybridizations
ISH on frozen sections was performed as previously described by (Schaeren-Wiemers and Gerfin-Moser 1993) with the modifications previously reported (Colombo et al. 2007). Cdkn1c in situ probe was a kind gift of L. Muzio (Muzio et al. 2002).
Immunohistochemistry
Whole heads (E11.5 and E12.5) or brains were dissected from embryonic and P1 mice and fixed overnight in 4% PFA at 4 °C. P14 and P30 mice were perfused with 4% PFA, then the brains were removed and fixed in 4% PFA overnight at 4 °C. Fixed brains were frozen and 10-μm-thick coronal sections were obtained. Antigen retrieval was performed in citric acid-based Antigen Unmasking Solution (Vector Laboratories) autoclaved at 105 °C for 10 min; for BrdU staining slides were also treated with 2N HCl for 20 min. No antigen retrieval was performed for ARX and TBR2. Sections were then blocked for 1 h at room temperature with 10% normal goat serum (Gibco) and 0.1% triton in PBS. Primary antibodies against ARX (rabbit, gift provided by Professor Kunio Kitamura, 1:500), KI67 (rabbit, Immunological Sciences, 1:300 and mouse, BD Pharmingen, 1:200), BrdU (rat, Accurate Chemical and Scientific, 1:200), PH3 (rabbit, Chemicon, 1:100), TBR2 (rabbit, Abcam, 1:200 and Chemicon, 1:300), PAX6 (mouse, Developmental Studies Hybridoma Bank, 1:1000), CUX1 (rabbit, Santa Cruz Biotechnology, 1:50), SATB2 (mouse, Bio Matrix Research, 1:200), CTIP2 (rat, Abcam and Beckton-Dickinson 1:300), GFP (chicken, 1:2000), Caspase3 (rabbit, Cell Signaling, 1:200), and TBR1 (rabbit, Abcam, 1:25 and Chemicon, 1:400) were diluted in 10% normal goat serum and incubated on slides overnight at 4 °C. Secondary antibodies were conjugates of Alexa Fluor 488, Alexa Fluor 594, and Alexa Fluor 647 (1:500, Invitrogen), biotinylated goat anti-mouse and anti-rabbit (Dako, 1:100). They were diluted in 10% normal goat serum and incubated on slides for 2 h at room temperature. Biotinylated secondary antibodies were subsequently incubated with strepdavidin-Cy3 (Jackson ImmunoResearch, 1:300) in PBS. Slides were then counterstained with DAPI (Invitrogen, 1:1000), washed and mounted in Fluorescent Mounting Medium (DakoCytomation).
For the ARX/TBR2 double labeling, sections were blocked in 10% normal donkey serum (Jackson ImmunoResearch) with 0.3% triton in TBS (Tris buffered saline pH 7.4, USB Cleveland, OH, USA; the buffer used throughout the procedure) for 30 min at room temperature. Anti-TBR2 antibody (Abcam, 1:200) in 10% donkey serum next was applied for 2 h at room temperature followed by washing with TBS and then incubation with goat anti-rabbit monoclonal Fab fragments (Jackson ImmunoResearch,, 1:25) in 10% donkey serum for 30 min at room temperature. They were then washed and secondary Alexa 594 donkey anti-goat 1:200 in 10% donkey serum in TBS was applied for 30 min at room temperature. For the second primary antibody, the sections were again washed and blocked with the goat anti-rabbit monoclonal Fab fragments 1:25 in 10% donkey serum. After washing, the anti-ARX antibody was applied overnight at 4 °C in 10% donkey serum. Finally, the slides were washed and the secondary Alexa 488 donkey anti rabbit was applied at 1:200 in 10% donkey serum for 30 min at room temperature. The slides went through on final wash, were counterstained with DAPI and mounted.
Quantifications and Statistical Analysis
All quantifications on P14 cryostat brain sections were performed using a 200-μm section of the dorsomedial cortex at the midhemispheric level from pia to white matter. For quantifications on E11.5 and P1 brain sections, cell counts were derived from 25% of the neocortex. The area of interest was derived by measuring the linear distance along the pial and ventricular surfaces of the entire lateral cortex, dividing those into quarters, and then using the total area, from pia to ventricle, in the third quartile from the dorsal midline. Quantifications on E14.5 brain sections were performed using a 150-μm section of the dorsomedial and lateral cortex. At least 3 serial sections from 3 different animals for each genotype were photographed using a Leica DMR fluorescent microscope. Images were imported into Photoshop CS3 and double-positive cells were overlaid manually by color-coded dots in new layers. The number of labeled cells (dots) was calculated using the Record measurements of Photoshop CS3 and imported into Excel 2008. The percentage of labeled cells per region per section was calculated for each brain, and the final mean percentage calculated across all brains. Results were expressed as mean value ±SD and were tested for statistical significance by the 2-tailed, unpaired Student's t test (P < 0.05 level of significance).
RNA Isolation, Processing, and Microarray Analysis
The experiment was performed as previously described (Colasante et al. 2009). Briefly, 3 pregnant Arx heterozygous dams crossed with C57Bl/6 males were sacrificed at E14.5. A total of 5 wt and 5 Arx KO embryos were harvested and after brain isolation the cerebral cortices were separated from the ventral telencephalon. Total RNA was extracted from the cortices using the Qiagen RNA micro kit (Qiagen, Valencia, CA, USA). cRNAs were generated and hybridized on a total of 10 different MOE430v2 Affymetrix DNA chips according to the Affymetrix protocol. The chips were scanned with an Affymetrix scanner to generate digitized image data files. The data were deposited in the NCBI Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series (# GSE12956).
Luciferase Reporter Assays
P19 cells (105) were seeded and transfected 24 h after plating, with 400 ng of each of the following constructs in different combinations: pT81 luciferase reporter plasmids, control plasmid pCAG-iresGFP, pCAGArx-iresGFP, pCDNA3-Tle1, and 80 ng of pRL-TK-Renilla luciferase plasmid DNA (Promega) using Lipofectamine PLUS™ Reagent (Roche Diagnostics, Alameda, CA). Forty-eight hours post-transfection, cell lysis, and measurement of firefly and Renilla luciferase activity was performed using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer's instructions. The firefly luciferase activity was normalized according to the corresponding Renilla luciferase activity, and luciferase activity was reported as mean relative to pCAG-iresGFP/luciferase transfection. The 2 Cdkn1c −549/−68 and −1490/−720 enhancer regions were PCR amplified from E14.5 mouse genomic DNA using the following primers: HindIII-Cdkn1c-F1 5′-CCCAAGCTTGCGCGCGGGCCTCCTCAC-3′; BamHI-Cdkn1c-R1 5′-CGCGGATCCCCAGGACCAGGACCCAGCTG-3′; HindIII-Cdkn1c-F2 5′-CCCAAGCTTGAGATCTAAAAGATCTGTAG-3′, and BamHI-Cdkn1c-R2 CGCGGATCCTAGTGAATGAGGGGTTCATG-3′, respectively. The amplified products were cut with HindIII and BamHI and cloned into the BamHI and HindIII sites of pT81-TK-Luciferase, which contains the thymidine kinase minimal promoter upstream of luciferase. The (−549/−68) enhancer region carrying mutagenized ARX binding site was generated by site-directed mutagenesis with the following primers 5′-CTGAAAATACTACAGGATGCTCCTCGTGAGGAG-3′ and 5′-CTCCTCACGAGGAGCATCCTGTAGTATTTTCAG-3′ using the QuikChange II site-directed Mutagenesis Kit (Stratagene), according to the manufacturer's instructions.
