

Abstract
Lysosomal storage disorders (LSD) are a group of heterogeneous diseases caused by compromised enzyme function leading to multiple organ failure. Therapeutic approaches involve enzyme replacement (ERT), which is effective for a substantial fraction of patients. However, there are still concerns about a number of issues including tissue penetrance, generation of host antibodies against the therapeutic enzyme, and financial aspects, which render this therapy suboptimal for many cases. Treatment with pharmacological chaperones (PC) was recognized as a possible alternative to ERT, because a great number of mutations do not completely abolish enzyme function, but rather trigger degradation in the endoplasmic reticulum. The theory behind PC is that they can stabilize enzymes with remaining function, avoid degradation and thereby ameliorate disease symptoms. We tested several compounds in order to identify novel small molecules that prevent premature degradation of the mutant lysosomal enzymes α-galactosidase A (for Fabry disease (FD)) and acid α-glucosidase (GAA) (for Pompe disease (PD)). We discovered that the expectorant Ambroxol when used in conjunction with known PC resulted in a significant enhancement of mutant α-galactosidase A and GAA activities. Rosiglitazone was effective on α-galactosidase A either as a monotherapy or when administered in combination with the PC 1-deoxygalactonojirimycin. We therefore propose both drugs as potential enhancers of pharmacological chaperones in FD and PD to improve current treatment strategies.
Introduction
Lysosomes contain acid hydrolase enzymes, which are involved in the degradation and recycling of macromolecules. For example, the enzymes α-galactosidase A (GLA, α-Gal A, NM_000169.2, EC 3.2.1.22) and acid α-glucosidase (GAA, acid maltase, NM_000152.3, EC 3.2.1.3) belong to the family of exoglycosidases that catalyze the cleavage of terminal α-D-galactoside and α-D-glycoside residues respectively.1,2 Mutations within the genes encoding lysosomal acid hydrolases lead to accumulation of the corresponding substrates3 with subsequent development of phenotypically distinct diseases described by the umbrella term “lysosomal storage disorders” (LSDs).
Fabry disease (FD, OMIM #301500), an X-linked lysosomal storage disorder causing the accumulation of glycosphingolipids (mainly globotriaosylceramides), classically presents with angiokeratoma, chronic pain, major pain crisis, anhidrosis, and gastrointestinal problems in childhood or adolescence with a progressive course.4 Pompe disease (PD, OMIM #232300) is an autosomal recessive neuromuscular disorder typically fatal during the first 12 months of life due to respiratory insufficiency.5,6 Despite different clinical presentations, both diseases share some important analogies. The two lysosomal enzymes involved belong to the same GH-D clan of the O-Glycosyl hydrolase group of the glycosyl hydrolases superfamily possessing an α/β8 barrel fold in the domain containing the active site and a comparable catalytic mechanism (retaining aspartate acts as catalytic nucleophile) (www.cazy.org). Certain mutations in FD and PD are associated with a milder disease course, characterized by later onset and slower progression of symptoms.7,8,9 These “mild” genotypes are missense mutations that disrupt the structure and stability of the lysosomal enzyme, resulting in misfolding, premature degradation, and failure to reach the target organelle.10,11,12 This leads to loss of specific lysosomal hydrolytic activity. To date, the number of reported missense mutations associated with both diseases is high, ranging from >200 in Pompe to >400 in FD (HGMD Professional 2013.2, fabry-database).
Enzyme replacement therapy (ERT) based on the intravenous administration of human enzyme (Replagal, Shire Human Genetic Therapies; Fabrazyme, Lumizyme, Genzyme) is available for each disease. The effectiveness of ERT relies on mannose 6-phosphate residues being recognized by their widely distributed receptors in the plasma membranes of cells.13 One main shortcoming of ERT is limited tissue penetrance.14 Central nervous system manifestations such as cerebrovascular complications and neuropathic pain in FD cannot be addressed due to the blood–brain barrier, which is not penetrated by ERT. Another shortcoming is the risk of an immune response with potentially neutralizing antibodies generated against the therapeutic enzymes.15,16,17 This might partly explain differences in ERT efficacy between individuals observed in long-term safety studies. A positive correlation has been noted between the deleterious effect of a mutation and the titer of crossreactive immunological material;18 so, patients with damaging missense mutations are also at risk of developing antibodies, which can compromise the efficacy of ERT.19
A new strategy in the treatment of LSDs is the use of small molecules known as pharmacological chaperones (PC) to enhance lysosomal activity by binding to and stabilizing the mutant enzyme.9,11,12,20,21,22,23,24 The prerequisite for this treatment approach, therefore, is the presence of a misfolded enzyme, which is still capable of functioning. The PC binds to the mutant enzyme, corrects protein folding, and recovers its lysosomal activity. A large number of mutations are potential candidates for PC therapy, although this is not yet clinically approved. For example, about half of all mutations described in FD are missense mutations and among those, about 50% respond in vitro to the PC galactose analog 1-deoxygalactonojirimycine (DGJ, Migalastat hydrochloride).9,25,26 A recently published study revealed that 26 PD mutations responded to the PC glucose analogue 1-deoxynojirimycine (DNJ).27 In Gaucher disease, the potent PC Ambroxol (ABX), exists to treat mutant enzymes resulting from the common missense mutations p.N370S and p.L444P, which together account for about two thirds of cases worldwide.28,29 ABX was investigated in the current study as a potential PC for both FD and PD. The reason behind this approach is that lysosomal hydrolases share common structural and functional features. PC display little selectivity for their target due to promiscuity within the glycosidase enzyme family and therefore in theory may bind to various different lysosomal hydrolases. For instance, the imino sugar N-butyl-deoxynojirimycin (NB-DNJ) was shown to be a PC for both PD and Gaucher disease.23,24,30 In another example, DGJ potently inhibits α-Gal A and α-N-acetylgalactosaminidase.31
It has already been established that PC correct misfolding, stabilize protein structure, and prevent rejection in the quality control system of the endoplasmic reticulum (ER), thereby avoiding premature proteasomal degradation and facilitating transport to the lysosome. Another treatment approach used in Gaucher and Niemann-Pick type C disease, both aim to bypass early enzyme degradation in the ER by either upregulating molecular chaperones or inhibiting the ubiquitin-proteasome-system.32,33,34 We therefore systematically investigated a broad range of small molecules for their ability to avoid premature enzyme degradation by either of these two mechanisms, e.g., Tunicamycin, MG-132, Rosiglitazone (RSG), etc.
The most effective small molecules in enhancing mutant lysosomal enzyme function in our cell culture-based system were found to be Ambroxol, RSG/Pioglitazone, and Bezafibrate. The fact that Ambroxol has been shown to be effective in increasing activity of mutant enzymes in both FD (the current study) and GD (previous study) suggests that one single compound could potentially be used in the treatment of different LSDs.
Results
A screening system for mutant glycosylase enhancement
The purpose of this study was to produce different mutant forms of the lysosomal hydrolase α-galactosidase A (α-Gal A) to investigate their response to small molecules with a view to (i) elucidating cellular pathways that can potentially be modulated in order to increase mutant enzyme activity and (ii) to identify potential compounds for the treatment of LSD. Substances with distinct biological and biochemical functions were investigated in an in vitro model for FD. HEK-293H cells were cultured and transfected with various mutant GLA cDNAs to produce α-Gal A with defects in folding but residual enzyme activity. These α-Gal A mutants were previously shown to be responsive to the pharmacological chaperone DGJ, which was used as an indicator of the capacity of the enzymes to gain functional recovery (Supplementary Figure S1). From the 32 mutations depicted in Supplementary Figure S1, a set of nine mutations was selected for further testing based on (i) residual activity (>1 % of wild type) and (ii) DGJ responsiveness (>1.5-fold increase, overall >5% of wild type), as established in an earlier article.9
The first candidate substance: ambroxol, a pharmacological chaperone effective in Gaucher disease
For FD, mutant misfolded α-Gal A enzymes were tested with Ambroxol (ABX), a recently identified PC for Gaucher disease. Several of the mutant α-Gal A enzymes appeared to show slightly elevated function after administration of 40 µmol/l ABX to the cell-culture medium, but a significant effect was only seen for wild-type α-Gal A and two specific mutants p.A156V and p.R301Q (Figure 1a). The concentration–response relationship was recorded for the wild-type enzyme (Figure 1b). ABX was effective at a concentration range of 10–60 µmol/l while displaying a decline to about 40% of the maximal effect at 120 µmol/l ABX; we used sigmoidal curve fit and calculated an EC50 of 17.4 µmol/l. The drop in activity detected at concentrations >80 µmol/l could actually be caused by a harmful effect on the cultured cells that has formerly been reported for ABX28 rather than a specific inhibitory effect of the compound on the enzyme. A concentration–response curve was recorded for one mutant (p.A156V), resulting in a similar EC50 to that of the wild-type, of 13.0 µmol/l (Supplementary Figure S2). In the following experiments, ABX was also used at 40 µmol/l, which represents approximately twice EC50. The mutations from Figure 1a were tested using a combination of 20 µmol/l DGJ and 40 µmol/l ABX, which resulted in increased enzyme activity for all nine mutations tested (p.E59K, p.A73V, p.A143T, p.A156V, p.I232T, p.R301G, p.R301Q, p.R356W, and p.R363H) when compared to treatment with DGJ alone (Figure 1c). The mutants p.A73V, p.I232T, and p.R363H attained close to normal enzyme activity, while the mutants p.A143T and p.R301Q exceeded 50% activity thereby crossing the estimated threshold for the normal range.35 This increase in activity was associated with a parallel increase in the level of α-Gal A protein in the cells (Figure 1d). The stronger α-Gal A signals suggest a potential stabilizing effect of the double treatment and/or enhanced transport into the lysosome. In summary, double treatment with DGJ and ABX resulted in increased enzyme activity for all mutations tested. This in turn prompted a similar double-treatment study using galactose and ABX (galactose, like DGJ, is also a PC in FD). A subset of six mutations responded with an elevated α-Gal A activity using this particular double treatment (Supplementary Figure S3).
Figure 1.