Chromatin Immunoprecipitation
E14.5 mouse embryonic cortices were isolated and single-cell suspension was derived by enzymatic treatment. Cells were crosslinked with 1% formaldehyde for 10 min and chromatin prepared essentially as described previously (Colasante et al. 2008; Ferrai et al. 2007). The chromatin aliquots (1 mL) were incubated overnight with 1 μg of anti-GFP (Chemicon) as mock and anti-ARX(C-14) (Santa Cruz). PCR primers used are the following: ChIPEbf3-F, 5′-GTCTATAAGTACAATGGTGACAC-3′; ChIPEbf3-R, 5′-CTCCATCAAGATCCTTCTC-3′ (amplification product, 240 bp); ChIPCdkn1c-F, 5′-GCGCCCCTTTATACGCGCTG 3′; ChIPCdkn1c-R, 5′-TCACGTTACCGCCCGCAGAG-3′ (amplification product 200 bp); ChIPCdkn1a-F 5′-GCTGTCAAAACGACCTTGAATG-3′; ChIPCdkn1a-R: 5′-GGAAGGACTAACTCTTTTCCAG-3′ (amplification product 220 bp); ChIPCdkn1b-F 5′-CCCTGATAAGAGCGGTCAGTC-3′; ChIPCdkn1b-R 5′-TTACGGAGCTTCGGTGGCTAG-3′ (amplification product 250 bp). PCR products were analyzed on 2% agarose gels in TBE buffer.
Results
Reduced Cerebral Cortex and Olfactory Bulbs after Deletion of Arx from the Dorsal Telencephalon
The germline loss of Arx in mice results in a thinner and disorganized cortex that was attributed to a primary defect in cell proliferation (Kitamura et al. 2002). To further define the role of Arx in cortical progenitor cell proliferation, we conditionally deleted Arx from the E9.5 dorsal telencephalon by crossing our Arxfl/+ mice to Emx1-Cre mice (Jin et al. 2000) (Fig. 1A,B). In Arxfl/y; Emx1-Cre mice (here after Arx cKO), no ARX protein was observed in the VZ and SVZ of the cortex by E11.5 and throughout the rest of development (Fig. 1A,B and data not shown). Morphological analyses performed at E14.5 revealed that, at this developmental stage, alterations of the gross morphology of Arx cKO brains are already evident. The cerebral hemispheres are reduced in size and the olfactory bulbs are smaller and characterized by an abnormally wide interspace separating them (Fig. 1C,D). Nissl staining on E14.5 coronal sections highlighted the diminution of cortical thickness affecting Arx cKO cerebral cortex in its whole medio-lateral extension (E14.5 relative volume (rv) Arx cKO/ctrl = 0.86 ± 0.03; n = 3, P < 0.005 (Fig. 1E,F,O). Arx−/y mice die perinatally (Collombat et al. 2003; Kitamura et al. 2002), whereas crosses between Arxfl/+ female and Emx1-Cre male mice recovered all expected genotypes in Mendelian ratios (data not shown). Morphological assessments at P30 of Arx cKO and relative control brains confirmed the analysis made at E14.5. Indeed, the cerebral hemispheres and olfactory bulbs in Arx cKO mice are reduced in size, whereas the cerebellum and the brain stem do not appear to be affected (Fig. 1I,J). In addition, P30 Nissl-stained coronal sections highlight an overall reduction of the cerebral cortex, most evident at the level of pyriform cortex and the hippocampus (rv P30 = 0.84 ± 0.05; n = 3, P < 0.005) (Fig. 1K,L,O).
Figure 1.

Reduction in cerebral cortex and olfactory bulb dimensions is observed in Arx cKO brains. (A and B) At E11.5 Arx expression is lost in the cortical VZ of Arx cKO brains when compared with control brains. (C–H) At E14.5 the Arx cKO brains exhibit a reduced size of the cerebral hemispheres and the olfactory bulbs (C and D). Nissl-stained coronal sections (E and F) and corresponding high magnification of the cerebral cortex (G and H) highlight a diminution in the cortical thickness in the Arx cKO brains when compared with controls. (I–N) Morphological analysis of postnatal (P30) control and Arx cKO brains confirms the previous observations: cortical vesicles and olfactory bulbs are underdeveloped, whereas the cerebellum and the brain stem do not appear to be affected (I and J); Nissl-stained coronal sections reveal an overall reduction in the telencephalic structures in Arx cKO brains, involving the cerebral cortex and hippocampus (K and L). Magnifications of cortical areas show an increased nuclear density in the deeper layers (IV–VI) of Arx cKO compared with controls (M and N). (O) Quantifications of relative cortical volume (rv) at E14.5 and P30 of Arx cKO brains compared with controls. Error bars correspond to SD (P and Q) The corpus callosum is absent in Arx cKO brains. WM: white matter.
In the Arx cKO, the laminar structure cannot be clearly delineated (Fig. 1M,N) and the corpus callosum is absent in Arx cKO brains (Fig. 1P,Q); this feature, together with the microcephaly, strongly resembles the morphologic abnormalities observed in patients with the XLAG syndrome (Kitamura et al. 2002).
Our data indicate the loss of Arx in cortical progenitors recapitulates the morphologic defects observed with the germline loss of Arx. These data support the model by which the reduced brain size observed in Arx KO mice and children with loss of function ARX mutations, is largely or completely due to a loss of Arx in cortical progenitor cells.
Loss of Arx in the Pallial Progenitor Cells Reduces the Proliferation Rate of Cortical Progenitor Cells
Previous data suggested a decrease in cortical progenitor cell proliferation as the basis for the reduced cerebral cortical thickness in Arx KO mice (Kitamura et al. 2002). At E11.5 in cortical development, there are similar numbers of progenitor cells in wild-type and Arx cKO mice cortices as measured by the number of KI67-positive cells (ctrl: 392 ± 33.5; cKO: 368 ± 45.5, n = 3, P = 0.579: cells counted in 25% of the cortex from ventricular to pial surface) (data not shown). This suggests normal numbers of cortical progenitor cells exist early in development when the progenitor cells are being produced. However, when the same analysis was performed at E12.5, we observed a reduction in the percentage of proliferating progenitor cells (% KI67+/Hoechst, ctrl: 96.65 ± 2.90; cKO: 87.49% ± 5.58, n = 6, P = 0.016, cells counted in a 150-μm bin from the ventricular to the pial surface) (Supplementary Fig. 1C,D′,G), and an increase in ßIII-tubulin-positive young neurons in Arx cKO compared with control (% TUBB3+/Hoechst, ctrl: 10.88 ± 2.96; cKO: 20.75 ± 3.39, n = 6, P = 0.003) (Supplementary Fig. 1A,B′,H).
At E14.5, the cortical plate (CP) is already thinner (Fig. 1E,F) and the reduction in the total number of proliferative cells is evident in Arx cKO cortices (Fig. 2A,C) (ctrl: 259 ± 21.8; cKO: 202.5 ± 16.3; n = 6, P < 0.001, cells counted in a 150-μm bin from the ventricular to the pial surface). Moreover, both the types of cortical progenitor cells, RGCs and IPCs, are diminished in Arx cKO cortices at this embryonic stage. Indeed, we observed a reduced PAX6-labeling intensity in Arx cKO mice (Fig. 2D′,E′) in addition to a 27% reduction in the number of PAX6+ cells (ctrl: 323.6 ± 13.3; cKO: 237.5 ± 26.8; n = 3, P < 0.001; Fig. 2J) as well as a 20% reduction of TBR2+ cells (ctrl: 223.6 ± 16.4; cKO: 179.3 ± 10.8; n = 3, P < 0.001) (Fig. 2F′,G′).
Figure 2.

Proliferation rate of radial glia cells and intermediate progenitors is reduced in Arx cKO cerebral cortex. E14.5 coronal sections from control (left) and Arx cKO cortices (right) are directly compared. Ki67 stained sections (A and B) and corresponding higher magnification (E and F) reveal an overall reduction of proliferating cells in Arx cKO cortex compared with wild type; (C) quantifications of Ki67+ cells in a 150-μm bin extending from the ventricular to pial surface: control 259 ± 21.8 and cKO 202.5 ± 16.3; n = 6 for each, P value < 0.001. (D–I′) PAX6, TBR2, BrdU, and PH3 immunolabelings performed on wild-type and Arx cKO sections from embryos treated with 1 h pulse of BrdU. (D′ and E′″) Reduction in the number and in the signal intensity of PAX6+ cells (D′ and E′) and in the number of BrdU+ (D″ and E″) in Arx cKO cortices compared with the control. (F′–G′″) A reduction in the number of TBR2+ cells (arrowheads, F′ and G′) and a decrease in TBR2+/BrdU+ double-positive cells (arrowheads, D′″ and E′″) in Arx cKO cortices compared with the control are shown (H′ and I′). Reduction of PH3+ cells is observed in the Arx cKO cerebral cortex, greater in the basal rather then in the apical region. (J) Quantifications of total PAX6+ and TBR2+ cells, dual-labeled PAX6+/BrdU+ and TBR2+/BrdU+ double-positive cells and PH3+ in E14.5 wild-type and Arx cKO cortices are shown.
In order to exclude an increase in cell-death contributing to the reduced cortical thickness in Arx cKO cortices, the apoptotic marker Caspase3 was analyzed at different embryonic stages (E13.5, E15.5, and E17.5). We found no difference between the Arx cKO and control cortices (Supplementary Fig. 2).
To determine if the reduction in proliferation was affecting both types of cortical progenitor cells (RGCs and IPCs) in the same way, their rate of BrdU incorporation was measured in control and Arx cKO cerebral cortices after a 1-h BrdU pulse. As expected, general reduction in BrdU incorporation (∼24%) (arrowheads in Fig. 2F″), was found in Arx cKO cortices with respect to controls (ctrl: 142.3 ± 18.2; cKO: 109.0 ± 10.2; n = 3, P < 0.001 and Fig. 2J). Interestingly, double labeling with PAX6 and BrdU revealed little differences in the PAX6+/BrdU+ cell number between Arx cKO and control (ctrl: 125.6 ± 19; cKO: 103.7 ± 7.17; in both cases about 40% of the total PAX6+ cells; n = 3, P = 0.012) (Fig. 2D′″,E′″,J). Conversely, double labeling with TBR2 and BrdU revealed a reduction of dual-labeled cells (ctrl: 52.9 ± 12.9, about 25% of the total TBR2+; cKO: 17.5 ± 2.9, about 10% of the total TBR2+; n = 3, P < 0.001) (arrowheads in Fig. 2F′″,G′″,J). Consistent with these data, staining with the M-phase marker phosphohistone 3 (PH3) revealed an overall reduction of positive cells in the Arx cKO cerebral cortex with a greater loss in the basal area (IPCs) than the apical area (RGCs) (apical, ctrl: 122.0 ± 5.6, cKO: 107.5 ± 3.5; basal, ctrl: 39.0 ± 4.8; cKO: 11.5 ± 2.12 n = 3, P < 0.001, Fig. 2H,I,H′,I′,J).
Taken together, these data indicate that at E14.5 both progenitor populations (RGCs and IPCs) are reduced in Arx cKO cerebral cortices and their proliferation is affected. However, BrdU incorporation rates and PH3 staining indicate that there is a disproportionate deficit in the proliferation of IPCs compared with RGCs in Arx cKO mice.
Cortical Progenitors Show a Normal Cell Cycle Length, but IPC Mitoses Are Significantly Reduced in Arx cKO Mice
To further understand the mechanism by which loss of Arx resulted in fewer proliferating cells, we tested 2 nonmutually exclusive hypotheses: that the cell cycle changes are related to lengthening of the cell cycle or to precocious exit of cortical progenitors from the cell cycle.
An increased cell cycle length during the neurogenic period would result in fewer cellular divisions, fewer progenitor cells and the observed reduction in BrdU incorporation. To examine neocortical cell cycle dynamics in the absence of Arx, the length of the cell cycle phases was estimated between E13.5 and E14.5 in wild-type and Arx cKO mice using a cumulative BrdU pulse-labeling paradigm (Takahashi et al. 1995). BrdU was injected intraperitoneally in gravid females every 3 h up to 6 time points with survival time being 0.5 h after the last injection. At least 2 brains of each genotype were assayed at each time point for BrdU labeling to evaluate the cell cycle parameters in the VZ precursors, where VZ was defined by 2 criteria: the region under the area with high density of TBR2+ cells and where cells with vertically oriented nuclei were present. Moreover, TBR2/BrdU double staining was performed in order to gain information regarding the BrdU incorporation kinetic of IPCs. Representative cortical fields of wild-type and Arx cKO BrdU- and TBR2-labeled coronal sections are shown for 5 out of the 6 time points (Fig. 3A–J).
Figure 3.

Cell cycle kinetic is not affected in both Arx cKO VZ and SVZ progenitors, but IPC growth fraction is severely reduced. (A–J″) Cortical fields of BrdU and TBR2 labelings performed on control and Arx cKO cortices at 5 (0.5, 3.5, 6.5, 9.5 and 12.5 h) of the 6 time points of the BrdU cumulative labeling experiment are shown. (K–M) Double labeling for TBR2 and KI67 and quantification of KI67+TBR2+ normalized on total TBR2+ cells reveals a severe reduction of the IPC proliferating fraction in Arx cKO with respect to control. (N and O) Labeling indices (LI) for both control and cKO RG and IPC progenitor (calculated as the ratio between TBR2+ BrdU+ cells on the total of TBR2+ and normalized on the IPC growth fraction in the latter case), are plotted against correspondent BrdU-labeling times and the interpolating lines are drawn. (P) Evaluation of cell cycle duration (Tc) and S-phase duration (Ts) are reported.
The GF in the VZ—defined as the maximum percentage of VZ progenitors entering the S-phase on the total of nuclei in the VZ—did not change between the 2 genotypes, and in both cases, almost 100% of VZ cells were BrdU+ at the 9.5 h time point (Fig. 3G,H). The LI of VZ (BrdU+cells/total cell number) at each time point was plotted against the corresponding time after BrdU injection, and, from the slope of the interpolating lines, the cell cycle parameters Tc and Ts were calculated (Fig. 3N,P). Interestingly, no significant difference was found in both parameters between control and Arx cKO brains (ctrl: tc = 14.5 h and Ts = 4 h; cKO: tc = 14.1 h and Ts = 4.1 h).
A similar analysis of the cell cycle kinetic was performed in the SVZ. First, the GF at E13.5 was evaluated as the percentage of proliferating IPC (KI67+ TBR2+) divided by the total number of IPCs (TBR2+) (Fig. 3K,L′). Interestingly, while ∼75% of IPCs in the control is proliferating, only 45% of Arx cKO IPCs enter any cell cycle phase (% KI67+ TBR2+/TBR2, ctrl: 74.30 ± 1.99; cKO: 45.17 ± 2.64, n = 4, P < 0.0004, Fig. 3M). A similar reduction in the SVZ GF of Arx cKO was noticed also later in cortical development (Supplementary Fig. 3; E17.5% KI67+ TBR2+/TBR2, ctrl: 16.57 ± 2.49; cKO: 7.58 ± 0.26). These data confirmed and completed the previous observations relative only to S and M phases of the cell cycle.
Then, LI in the SVZ was calculated as the ratio between the TBR2+ cells incorporating BrdU on the total of TBR2+ proliferating cells (BrdU+ TBR2+/Ki67+ TBR2+) and plotted against the corresponding time after BrdU injection and interpolating lines generated for both control and Arx cKO (Fig. 3A″–J″,O). The slope of the Arx cKO interpolating line resulted slightly higher; indeed, cKO IPCs seem to reach the value of GF = 1, a little before the control (Tc-Ts, control: 7.8 h; cKO: 6.3 h). As a consequence, Tc and Ts is slightly shorter in cKO IPCs with respect to control (Fig. 3P). The ratio between Ts and Tc is not significantly altered (Ts/Tc, ctrl: 0.25; cKO: 0.24), suggesting that the relative length of the different cell cycle phases is constant.
Interestingly, the same conclusions were reached through a different approach. We measured the BrdU LI (0.5 h pulse) in SVZ precursors (Ki67+) at E14.5 (Arnold et al. 2008), finding significant decrease in the number of proliferating Ki67+ cells/100 μm2 area in cKO SVZ with respect to control (ctrl: 19.83 ± 6.04 and cKO: 3.83 ± 1.94; n = 4, P < 0.005; Fig. 4A–C), comparable to the reduction in BrdU+ (Supplementary Fig. 4) and PH3+ cells in cKO SVZ (Fig. 2J). Consequently, the ratio between the number of S-phase cells (BrdU+) and the total of those proliferating (Ki67+) show no significant difference (ctrl: 0.44 ± 0.02 and cKO: 0.40 ± 0.10; n = 4, P = 0.34; Supplementary Fig. 4C). This last set of data again indicates that Ts/Tc ratio of Arx cKO SVZ precursors is comparable to control.
Figure 4.