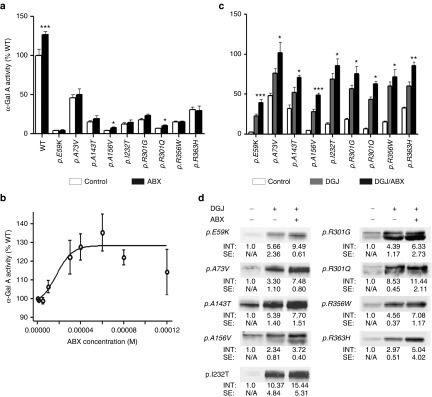
Effect of ABX on overexpressed mutant forms of α-Gal A in HEK-293H cells. ABX was administered 6 hours after transfection of the GLA cDNA-containing plasmids and then cultured for 60 hours changing the media any other day adding fresh treatment as described in the Materials and Methods section (a) Bona fide analysis with 40 µmol/l ABX revealed a tendency to mildly increase intracellular activities of several mutant α-Gal A forms that was significant for p.A156V and p.R301Q. Wild-type enzyme increased markedly upon the addition of the compound as well. (b) Concentration–response relation analysis showed increasing wild-type α-Gal A activity. An EC50 of 17.4 µmol/l was calculated by a nonlinear regression analysis A maximum stimulatory effect was obtained at 60 µmol/l. At concentrations ≥80 µmol/l, the α-Gal A activity dropped back to normal. (c) In the same culture system as under (a), the GLA expressing HEK-293H cells were DGJ or DGJ/ABX combination-treated. The DGJ-responsive α-Gal A forms (see also Supplementary Figure S1) displayed considerable gains from the additional administration of ABX. (d) Western blot of the mutant α-Gal A forms indicated higher levels of intracellular enzyme after treatment with DGJ in combination with 40 µmol/l ABX compared to the monotherapy. Western blots were repeated at least three times. In each lane 30 µg of total protein was loaded and separated by SDS-PAGE. Semiquantitative analysis was carried out using the Odyssey software v1.2. Calculated intensities were normalized for GAPDH internal loading control (not shown). The average intensities (“INT”) are given as fold change ± standard error. Enzyme activity values are shown as mean ± SEM (n ≥ 5). Results were considered significant if *P < 0.05, **P < 0.01, ***P < 0.005. Control treatment denotes the respective carrier solvent used for the compounds (DGJ was diluted in hypure H2O as a 10 mmol/l stock solution, ABX was typically diluted in DMSO (100 mmol/l).
Ambroxol stabilizes α-Gal A in combination with DGJ in vitro
We wanted to test the previous suggestion that the DGJ/ABX double treatment stabilized the enzyme. We therefore carried out a thermal denaturation test in a cell-free environment using human α-galactosidase A produced in a human fibroblast cell line (agalsidase alfa, Shire Human Genetic Therapies, Berlin, Germany), to examine the hypothesis that ABX could act via pharmacological chaperoning (i.e., bind to the enzyme and in turn lead to stabilization). Briefly, the method involves incubation of agalsidase alfa at 51 °C for 60 minutes in a 96-well plate with or without the respective additive (DGJ, ABX or a combination of both) as previously described.26 A control plate with the same sample set up was kept on ice. Enzyme activity was measured with the artificial substrate 4-methylumbelliferyl-α-D-galactopyranoside (4-MUG). The thermal incubation of mock-treated (DMSO) α-Gal A at 51 °C led to a decreased active enzyme fraction of about 29.9% compared to control incubation on ice (Figure 2). Increasing DGJ concentrations attenuated α-Gal A thermal denaturation (restoring up to 63.4% of normal activity at 2.5 µmol/l) whereas increasing concentrations of ABX led to an accelerated loss of enzyme activity (reducing to 18.8% of normal activity at 2.5 mmol/l). Coadministration of DGJ and 2.5 mmol/l ABX led to a further stabilization compared to DGJ alone. The conclusion drawn is that following heat denaturation, ABX alone does not preserve enzyme activity but when used with DGJ has a synergistic positive effect.
Figure 2.

Thermal denaturation of α-Gal A. Replagal was mixed with increasing amount of compound. The mixture was incubated at 51 °C for 60 minutes and the enzymatic reaction was started upon the addition of substrate directly thereafter. The decrease of activity was compared to a reference sample kept on ice for 60 minutes before the enzymatic assay. The curve obtained for DGJ showed attenuated denaturation heat-treated enzyme in a concentration-dependent manner (light-grey graph) and significant stabilization at 500 nmol/l onwards. ABX showed no change compared to the untreated state at concentrations up to 200 µmol/l (dark–grey graph). In millimolar concentrations ABX accelerated the denaturation resulting in lowered enzyme activity compared to the untreated enzyme. In combination with a constant addition of 2.5 mmol/l ABX, the DGJ treatment led to enhanced stabilization of the enzyme at higher DGJ concentrations. At 2.5 µmol/l DGJ, the enzyme was more effectively stabilized under combination compared to monotherapy. Data was obtained from at least five independent experiments, each experiment included duplicate measurements. Values are shown as mean ± SEM (n ≥ 5). Results were considered significant if *P < 0.05, **P < 0.01, ***P < 0.005.
Can the effect of ABX be applied mutatis mutandis to other LSD cell culture models?
In a similar heterologous expression system as described for GLA mutations in FD, ABX was combined with a PC to analyze the effect on mutant GAA in PD. Mutations with a known ability to respond to PC treatment were investigated in this study.23,24 For example, p.Y455F, p.P545L, and p.L552P showed a significant benefit from the 60-hour N-butyl-deoxynojirimycine (NB-DNJ) treatment, whereas no effect was achieved with the ABX (Figure 3, upper part). Surprisingly, mutant p.L552P showed a significant benefit from the combination of NB-DNJ and ABX indicated by an increase of activity from 6.9 to 15.3% of wild type, compared to monotherapy with NB-DNJ. The same did not hold true for p.Y455F and p.P545L. The effect of another pharmacological chaperone DNJ, was also triggered by the addition of ABX, increasing activity of the mutant p.L552P from 11.4 to 25.1% (Figure 3, lower part), which corresponds to the ratio observed with NB-DNJ (2.2-fold increase). A significant improvement in enzyme activity using a combination of DNJ and ABX was also seen for mutants p.Y455F (1.6-fold) and p.P545L (2.3-fold). Apparently, in the case of the GAA enzyme, the success of combined administration with ABX strongly depended on the chaperone used and the type of mutation. The combination DNJ/ABX was efficient on all tested mutations except p.Y575S.
Figure 3.

Acid α-glucosidase (GAA) activity in HEK-293H cells expressing mutant forms of the enzyme treated with a pharmacological chaperone (NB-DNJ or DNJ) and a combination consisting of NB-DNJ/ABX and DNJ/ABX. Upper: PC-responsive mutations of GAA were treated with 40 µmol/l ABX, 20 µmol/l NB-DNJ and a combination of 20 µmol/l NB-DNJ and 40 µmol/l ABX. The monotreatment with ABX did not beneficially influence mutant GAA activity. The NB-DNJ was effective on p.Y455F, p.P545L, and p.L552P, but not p.Y575S. The mutant p.L552P was amenable to the double treatment whereas p.Y455F and p.P545L did not display synergistic effects from the combination of the PC with ABX. Lower: In the same set of mutations, DNJ provoked a similar response compared to NB-DNJ. In combination with ABX, the mutations (except for p.Y575S) showed a siginificant synergistic effect with the imino sugar compared to the monotherapy. Values are shown as mean ± SEM (n ≥ 3). Results were considered significant if *P < 0.05, **P < 0.01, ***P < 0.005.
RSG, a known inhibitor of the ubiquitin-proteasome-system, acts as enhancer of intracellular mutant α-Gal A activity
ER stress inducers and ubiquitin-proteasome-system inhibitors have both been proposed as potential drugs in several LSDs.32,33,34 Both strategies aim to modulate cellular proteostasis in order to beneficially influence enzyme folding and subsequently deliver mutant enzymes to the lysosome. We treated mutant α-Gal A (p.R301Q), overexpressed in HEK-293H cells, in the presence or absence of DGJ, with different ER stress inducing agents: Kifunensine (an α-mannosidase inhibitor), Thapsigargin (an ER Calcium releaser and reuptake inhibitor), and Tunicamycin (an inhibitor of N-glycosidic linkage formation). None of these three agents produced a positive effect on cellular enzyme activity (Supplementary Figure S4). Thapsigargin even had a negative effect on α-Gal A activity in a concentration-dependent manner and at concentrations >50 nmol/l, it appeared to be toxic to the cells. MG-132, an inhibitor of proteasomal activity, showed a modest, but not statistically significant increase on p.R301Q enzyme activity at 50 nmol/l (1.2-fold) (Supplementary Figure S4). The effect was also detected when MG-132 was administered in combination with 20 µmol/l DGJ leading to a 1.1-fold increase over DGJ monotherapy (from 8.6- to 9.5-fold of the untreated control). Other inhibitors of proteasomal activity, Lactacystin, Bortezomib, and Ritonavir, were also tested, but did not show an effect on mutant α-Gal A function (data not shown). Supplementary Table S1 shows the effect on the distinct proteasomal activities (Chymotrypsin-, Trypsin-, and Caspase-like activity) in the cell system used at different concentrations. Remarkably, MG-132 had no inhibitory effect on the proteasome when applied at a concentration of 100 nmol/l. Only at a concentration of 1 µmol/l, an inhibitory effect on all three proteasomal activities was noticeable. However, due to the cytotoxic effect of MG-132, it was not feasible to apply concentrations above 100 nmol/l over long periods (such as 60 hours) in the present cell system. A short-term treatment with 1 µmol/l MG-132 for 12 hours preceding cell harvest did not significantly change α-Gal A activity either (data not shown).