Increased cell cycle exit affects Arx cKO progenitors and the progeny of TBR2+ cells is reduced. (A and B) E14.5 control and Arx cKO cortical sections were double labeled for Ki67 and BrdU, following BrdU injection at E13.5. (C) Quantification of the proportion of cells leaving the cycle (leaving fraction: BrdU+Ki67-/tot BrdU+ cells). (D and E) E15.5 coronal sections and relative higher magnifications (D′ and E′) of control and Arx cKO in the transgenic background Tbr2::GFP analyzed for GFP expression reveal a reduction in intensity and number of GFP+ cells in Arx cKO SVZ, intermediate zone (IZ), and cortical plate (CP) as quantified in F.
Consequently, the reduced BrdU incorporation observed in cumulative labeling experiment of TBR2+ cells can be attributed to a consistent reduction in the proliferation ability of Arx cKO SVZ progenitors and not to an alteration of their cell cycle length.
Precocious Cell Cycle Exit in Arx cKO Cortical Progenitors
Since only about half of the pool of IPCs is able to proliferate in Arx cKO cortex, we predicted precocious cell cycle exit of these cells. Thus, we assessed the fraction of cells exiting the cell cycle 24 h after a BrdU pulse (between E13.5 and E14.5). Cells remaining in the cycle are BrdU+/Ki67+, while those that have stopped cycling exhibit BrdU labeling only (Chenn and Walsh 2002). At E13.5, an increase of 18% in the cells leaving the cycle is observed in Arx cKO progenitors (leaving fraction in ctrl: 0.49 ± 0.06; in cKO: 0.58 ± 0.05; n = 8 for each, P < 0.0005; Fig. 4A″,B″,C) suggesting precocious neurogenesis. Consistent with that, more cells with undiluted BrdU exist in the Arx cKO when compared with control (arrowheads, Fig. 4A′,B′). Although it is not possible to distinguish the contribution of RG cells and IPCs to the fraction of precociously differentiating cells, it is likely that most of those cells are IPCs that may exit cell cycle without any round of amplification.
In order to evaluate the effects of IPC reduced proliferation in Arx cKO, we analyzed GFP expression in E15.5 Arx cKO and relative control mice crossed with Tbr2::GFP transgenic animals (Fig. 4D,E′), where GFP is under the control of the Tbr2 endogenous regulatory sequences (Kwon and Hadjantonakis 2007). Due to the stability of the GFP protein in vivo, GFP fluorescence in Tbr2::GFP mice can be used as a lineage tracer of the IPC progeny (Sessa et al. 2008). We noticed an overall decrease in GFP+ cells in Arx cKO SVZ and CP with respect to control consistent with a reduction in the amplification of IPCs in the cKO (GFP+ cells/150 um area unit, ctrl: SVZ, 55.7 ± 5.13; IZ, 80.00 ± 16.7; CP, 180.33 ± 6.51; total, 316.00 ± 15.39; cKO: SVZ, 27.70 ± 7.37; IZ, 63.33 ± 26.63; CP, 83.00 ± 2.64; total 181.70 ± 21.01; n = 4 P < 0.005 (Fig. 4F).
Given the more pronounced effect of Arx ablation on IPCs than on RGCs, we sought to assess if Arx is also expressed in IPCs in addition to RGCs (Friocourt et al. 2006). We performed immunofluorescence for ARX and TBR2 in wild-type cortex at E14.5 and showed that ARX colocalizes with TBR2 in many cells of the VZ and SVZ (Supplementary Fig. 5).
Arx cKO Mice Have a Reduced Number of Upper Cortical Layer Neurons with Normal Number of Deeper Layer Neurons
In order to determine if the reduced proliferation of cortical progenitors has an effect on cortical lamination, we evaluated the cortical layers using molecular markers at different developmental stages. The expression of TBR1, which labels neurons in the subplate, layer VI, and few in layer V (Molyneaux et al. 2007), was evaluated in control Arx cKO mice at E16.5. In Arx cKO, the number of TBR1+ cells in layers V–VI was slightly decreased with respect to control (TBR1, ctrl layers V–VI = 98.2 ± 6.5; cKO layers V–VI = 82.0 ± 7.8) (Fig. 5A,B). Interestingly, more neurons were found in Arx cKO subplate, still detectable at this stage (TBR1, ctrl: SP = 36.5 ± 4.5; cKO: SP = 60.0 ± 3.5) (arrowheads in Fig. 5A,B).
Figure 5.

Cortical layering is altered in Arx cKO brains. Immunohistochemistry for molecular markers identifying deeper cortical layers at E16.5 and both deeper and upper cortical layers at P1 and P14, in control and Arx cKO cortices (A–T). (A and B) TBR1+ cells are slightly decreased in layer V–VI neurons and increased in subplate in E16.5 Arx cKO cortices with respect to the controls (aarowheads). (C and D) CTIP2 immunolabeling shows the same alterations observed with TBR1 although more prominently (arrowheads). (E and F) At P1, similar numbers of TBR1+ cells in Arx cKO cortices with respect to control are detected as well as a similar number of CTIP2+ cells in both layers V and VI (G and H). (I and J) SATB2 is decreased in upper layer neurons (II, III) in the Arx cKO cortices with respect to the controls (arrowheads). (K and L) Similarly, staining for CUX1 (layers II–IV) shows a diminution of upper cortical layer neurons in Arx cKO cortices with respect to controls (arrowheads). (M–P) At P14, there are still similar numbers of TBR1+ and CTIP2+ cells in the Arx cKO cortices when compared with controls but still a decrease in SATB2+ cells in layers II/III (Q, R). Similarly, there are also still fewer CUX1+ cells in the Arx cKO cortices (S and T). (U) Quantifications of TBR1+ and CTIP2+ cells in layers VI and V and of SATB2+ and CUX1+ at E16.5, P1, and P14 in both control and Arx cKO cortices. TBR1+ and CTIP2+ cells at P14. Cells positive for each molecular marker were counted 25% of the cortex in P1 and in a 200-μm bin in P14, both from the ventricular to the pial surface in both control and Arx cKO cortices.
A similar difference, although more dramatic, was observed staining for CTIP2, which labels predominately layer V, but also layer VI neurons (Molyneaux et al. 2007). CTIP2+ cells are already well organized in control layers V–VI, with some positive cells localized in the subplate, whereas in the Arx cKO, they appear less compact and completely disorganized (arrowheads in Fig. 5C,D). However, the total number of CTIP2+ cells does not change between Arx cKO and control (CTIP2, ctrl: 166.9 ± 4.2 and cKO: 155.7 ± 5.7) (Fig. 5U). To assess upper layer stratification, we used P1 and P14 animals. At P1 Arx cKO mice had a decreased number of cells labeled with SATB2 in layer II/III in the Arx cKO cortices with respect to controls (SATB2 ctrl: layer II/II = 131.4 ± 18.5 and SATB2 cKO: layer II/III = 96.9 ± 12.6; n = 5, P = 0.0126) (arrowheads in Fig. 5I,J), but not in layer IV/V (SATB2 ctrl: layer IV/V = 84.0 ± 6.8 and SATB2 cKO: layer IV/V = 77.9 ± 7.6; n = 5, P = 0.547) (Fig. 5I,J). In addition, the number of cells labeled with CUX1, a marker of layers II–IV, is also decreased in the Arx cKO cortices (CUX1 ctrl: layers II–IV = 125.9 ± 7.0 and CUX1 cKO: layers II–IV = 95.9 ± 7.8; n = 5, P = 0.0327) (arrowheads in Fig. 5K,L). However, no significant difference between the Arx cKO and control cortices was detected for layers V and VI markers (CTIP2 ctrl: total = 97.5 ± 7.5 and CTIP2 cKO: total = 93.2 ± 9.6; n = 3, P = 0.587; TBR1 ctrl: total = 116.9 ± 4.9 and TBR1 cKO: total = 92.1 ± 14.2; n = 3, P = 0.132) (arrowheads in Fig. 5E–H).