Contrary to the findings for MG-132, a major response to RSG, a Peroxisome proliferator–activated receptor-γ (PPAR-γ) agonist reported to inhibit global cellular ubiquitination,36 was observed for p.R301Q. The administration of 50–140 µmol/l RSG led to approximately threefold increase of p.R301Q activity. RSG in combination with DGJ was even more effective in enhancing activity of the mutant p.R301Q enzyme (Figure 4). Therefore, other mutant α-Gal A forms, which had previously been classified as DGJ nonresponders were also tested for responsiveness (compare Supplementary Figure S1). Mutant α-Gal A forms p.R118C, p.A156V, p.R301Q, and p.T385A were exposed to 20 µmol/l DGJ, 100 µmol/l RSG, and a combination of 20 µmol/l DGJ and 100 µmol/l RSG (Figure 5a). The DGJ nonresponding mutants p.R118C and p.T385A showed a 1.2- and 1.1-fold increase in activity when DGJ was administered, which increased to a 1.4- and 1.3-fold increase when RSG was added, respectively. According to the established responder criteria, both drugs failed to increase the enzyme activity >1.5-fold. However, taking into account the fact that p.R118C and p.T385A retain intrinsic activities of 22.9% and 47.0% of wild type respectively, both mutations can be considered as RSG responders, because activity was increased by over 5% up to 30.7 and 62.5%, respectively. The efficiency of the combinational treatment exceeded the monotherapy. p.A156V and p.R301Q are strong DGJ responders and the addition of RSG boosted the monotherapy by 1.8-fold (p.R301Q) and 1.3-fold (p.A156V). Pioglitazone is a structural and functional analogue of RSG. The combination of DGJ/Pioglitazone was equally effective as DGJ/RSG on p.R301Q activity.
Figure 4.

Mutant p.R301Q α-Gal A activity under treatment with Rosiglitazone. The GLA mutation p.R301Q was overexpressed in HEK-293H cells and treated with the known ubiquitination modifying agent RSG in the presence or absence of 20 µmol/l DGJ. Significant enzyme activity enhancement was gained at concentrations ≥50 µmol/l. In a therapeutic margin of 50–140 µmol/l RSG proved to be an effective enhancer of mutant α-Gal A activity.
Figure 5.

Mutant α-Gal A forms respond to PPAR-γ agonist drugs. HEK-293H cells were transfected and treated as described in Materials and Methods (“Transient GLA/GAA expression in HEK-293H cells”). (a) The administration of 100 µmol/l RSG increased the enzyme activity of mutant forms of α-Gal A. In the case of p.A156V and p.R301Q the DGJ treatment is an efficacious single treatment, but the combination with RSG leads to a further increase of activity. A structural and functional analogue of RSG, Pioglitazone (Pio), administered at 100 µmol/l is similarly effective on p.R301Q in combination with DGJ. RSG had a distinct effect on mutants p.R118C and p.T385A that did not respond to DGJ (see Supplementary Figure S1). The combination of both drugs was most effective. Values are shown as mean ± SEM (n ≥ 3). Students T-test was used to investigate significance of the combined treatment compared to control and to DGJ monotherapy. Results were considered significant if *P < 0.05, **P < 0.01, ***P < 0.005. (b) Western blot analysis of different mutant α-Gal A forms under DGJ, RSG and DGJ/RSG combination treatment. In accordance with (a) the mutant enzyme forms displayed best responsiveness towards the combination of pharmacological chaperone DGJ and RSG. Experiments were repeated at least 3 times for reliability. Western blot analysis was taken out essentially as described under Fig. 1d. (c) E1 ubiquitin activating enzyme inhibitor Pyr-41 and PPAR-α/γ agonist Bezafibrate were tested on overexpressed p.R301Q mutant in HEK-293H cells. No change of α-Gal A activity was observed for Pyr-41 over a concentration range of 0.1–40 µmol/l. By contrast, treatment with Bezafibrate led to an increase from 8.4- to 12.7-fold and from 1.0 to 2.3 with or without additional administration of DGJ, respectively.
The corresponding effect on the protein level is shown in Figure 5b. As shown before, the mutant enzyme forms responded to the administration of 20 µmol/l DGJ, 100 µmol/l RSG, and the combination of both. In the case of p.R118C and p.T385A, the band signal of 100 µmol/l RSG-treated mutants was stronger than for DGJ-treated mutants, but the combination revealed the strongest effect. For mutants p.A156V and p.R301Q, RSG alone increased the amount of the 50 kDa immature form of the enzyme, whereas DGJ apparently enhanced maturation resulting in higher levels of the 46 kDa mature form. With the combination therapy, an enhancement of the effect of DGJ on the 46 kDa mature form was observed. In order to clarify whether the inhibitory effect on global cellular ubiquitination or the PPAR-γ agonism was key for the α-Gal A activity elevation, Ubiquitin-activating enzyme (E1) inhibitor Pyr-41 and PPAR-α/γ agonizing agent Bezafibrate were tested. Pyr-41 did not enhance α-Gal A activity, whereas Bezafibrate increased p.R301Q activity significantly in 100, 200, and 500 µmol/l with or without the simultaneous administration of DGJ (Figure 5c). This evidence shows that PPAR function may play a role in mutant α-Gal A activity elevation rather than ubiquitination inhibition alone.
Discussion
To date, large scale screenings to identify compounds, which enhance enzyme activity in LSD have been based on direct interaction with the lysosomal enzyme in vitro.28,37,38 Our approach was to instead use HEK-293H cells and screen for compounds that enhanced activity of mutant lysosomal enzymes within a cellular environment. The advantage of a cell-based system over a cell-free in vitro system is that any beneficial compounds identified may be (i) more relevant for clinical applications and (ii) generally applicable to different diseases sharing the same biochemical basis. A particular problem however, is the relatively low throughput of this system due to the time and labor demands of cell culture with a high number of repetitions required for each compound.
In order to discover substances counteracting the loss of enzyme activity in lysosomal storage disorders, Ambroxol (ABX) was tested in the HEK-293H cellular model of Fabry and PD. ABX is a compound formerly tested in Gaucher disease that was shown to interact with GCase, causing diminished enzyme activity in vitro but increased activity of mutant enzymes in fibroblasts,28 (p.N370S in non-neuronopathic type I GD and p.L444P29 in subacute neuronopathic type III GD). In the current study, ABX alone was applied to HEK-293H cells expressing either normal (wild type) or mutant α-Gal A (Figure 1a,b). Combined treatment with ABX and synthetic α-Gal A PC DGJ was also tested (Figure 1c). Wild-type α-Gal A showed a 1.4-fold increase in activity after the addition of 60 µmol/l ABX, but its effectiveness was less pronounced at concentrations ≥100 µmol/l. Mutants of α-Gal A, which were DGJ-responsive, showed an additional benefit from combined treatment with ABX (Figure 1c) leading to higher enzyme levels after 60 hours of treatment (Figure 1d). This was due to a stabilizing effect of DGJ/ABX on the enzyme as demonstrated by an in vitro heat-treatment experiment using the pharmaceutical form of α-Gal A (agalsidase alfa). DGJ monotherapy was not as effective in stabilizing the enzyme as when used in combination with ABX (Figure 2). In contrast, ABX monotherapy in vitro led to accelerated loss of enzyme activity. Hypothetically, binding of ABX to the enzyme and stabilization could be enhanced by structural changes induced by DGJ binding. However, ligand binding to the protein has been reported to barely affect the conformation of the enzyme.39 Moreover, ABX monotherapy provoked a significant, though modest, activity elevation of α-Gal A wild-type, p.A156V and p.R301Q enzyme forms in HEK-293H cells, which could argue for an alternative work mechanism of ABX via a general cellular process, affecting protein maturation and transport. For example, it has been reported that ABX regulates the efflux of extracellular Ca2+ in dorsal root ganglion cells40 possibly mediated by inhibition of voltage-gated sodium and calcium channels.41 Calcium is a critical agent for protein folding. Manipulating the release of Ca2+ from the ER, the largest intracellular Ca2+ store, was shown to have an influence on mutant GCase enzyme in Gaucher disease.33 Since α-Gal A mutants show an increased propensity to aggregate within the cell,42 we can speculate that ABX perhaps enhances the effect of the pharmacological chaperone via modulation of the protein folding conditions in the ER. Besides the chaperoning effect, ABX was recently speculated to impact on GBA expression and function in Gaucher disease fibroblasts.43 Therefore, the utilized cell culture system cannot elucidate the specific mode of action of substances that have a diverse functional spectrum.
Anyhow, ABX did not act as an in vitro inhibitor of α-Gal A activity (Supplementary Figure S5), so evidently does not bind the active site in a substrate competitive manner. Allosteric binders of mutant lysosomal enzymes are regarded as promising treatment options, because the risk of undesired side-effects and overdosage is minimized.44
Mutant GAA activity was also beneficially influenced by the combination of a PC and ABX (Figure 3). ABX alone had no effect on mutant GAA; a differential effect was shown for both PCs, NB-DNJ and DNJ, when used in combination with ABX. While DNJ and NB-DNJ as a monotherapy had a similar outcome, it is not yet clear why DNJ/ABX had a synergistic effect on mutants p.Y455F and p.P545L and NB-DNJ/ABX did not.
PPAR-γ agonist RSG increased mutant α-Gal A activity (p.R301Q, p.A156V, p.R118C, p.T385A). It was assumed that RSG mediates its effect via inhibition of cellular ubiquitination, thereby avoiding α-Gal A degradation. However, the ubiquitination inhibitor Pyr-41 did not increase α-Gal A activity. Interruption of the ubiquitin–proteasome system by the use of proteasomal inhibitors (e.g., MG-132) was also ineffective. An earlier report suggested that treatment with the proteasomal inhibitor Lactacystin led to increased amounts of α-Gal A in COS7 cells expressing several mutant forms of the enzyme, but without a concurrent enhancement of enzyme activity.45 Western blot analysis performed in our laboratory did indeed show that Lactacystin and MG-132 increased the amount of p.R301Q, but not p.A156V (Supplementary Figure S6), which is in accordance with the results from Ishii et al. Since only the immature 50 kDa band of p.R301Q is markedly increased, we conclude that this α-Gal A form does not significantly contribute to enzyme activity under the applied assay conditions.
On the other hand PPAR-γ agonist Bezafibrate did enhance activity of p.R301Q which suggests that increased PPAR-γ activity may instead play a role. However, it cannot be excluded that both increased PPAR-γ activity and inhibition of ubiquitination enhance enzyme activity since there is crosstalk between PPAR-γ-mediated transcriptional regulation and the ubiquitination machinery.46 For example, PPAR-γ itself is an E3 ubiquitin ligase that facilitates the degradation of NF-κb.47 Certain players in the ubiquitin machinery have been shown to interact with mutant GCase48,49 indicating a role in its premature degradation. Elucidation of why RSG was operative demands further investigation. The use of more specific drugs could reveal the pivotal mechanism of action. Interestingly, RSG did not display an effect on GAA (data not shown).