At P14, a time in which laminar organization is complete, a decrease in the number of SATB2- and CUX1-positive cells persisted in layer II/III in the Arx cKO cortices when compared with control cortices (SATB2 ctrl: layer II/II = 29.5 ± 2.4, layer IV = 41.4 ± 3.8 and SATB2 cKO: layer II/III = 21.2 ± 3.9, layer IV = 24.5 ± 3.0; n = 4, P = 0.0286; CUX1 ctrl: layers II–IV = 30.5 ± 2.1 and CUX1 cKO: layers II–IV = 24.9 ± 3.5; n = 4, P = 0.0482) (arrowheads in Fig. 5Q–T). However, similar numbers of CTIP2-positive and TBR1-positive cells are present in the Arx cKO cortices as compared to control (CTIP2 ctrl: total = 35.5 ± 3.7 and CTIP2 cKO: total = 29.2 ± 1.7, n = 3, P = 0.272; TBR1 ctrl: total = 32.7 ± 2.1 and TBR1 cKO: total = 32.5 ± 2.5, n = 3, P = 0.406) (Fig. 5M–P).
In summary, we find a reduction of upper layer (II–IV) neurons with no change in deeper layer (V–VI) neurons in Arx cKO mice.
Identification of Genes Differentially Expressed in Arx KO Versus Wild-Type Cerebral Cortex by Transcriptome Analysis
To gain insight into the Arx-dependent mechanism responsible for the reduced proliferation in Arx cKO cortical progenitor cells observed at E14.5, we performed a gene expression profile analysis. Since all the proliferation defects observed in the Arx cKO cortices were also detected in Arx KO mouse model (data not shown), we decided to perform this experiment in Arx KO dorsal telencephalon. E14.5 embryonic stage was selected for analysis, as the proliferation defect was clearly detectable. Brains were harvested, and the cerebral cortices were carefully dissected and processed for gene expression profiling (Fig. 6). RNA was separately extracted from the isolated tissues and used to hybridize Affymetrix microarrays (MOE430v2) containing 45 101 probe sets. Cortical tissues from Arx KO (n = 5) and control (n = 5) embryos were independently processed on 10 different gene-chips in order to minimize individual biological differences. Datasets obtained were then used to identify differentially expressed transcripts between Arx KO and wild-type dorsal telencephali (Fig. 6A–I).
Figure 6.

Outline of the gene expression profile analysis and hierarchical clustering of the transcripts found differentially expressed between control and Arx KO dorsal telencephalon. (A–I) The 86 genes found differentially expressed between control and Arx KO cerebral cortices were grouped according to the protein function they codify for, in: (A) transcription factors; (B) receptors; (C) intracellular regulatory proteins; (D) structural proteins; (E) extracellular matrix proteins; (F) extracellular regulatory proteins; (G) junctions and adhesion molecules; (H) carriers and transporters; (I) Y-linked genes. (J) 7 genes found misregulated by microarray analysis were selected for quantitative real-time RT-PCRs (qPCRs) validation.
Using a log2 (fold-change ≥ 1 and a FDR ≤ 0.05) linear model, the analysis revealed that a total of 147 probe sets corresponding to 86 different Entrez Gene Identifiers presented significantly different expressions between Arx KO and wild-type cortices (Fig. 6A–I).
Among the full set of identified genes, 3 (DDX3Y, KDM5D, and EIF2S3Y) (Fig. 6I) were considered as Arx-independently misregulated. In fact, they correspond to Y-linked expressing units and their upregulation in Arx KO tissues is indicative of a sex-biased harvest of the embryos. This was expected because all Arx KO embryos were males (Arx−/Y), while wild-type embryos displayed an equal probability to be males or females. The remaining 83 differentially expressed genes were grouped according to the known or predicted protein function they coded for: transcription factors (Fig. 6A); receptors (Fig. 6B); intracellular regulatory proteins (Fig. 6C); structural proteins (Fig. 6D); extracellular regulatory proteins (Fig. 6E); extracellular junctions and adhesion molecules (Fig. 6F); carriers and transporters (Fig. 6G); extracellular matrix proteins (Fig. 6H).
To ensure the reliability of our analysis, a subset of the misregulated genes were selected and analyzed in control and KO tissues using quantitative real-time RT-PCRs (qPCRs). In this experiment, only RNA extracted from male control embryos was used, in order to avoid any sex-biased gene expression between males and females. In all cases analyzed, qPCR results confirmed previous gene-array analyses both for the upregulated genes RARb, Id1, Id3, Tub6, Olig2, and Cdkn1c as well as the downregulated Pax6 gene (Fig. 6J, all normalized to ß-actin). However, the magnitude of the changes in expression levels detected by qPCRs was, in some cases, significantly higher (up to 2.5 fold) when compared with the microarray analyses (RARb, Olig2, Cdkn1c), while, in other instances, the results were comparable (Id1, Tub6, Pax6). Interestingly, the expression of Tbr2 was decreased in Arx KO compared with control (20% reduction) in agreement with our immunohistochemistry data, despite the microarray analysis did not show a significant alteration.
Cdkn1c is Overexpressed in Arx KO Cerebral Cortex
Among the genes found to be misexpressed in Arx KO cerebral cortices, we noticed the upregulation of Cdkn1c. Cdkn1c encodes for CDKN1C (also known as p57/KIP2), a member of the Cip/Kip family of cyclin-dependent kinase inhibitors (CKIs). Together with the 2 other members of this family, CDKN1A and CDKN1B, it exerts a basilar role in the regulation of cell cycle progression of cortical progenitors at level of G1/S transition by inhibiting the cyclin/CDK complexes (Sherr and Roberts 1999). Recently, Mairet-Coello and coworkers showed that CDKN1C overexpression in the cerebral cortex elicits precursor cell cycle exit and promotes a transition from proliferation to neuronal differentiation, while the opposite effects occur in CDKN1C-deficient precursors. Indeed, they reported that, in mutant mice, proliferation of RGC and intermediate precursors (IPC) was increased, expanding both populations, with greater effect on IPCs (Mairet-Coello et al. 2012).
As this function is particularly consistent with the phenotype observed in Arx cKO brains and might underlie the reduction in the cerebral cortex volume, we focused further analyses on this gene.
In order to ascertain if Cdkn1c misregulation plays a role in Arx KO tissues, we first assessed its expression by in situ hybridization throughout embryonic development (E12.5–E18.5) in wild-type and Arx KO mouse brain sections (Fig. 7A,B,D,E and Supplementary Fig. 6). The expression of Arx was evaluated on adjacent wild-type brain sections (Fig. 7C,F and Supplementary Fig. 6). At E12.5, Cdkn1c transcript was barely detectable in the SVZ of control cortices whereas it was significantly overexpressed in both VZ and SVZ of Arx KO (Fig. 7A,B, arrowheads A′,B′). Interestingly, the upregulation of Cdkn1c seemed to follow a gradient in Arx KO cortices, higher lateral lower medial (Fig. 7B). Similarly, at E14.5, Cdkn1c is still expressed in the SVZ in control tissues (Fig. 7D, arrowheads D′) and overexpression persists in Arx KO cortices (Fig. 7E, arrowheads E′). In contrast to E12.5, Cdkn1c expression clearly followed a higher medial-lower lateral gradient in both control and KO cortices (Fig. 7D,E). Beginning on E16.5, Cdkn1c transcript was additionally detected in CP cells of both control and KO brains and, at E18.5, the level of Cdkn1c upregulation in Arx KO cortices was significantly diminished, inversely correlating with the Arx expression pattern at the same developmental stages (Supplementary Fig. 6). Similar results were also observed in Arx cKO cortices.
Figure 7.