Prospectively, these compounds need to be tested in patient-derived cell systems (i.e., lymphoblastoid or fibroblast cell lines) in order to verify the suspected effects. Such cell systems do not require previous transfection of plasmid vectors harboring cDNA of the mutations and allow for more flexible and optimized treatment regimes.50 This is an advantage over more “synthetic” cell systems such as the HEK-293H system used here, in which transient expression (de novo enzyme synthesis) confounds the beneficial effect of small molecules. Patient cell lines are therefore a convenient solution for temporal-oriented studies and should be applied as a second step. Testing findings in a humanized mouse model is also practical.51 Regrettably, to our knowledge, a mouse model does not yet exist for PD.
This work delivers a novel step forward in drug development for LSD. Our use of FDA-approved compounds renders this study an attractive target for further research to elucidate the role of proposed pathways in the enhancement of mutant enzyme stability and activity as a prelude to preclinical and clinical testing.
In particular, ABX and RSG could potentially be suitable compounds as additional treatments in Fabry (or Pompe) patients where monotherapy with a pharmacological chaperone is not sufficiently effective. As described above, the combination of DGJ and ABX was successful in restoring either near normal α-Gal A activity (e.g., p.A73V) or boosting activity to >50% to cross the threshold into the normal range (e.g., p.R301Q). Both outcomes are potentially associated with an amelioration of the disease (Fabry) not obtained using PC monotherapy. Moreover, for mutants with residual activity but not responsive to DGJ, like p.R118C and p.T385A, the usage of RSG could be an alternative treatment strategy because it exceeds the effect of DGJ (Figure 5).
In PD, a genotype/phenotype correlation model is not available, but it can be assumed that an elevation of GAA activity from 11.4% (DNJ) to 25.1% (DNJ/ABX) of normal, as observed for the mutant p.L552P, is likely to be clinically relevant.
Lately, it was demonstrated that small molecules can also have synergistic effects in ERT.52 Evidently, we must endeavor to find the most effective course of treatment for patients with FD and PD, not limited to just one active agent. It is important to note that combination treatments have possible implications for other LSDs as well.40,53
Materials and Methods
Materials. HEK-293H cell line was purchased from Invitrogen (Carlsbad, CA). Ambroxol hydrochloride, Bezafibrate, 1-deoxygalactonojirimycine hydrochloride, 1-Deoxynojirimycin, α-Lobelin, RSG, Thapsigargin, Tunicamycin, and the synthetic fluorogenic substrates for α-Gal A and GAA were purchased from Sigma Aldrich (Steinheim, Germany). Kifunensine, MG-132 were obtained from Merck (Darmstadt, Germany). Lactacystin and bortezomib were purchased from Biomol (Hamburg, Germany). A rabbit polyclonal antibody targeted against human α-galactosidase A was custom-made (Eurogentec, Liège, Belgium)9 and mouse anti-GAPDH (ab8245) was purchased from abcam (Cambridge, UK). Secondary antibody Alexa Fluor 680 Goat Anti-Rabbit IgG (H+L) was purchased from Molecular Probes (Eugene, OR) and Anti-Mouse IgG (H&L) IRDye800 was purchased from Rockland (Gilbertsville, PA).
All primers were obtained from MWG Operon (Ebersberg, GER). A register of used primers is shown in Supplementary Table S2.
Cultivation of HEK-293H cells. HEK-293H were maintained in Dulbecco's modified Eagle medium containing 4,500 mg glucose/l supplemented with 10% fetal bovine serum (both from Gibco, Carlsbad, CA) and 1% Penicillin/Streptomycin.
Cloning. Human cDNA clones of α-Gal A (IRAUp969H0320D, GLA, NM_000169.2) and GAA (IRATp970C0971D, GAA, NM_000152.3) were obtained from Source BioScience (Berlin, GER). cDNAs were amplified by PCR (for primer sequences refer to Supplementary Table S2). After subcloning into pGEM-T (Promega, Mannheim, GER) or pCRII TOPO (Invitrogen), respectively, the fragments were obtained via restriction digestion using the respective restriction endonucleases indicated in Supplementary Table S2 and ligated into a mammalian expression vector (pcDNA3.1/V5-His TOPO, Invitrogen). Gene sequence integrity was confirmed by standard sequencing and alignment analysis as described earlier.9
Mutagenesis. Site-directed mutagenesis of GLA and GAA plasmids were carried out as described before.9 Supplementary Table S2 reports the list of applied primers for all GLA and GAA mutations.
Transient GLA/GAA expression in HEK-293H cells. Commonly, HEK-293H cells were seeded at a density of 1.5 × 105 cells/well of a 24-well plate (Greiner Bio-One, Frickenhausen, GER) the day before transfection. On the day of transfection, mutant GLA or GAA plasmid was transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's guidelines. The compounds were added 6 hours after transfection and left for a 60-hour cultivation unless indicated otherwise. Fresh medium and treatment was added every other day. Transfection control was carried out by parallel transfections of the wild-type and mutant GLA/GAA, which differ by just one base pair.25 Additionally, enzyme activity of every mutant was checked for coherence and stability, indicating maintained plasmid quality.