The cyclin-dependent kinase inhibitor (CKi) Cdkn1c is ectopically expressed in Arx KO cortical VZ and SVZ and it is a direct transcriptional target of ARX. In situ hybridizations for Cdkn1c on telencephalic coronal sections at different embryonic stages (E12.5–E14.5) of control (left) and Arx KO (right) brain tissues; in situ hybridizations for Arx on control coronal sections at the corresponding developmental stages are also showed. (A–C) At E12.5 Cdkn1c transcript is detected in the neo-formed SVZ in control brains (arrowheads, A′); differently, in Arx KO, it is significantly upregulated in both VZ and SVZ (arrowheads, B′), where Arx is normally expressed at this stage (C). Similarly, Cdkn1c is found upregulated in Arx KO VZ and SVZ at E14.5 (arrowheads, E′) whereas in the relative control sections it is still barely detected in the SVZ (arrowheads, D′). (G) A 5-kb genomic region upstream to the murine Cdkn1c gene locus was screened for the presence of paired homeodomain binding sites: a palyndromic sequence 5′-ATTAGCATAAT-3′, highly conserved in mouse, rat, human, and orangutan, was found between −71/−81 bp before the Cdkn1c transcription start site (+1). (H) A <500 bp (−549/−68) genomic region containing the putative ARX binding site, a <750 bp (−1490/−20) containing some TA rich repeats and the same 500 bp (−549/−68) genomic region with the binding site mutagenized 5′-AGGAGCATCCT-3′ were separately cloned in a construct containing the Luciferase gene and tested in transcriptional activity assays in P19 cells. When the 500 bp (−549/−68)-Luc construct is cotransfected with pCAG-ArxIRESGFP a reduction in Luciferase expression compared control plasmid pCAG-IRESGFP cotransfection was appreciable (0.67 ± 0.04; n = 3, P < 0.005,); similar reduction was observed when the corepressor TLE1 was cotransfected (0.60 ± 0.07, n = 3); finally, higher reduction was detected when both ARX and TLE1 were cotransfected (0.42 ± 0.06, n = 3). Differently, ARX cotransfected with the 750 bp (−1490/−720)-Luc induced only a slight reduction in the basal transcriptional activity (0.94 ± 0.01, n = 3, P < 0.05,), even in combination with TLE1. When the binding site 5′-ATTAGCATAAT-3′ was mutagenized in 5′-AGGAGCATCCT-3′, no significant reduction in Luciferase expression was appreciable in cotransfection experiments. (I) Immunoprecipitation of E14.5 telencephalic chromatin with anti-ARX, anti-GFP, and IgG only. Primers spanning the 500 bp (−549/−68) genomic region upstream to the Cdkn1c, and Ebf3, Cdkn1b, and Cdkn1a regulative regions were used for PCR analysis of the immunoprecipitated DNA. Enrichment of the PCR products in anti-ARX-immunoprecipitated chromatin sample compared with the anti-GFP and IgG only samples was evaluated.
Unexpectedly, although Arx is strongly expressed in the ganglionic eminences, in particular at E12.5, E14.5, and E16.5 (Fig. 7C,F and Supplementary Fig. 6), its ablation in Arx KO mice does not induce Cdkn1c upregulation in this compartment (Fig. 7B,E and Supplementary Fig. 6).
In conclusion, our expression analysis of Cdkn1c in wild-type and Arx KO cerebral cortices throughout the neurogenic period, showed that this gene is upregulated in Arx KO cortical VZ and SVZ, with a pattern that parallels Arx expression in control tissues. This correlation raises the possibility that Arx depletion directly promotes Cdkn1c upregulation in cKO tissues.
Cdkn1c is a Direct Transcriptional Target of ARX
To determine if Cdkn1c is a direct transcriptional target of ARX, a 5-kb genomic region upstream to the murine Cdkn1c gene locus was screened for the presence of putative ARX binding sites. Previous studies identified palindromic sequences TAAT/ATTA spaced by 3 nucleotides (5′-ATTANNNTAAT-3′) as favored binding sites for paired homeodomain proteins (Wilson et al. 1993). Indeed, our search highlighted the presence of a unique palindromic sequence (5′-ATTAGCATAAT-3′) located between bases −71/−81 upstream of the Cdkn1c transcription start site (named +1) (Fig. 7G). As binding sites for transcriptional regulators are usually evolutionary conserved, we performed a multiple alignment of the aforementioned genomic region among different species, finding a high level of conservation in the ARX putative binding site in mouse, rat, human, and orangutan, except for the 3 spacing nucleotides that were not conserved in orangutan (Fig. 7G). A genomic region of about500 bp (−549/−68) containing the putative ARX binding site and another of about 750 bp (−1490/−720 containing some TA repeats, were separately cloned, upstream to a minimal promoter, in a construct containing the Luciferase gene and tested for transcriptional reporter activity (Fig. 7H). The 500 bp (−549/−68)-Luc and 750 bp (−1490/−720)-Luc constructs were separately cotransfected with control plasmid pCAG-IRESGFP or pCAGArx-IRESGFP in P19 cells and the effect of ARX expression on the basal transcriptional activity of the 2 putative regulative sequences was evaluated. ARX cotransfection reduced the basal transcriptional activity of the 500-bp regulative region by 30% (fold increase/decrease in transcriptional activity of pCAG-ArxIRESGFP coinfected with respect to pCAG-IRESGFP coinfected: 0.67 ± 0.04; n = 3, P < 0.005). However, higher reduction was detected when the corepressor Tle1 (McKenzie et al. 2007) was cotransfected in association with ARX (0.42 ± 0.06, n = 3, P < 0.005). Surprisingly, ARX and in particular TLE1 alone are able to repress luciferase transcription; this is due to the fact that both proteins are already expressed at low levels in P19 cells (data not shown). Conversely, when pCAG-ArxIRESGFP was cotransfected with the Luciferase construct regulated by 750 bp enhancer, only a slight reduction in the basal transcriptional activity was assessed (fold increase/decrease in transcriptional activity of pCAG-ArxIRESGFP coinfected with respect to pCAG-IRESGFP coinfected (transcriptional activity; 0.94 ± 0.01, n = 3, P < 0.05), even in combination with TLE1 (Fig. 7H). When the binding site 5′-ATTAGCATAAT-3′ was mutagenized to 5′-AGGAGCATCCT-3′, no significant reduction in Luciferase expression was appreciated in cotransfection experiments (Fig. 7H).
These results indicate that ARX exerts a specific transcriptional repression activity on this 500 bp (−549/−68) genomic region; in order to assess if this repression is direct or mediated by other factors expressed in P19 cells, we performed chromatin immunoprecipitation experiments. Anti-ARX antibody was used to immunoprecipitate chromatin isolated from E14.5 telencephali; then, PCR amplification reactions, with primers designed to span the 500 bp (−549/−68) region upstream to Cdkn1c gene, allowed us to evaluate an enrichment of that region in the ARX-immunoprecipitated chromatin with respect to IgG only and anti-GFP immunoprecipitated chromatin (Fig. 7I). Amplifications with primers spanning Ebf3 regulatory region were also performed and used as a positive control, as Ebf3 was previously showed to be a direct target of ARX in the ventral pallium (Fulp et al. 2008). We found enrichment of the Cdkn1c (and Ebf3) regulatory regions when pulled down with an anti-ARX antibody but not with the anti-GFP or no antibody controls. No enrichment in ARX-immunoprecipitated chromatin samples was detected amplifying for Cdkn1a and Cdkn1b regulative regions (Fig. 7I), as expected by the absence of any alteration of the 2 gene transcripts in Arx KO cortices. Taken together, these results suggest that ARX is a direct repressor of Cdkn1c transcription, and it likely exerts this role by direct interaction with the palindromic binding site 5′-ATTAGCATAAT-3′ located in the 500 bp enhancer region.
Discussion
Loss of ARX/Arx in both humans and mice results in a reduced size of the cerebral hemispheres. In this study, by crossing Arxfl/y mice with Emx1-Cre mice, we have confirmed that the reduced brain size is largely if not completely due to the loss of Arx in cortical progenitor cells. Indeed, in E14.5 Arx cKO, we observed a reduction in the number of cortical progenitor cells, both PAX6+ and TBR2+, that might be the result of the precocious RG progenitor differentiation observed at E12.5 in Arx cKO.
However, this cannot be the only underlying mechanism. In fact, the proliferation rate of cortical progenitor cells is also affected in Arx cKO and our data suggest that the IPC pool is more effected than the RGC compartment: most of S-phase progenitors in E14.5 Arx cKO cortices are PAX6+ cells and only a little percentage is TBR2+. Consistent with this, basal mitoses are dramatically reduced. Interestingly, the determination of the GF at E13.5 allowed us to understand that, whereas all the cKO RG progenitors were able to proliferate at that stage, only 45% of TBR2+ cells in Arx cKO could enter the cell cycle (vs. the 75% of the control). These observations support the hypothesis that the drastic effect on the IPC proliferation is not only due to a premature differentiation among VZ progenitors but essentially to a direct effect of ARX ablation on IPCs. Consistent with this, we found ARX expression in many TBR2+ cells of the VZ/SVZ.
The reduction of cortical progenitor cells led to an alteration of the cortical layering: a significant loss of cells from the superficial layers of Arx cKO cerebral cortex with relative preservation of the deeper layers. Our observations in this genetic model complement those found after an acute reduction of ARX through the in utero electroporation of an Arx shRNA in midgestation where an increased number of progenitors exiting the cell cycle and prematurely adopting the neuronal fate were found (Friocourt et al. 2008).
We also performed a gene expression profile analysis in wild-type and Arx KO cerebral cortices that identified 83 misregulated genes. Interestingly, this analysis found very little overlap with the results of the same analysis that we previously performed in the ganglionic eminences (Colasante et al. 2009; Fulp et al. 2008). These data reinforce the assertion that ARX exerts region-specific roles in the ventral telencephalon and in the cerebral cortex through the activation/repression of different transcriptional programs (Nasrallah et al. 2012). In agreement with the reduction of GABAergic interneurons migrating to the cerebral cortex in Arx KO brains (Colombo et al. 2007; Kitamura et al. 2002), markers of specific interneuron subclasses (NPY, Somatostatin and also LHX6) were downregulated in KOs. However, among the several misregulated genes, we focused our attention on Cdkn1c, as we considered it the most interesting ARX putative target, whose upregulation could account for the phenotype observed in Arx cKO cortices (Mairet-Coello et al. 2012).
Our characterization of the Cdkn1c expression pattern during normal corticogenesis found Cdkn1c to be expressed in the SVZ in the first phases of neurogenesis (E12.5–E14.5). At later stages it is also detected in the CP. Conversely, in Arx cKO, it is strongly upregulated in the VZ and SVZ starting at E12.5. The expression of Cdkn1c during corticogenesis has been debated for a long time. While early reports indicated that Cdkn1c mRNA is localized in the VZ, SVZ, and CP of embryonic rat cortex (Campagne and Gill 1998), CDKN1C protein has been localized in the CP, with faint expression in the IZ and no signal in the VZ/SVZ (Itoh et al. 2007; Nguyen et al. 2006). A few recent studies have helped clarify this issue (Mairet-Coello et al. 2012; Nguyen et al. 2006; Tury et al. 2011, 2012). Using an antibody against CDKN1C, they found it is expressed mainly in differentiating postmitotic neurons, but also it colocalizes with TBR2+ cells in SVZ and with some PAX6+ in VZ. These data are fully consistent with our data on Cdkn1c expression obtained by in situ hybridizations.
Recently, Tury et al. (2011) showed that CDKN1C overexpression in E14.5–15.5 rat embryos elicits precursor cell cycle exit and promotes transition from proliferation to neuronal differentiation. Comparing the neurogenic and astrogliogenic effects of CDKN1B and CDKN1C, they discovered that CDKN1C overexpression promotes more neuronal differentiation than CDKN1B. The specific neurogenic effect of CDKN1C is particularly interesting in relation to its expression in IPCs, which, distinct from RGCs, generate only neurons. However, they do not clearly analyze the specific effect of CDKN1C overexpression on the 2 cortical progenitor cell populations. In their follow-up work, Mairet-Coello et al. (2012), analyze the phenotype of Cdkn1c mutant mice, finding that proliferation of both RGCs and IPCs is increased, expanding both precursors with greater impact on IPCs.
In this context, our report is the first to describe the effect of CDKN1C overexpression on cortical progenitor cell proliferation, integrating the analysis previously performed. Interestingly, our data also support the hypothesis that CDKN1C is more relevant for IPC than RGC cell cycle exit, although CDKN1C is overexpressed in both cortical VZ and SVZ, the most dramatic effect is observed on IPC and not on RGC proliferation.
We propose a model for the ARX-dependent control of cortical progenitor cell proliferation (Fig. 8). ARX expression in the VZ and partially in the SVZ is able to repress Cdkn1c premature expression, allowing proliferation of RG cells and expansion of IPCs. When Arx is normally downregulated, the consequent upregulation of CDKN1C in the SVZ favors IP cell cycle exit. In contrast, in Arx KO cortex, CDKN1C expression is high in the VZ resulting in a mild proliferation effect on RG cells. More dramatic is the impact on TBR2+ IPCs; these progenitors have already such a high level of CDKN1C that directly exit the cell cycle without significant cell division.
Figure 8.
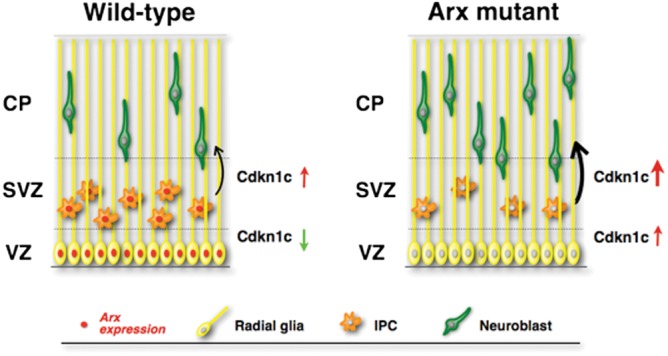
Schematization of the molecular mechanism through which ARX controls cortical progenitor expansion. ARX expression in the cortical VZ/SVZ normally represses CDKN1C expression allowing progenitor cell proliferation and expansion. Loss of Arx in the VZ/SVZ causes precocious CDKN1C upregulation in the cortical progenitors cells, which results in a mild decrease in proliferation of RGCs and a significant loss of IPC proliferation, with consequent cell cycle exit. This ultimately results in a loss of upper layer neurons in the adult cortex.
Certainly, the presence of other alterations or compensatory mechanisms cannot be excluded in Arx cKO cortices. For example, the microarray analysis highlighted the upregulation of Id1 and Id2 genes in Arx KO cortices. It is known that their targeted disruption in mice results in premature withdrawal of progenitor cells from the cell cycle and expression of neural-specific differentiation markers (Lyden et al. 1999). Thus, it is likely that their overexpression in Arx cKO cortices might compensate for the Cdkn1c upregulation, mitigating the full phenotypic expression. Among the compensatory mechanisms, we must also consider the slight acceleration of cell cycle duration in IPCs observed in Arx cKO contributing to the observed phenotype.
We also demonstrated that Cdkn1c is a direct transcriptional target of ARX. The genomic region 5′ to the murine Cdkn1c gene locus was screened for the presence of the ARX putative binding site and a unique palindromic sequence (5′-ATTAGCATAAT-3′) was located a few bases upstream of the Cdkn1c transcription start site. Luciferase assays allowed us to demonstrate that ARX, together with the corepressor TLE1, is able to repress transcription through binding at the identified 500 bp enhancer region. Furthermore, chromatin immunoprecipation revealed that ARX directly binds in this 500 bp Cdkn1c enhancer region.
Surprisingly, the ARX binding site that we identified in this study is different from the one described in our previous work (Fulp et al. 2008). In that work, we analyzed the transcription factor binding sites significantly enriched in the regulatory regions of genes whose expression increased within Arx KO subpallium, we identified the sequence TAATTA. In a recent study using the ChIP-chip methodology in ARX transfected N2a cells almost 50% of Arx-immunoprecipitated sequences N2a cells were unequivocally enriched for TAATTA binding site in comparison to controls (Quille et al. 2011). In this same study, only 20% of the genes ARX-immunoprecipitated in E15.5 mouse embryonic brains were found to have this binding site; furthermore, any other motif enriched in ARX-bound sequences by comparison to control sequences, could not be identified (Quille et al. 2011). These results suggest that whereas overexpressed ARX seems to be recruited primarily to TAATTA, in more physiological conditions such as in embryonic brain, ARX is recruited to other less common motifs or needs other factors for DNA binding. Based on these data, the ATTAGCATAAT site we identified might be one of the in vivo binding sites to which ARX is recruited. Surprisingly, the ChIP on chip analysis performed by Quillé and coworkers revealed that ARX binds the Cdkn1a (p21/Cip1) promoter but not Cdkn1c; however, this might be due to limitations of the ChIP-chip technique. Indeed, ChIP-seq experiments might allow further delineation of ARX direct transcriptional target genes.
In summary, our data indicate ARX plays a unique role in the progenitor cells of the cerebral cortex when compared with its role in the ventral forebrain. By genetically eliminating Arx from pallial progenitor cells, we find a reduction in the number of RGC due to a premature exit from the cell cycle and significant skipping of the IPC amplification stage of corticogenesis. This defect results in the predicted deficit in upper layer neurons of the cerebral cortex. We further elucidated the pathogenesis of this defect by identifying the cell cycle regulator CDKN1C as a downstream target of ARX. Our study extends our understanding of the aberrant molecular network responsible for the reduced proliferation observed in Arx cKO forebrains; indeed, this is a critical point for understanding the pathogenic mechanisms underlying these devastating neurologic disorders and in devising genetic or cellular therapeutic strategies to ameliorate Arx mutation-induced pathological phenotypes.
Supplementary Material
Supplementary Material can be found at http://www.cercor.oxfordjournals.org/ online.
Funding
This work was supported by following grants: Telethon, the NIH NS46616 and HD26979 and the Neurodevelopmental Disabilities Training Grant T32NS007413.
Supplementary Material
Notes
We thank K. Kitamura for the anti-ARX antibody and L. Muzio for helpful discussion. The anti-PAX6 antibody developed by A. Kawakami was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242, USA. Finally, we also thank George Clement for his assistance with animal husbandry and genotyping. Conflict of Interest: None declared.
References
- Arnold SJ, Huang GJ, Cheung AF, Era T, Nishikawa S, Bikoff EK, Molnar Z, Robertson EJ, Groszer M. The T-box transcription factor eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 2008;22:2479–2484. doi: 10.1101/gad.475408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagne MVL, Gill R. Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21Waf1/Cip1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the proto-oncogene bax. J Comp Neurol. 1998;397:181–198. doi: 10.1002/(sici)1096-9861(19980727)397:2<181::aid-cne3>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Colasante G, Collombat P, Raimondi V, Bonanomi D, Ferrai C, Maira M, Yoshikawa K, Mansouri A, Valtorta F, Rubenstein JL, et al. Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J Neurosci. 2008;28:10674–10686. doi: 10.1523/JNEUROSCI.1283-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colasante G, Sessa A, Crispi S, Calogero R, Mansouri A, Collombat P, Broccoli V. Arx acts as a regional key selector gene in the ventral telencephalon mainly through its transcriptional repression activity. Dev Biol. 2009;334:59–71. doi: 10.1016/j.ydbio.2009.07.014. [DOI] [PubMed] [Google Scholar]
- Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo E, Collombat P, Colasante G, Bianchi M, Long J, Mansouri A, Rubenstein JL, Broccoli V. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;27:4786–4798. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo E, Galli R, Cossu G, Gecz J, Broccoli V. Mouse orthologue of ARX, a gene mutated in several X-linked forms of mental retardation and epilepsy, is a marker of adult neural stem cells and forebrain GABAergic neurons. Dev Dyn. 2004;231:631–639. doi: 10.1002/dvdy.20164. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrai C, Munari D, Luraghi P, Pecciarini L, Cangi MG, Doglioni C, Blasi F, Crippa MP. A transcription-dependent micrococcal nuclease-resistant fragment of the urokinase-type plasminogen activator promoter interacts with the enhancer. J Biol Chem. 2007;282:12537–12546. doi: 10.1074/jbc.M700867200. [DOI] [PubMed] [Google Scholar]
- Friocourt G, Kanatani S, Tabata H, Yozu M, Takahashi T, Antypa M, Raguénès O, Chelly J, Férec C, Nakajima K, et al. Cell-autonomous roles of ARX in cell proliferation and neuronal migration during corticogenesis. J Neurosci. 2008;28(22):5794–5805. doi: 10.1523/JNEUROSCI.1067-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friocourt G, Poirier K, Rakic S, Parnavelas JG, Chelly J. The role of ARX in cortical development. Eur J Neurosci. 2006;23:869–876. doi: 10.1111/j.1460-9568.2006.04629.x. [DOI] [PubMed] [Google Scholar]
- Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet. 2008;17:3740–3760. doi: 10.1093/hmg/ddn271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gecz J, Cloosterman D, Partington M. ARX: a gene for all seasons. Curr Opin Genet Dev. 2006;16:308–316. doi: 10.1016/j.gde.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Masuyama N, Nakayama K, Nakayama KI, Gotoh Y. The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J Biol Chem. 2007;282:390–396. doi: 10.1074/jbc.M609944200. [DOI] [PubMed] [Google Scholar]
- Jin XL, Guo H, Mao C, Atkins N, Wang H, Avasthi PP, Tu YT, Li Y. Emx1-specific expression of foreign genes using “knock-in: approach. Biochem Biophys Res Commun. 2000;270:978–982. doi: 10.1006/bbrc.2000.2532. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- Kwon GS, Hadjantonakis AK. Eomes::GFP-a tool for live imaging cells of the trophoblast, primitive streak, and telencephalon in the mouse embryo. Genesis. 2007;45:208–217. doi: 10.1002/dvg.20293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O'Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- Mairet-Coello G, Tury A, Van Buskirk E, Robinson K, Genestine M, Dicicco-Bloom E. p57KIP2 regulates radial glia and intermediate precursor cell cycle dynamics and lower layer neurogenesis in developing cerebral cortex. Development. 2012;139:475–487. doi: 10.1242/dev.067314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie O, Ponte I, Mangelsdorf M, Finnis M, Colasante G, Shoubridge C, Stifani S, Gecz J, Broccoli V. Aristaless-related homeobox gene, the gene responsible for West syndrome and related disorders, is a Groucho/transducin-like enhancer of split dependent transcriptional repressor. Neuroscience. 2007;146:236–247. doi: 10.1016/j.neuroscience.2007.01.038. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Muzio L, DiBenedetto B, Stoykova A, Boncinelli E, Gruss P, Mallamaci A. Conversion of cerebral cortex into basal ganglia in Emx2(-/-) Pax6(Sey/Sey) double-mutant mice. Nat Neurosci. 2002;5:737–745. doi: 10.1038/nn892. [DOI] [PubMed] [Google Scholar]
- Nasrallah MP, Cho G, Simonet JC, Putt ME, Kitamura K, Golden JA. Differential effects of a polyalanine tract expansion in Arx on neural development and gene expression. Hum Mol Genet. 2012;21:1090–1098. doi: 10.1093/hmg/ddr538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts JM, Guillemot F. P27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Quille ML, Carat S, Quemener-Redon S, Hirchaud E, Baron D, Benech C, Guihot J, Placet M, Mignen O, Ferec C, et al. High-throughput analysis of promoter occupancy reveals new targets for Arx, a gene mutated in mental retardation and interneuronopathies. PloS One. 2011;6:e25181. doi: 10.1371/journal.pone.0025181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Sessa A, Mao CA, Hadjantonakis AK, Klein WH, Broccoli V. Tbr2 directs conversion of radial glia into basal precursors and guides neuronal amplification by indirect neurogenesis in the developing neocortex. Neuron. 2008;60:56–69. doi: 10.1016/j.neuron.2008.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK Inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995;15:6046–6057. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarabykin V, Stoykova A, Usman N, Gruss P. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development. 2001;128:1983–1993. doi: 10.1242/dev.128.11.1983. [DOI] [PubMed] [Google Scholar]
- Tury A, Mairet-Coello G, DiCicco-Bloom E. The cyclin-dependent kinase inhibitor p57Kip2 regulates cell cycle exit, differentiation, and migration of embryonic cerebral cortical precursors. Cereb Cortex. 2011;21:1840–1856. doi: 10.1093/cercor/bhq254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tury A, Mairet-Coello G, DiCicco-Bloom E. The multiple roles of the cyclin-dependent kinase inhibitory protein p57(KIP2) in cerebral cortical neurogenesis. Dev Neurobiol. 2012;72:821–842. doi: 10.1002/dneu.20999. [DOI] [PubMed] [Google Scholar]
- Wilson D, Sheng G, Lecuit T, Dostatni N, Desplan C. Cooperative dimerization of paired class homeo domains on DNA. Genes Dev. 1993;7:2120–2134. doi: 10.1101/gad.7.11.2120. [DOI] [PubMed] [Google Scholar]
- Zimmer C, Tiveron MC, Bodmer R, Cremer H. Dynamics of Cux2 expression suggests that an early pool of SVZ precursors is fated to become upper cortical layer neurons. Cereb Cortex. 2004;14:1408–1420. doi: 10.1093/cercor/bhh102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.