For the compound serial tests, HEK-293H cells were plated on 10 cm cell culture dishes and transfected with pcDNA3.1 p.A156V and p.R301Q mutated GLA inserts. Six hours after transfection, the cells were trypsinized, collected in a 50 ml tube, and spun down at 1,000 × g. The cells were then aliquoted in medium containing the respective compound or compound combination and plated at a density of 2 × 105/well on 24-well plates in order to obtain identical transfection efficiencies for all samples.
In vitro enzyme activity measurements in HEK-293H cell lysates. α-Gal A and GAA activities were measured in cell lysates of transiently expressing HEK-293H cells. On the day of cell harvest, 66 hours after transfection, medium was aspirated with a vacuum pump (KNF Neuberger, Freiburg i. Breisgau, GER) and washed in phosphate buffered saline. The cells were then collected in 150 µl sterile distilled water and transferred to fresh 1.5 ml centrifugation tubes. The proteins were yielded using five cycles freeze and thaw in liquid nitrogen. For normalization purposes, the protein was quantified using BCA protein assay kit (Thermo Scientific, Braunschweig, GER). The crude protein extract was subsequently used for enzyme activity measurement (0.5 µg for α-Gal A and 5 µg for GAA) employing synthetic fluorogenic substrates 4-methylumbelliferyl-α-D-galactopyranoside (for α-Gal A) and 4-methylumbelliferyl-α-D-glucopyranoside (for GAA), respectively.
α-Gal A enzyme thermal denaturation test. In vitro thermal denaturation was carried out using 200 ng of human α-Gal A (agalsidase alfa, Replagal, Shire Human Genetic Therapies, Berlin, Germany) in a whole volume of 25 µl (in 0.06 mol/l phosphate-citrate buffer, pH 6.7). The enzyme was pipetted in 96-well plates (Greiner bio-one GmbH, Frickenhausen, GER) and incubated at 51 °C for 60 minutes with the respective additive DGJ, ABX or a combination of both basically as reported elsewhere.26 A reference plate was kept on ice during that period. Thereafter, 4 volumes of 0.06 mol/l phosphate-citrate buffer (pH 4.7) were added to the wells and mixed. Twenty-five microliters of the mixture were transferred into a fresh 96-well plate. The enzyme activity measurement was started by addition of 20 µl of the substrate 4-methylumbelliferyl-α-D-galactopyranoside solution (stock concentration: 2.0 mol/l. Samples were kept under slight agitation in a 37 °C water bath for 20 minutes. To stop the reaction, 200 µl of glycine-NaOH buffer (pH 10.5) was added and substrate turnover was immediately surveyed as described above. Comparison of heat-treated versus reference sample was used to measure the reduction in enzyme activity.
Western blot analysis. Western blot analysis was carried out basically as described before.9
PPAR-γ transcriptional regulation reporter assay. The HEK-293H cells were seeded at a density of 4 × 104 in a 96-well plate in antibiotic free medium 24 hours before transfection. On the day of transfection, the medium was changed prior to adding the transfection solution (Lipofectamine 2000, Invitrogen) containing the reporter plasmid constructs (Cignal PPAR Reporter (luc) Kit, Qiagen, Hilden, GER). Six hours post-transfection, the compounds were added. The treatment was carried out for 18 hours before the cells were lysed in passive lysis buffer (Dual-Luciferase Reporter Assay System, Promega GmbH). The detected firefly luciferase activity was normalized against intrinsic renilla luciferase contained in the transfection solution at a plasmid ratio of 1:40. Samples were diluted to ensure that the signal was in the linear assay range, transferred to polypropylene round bottom tubes and measured in a luminometer (Lumat LB9507, Berthold Technologies, Bad Wildbad, GER) with the respective substrates.
SUPPLEMENTARY MATERIAL Figure S1. Excerpt of pharmacological chaperone- (DGJ-) treated GLA mutations. Figure S2. ABX concentration-dependent increase of PC-treated p.A156V α-Gal A activity. Figure S3. Synergistic effect of a galactose/ABX combination on galactose-responsive GLA mutations. Figure S4. ER protein homeostasis re-modeling agents did not have a significant effect on p.R301Q α-Gal A activity. Figure S5. Agalsidase alfa inhibition assay with DGJ and ABX. Figure S6. Proteasomal inhibitors MG-132 and Lactacystin increased cellular α-Gal A mutants. Table S1. Proteasomal activities in compound-treated HEK-293H cells. Table S2. Primer registry.
Acknowledgments
This work was conducted with excellent technical support by Tina Czajka, Mandy Loebert, and Sebastian Rost (Albrecht-Kossel-Institute, Medical University Rostock). We greatly thank Ulrike Grittner (Charité Berlin) for her appreciated advice and statistical support and Jenny Creed Geraghty (Centogene AG, Rostock) for critical manuscript revision. This work was supported by the Medical University Rostock by an internal funding program. Replagal was a kind gift of Werner Foeller (Shire Human Genetic Therapies Inc., Berlin, GER). The International patent “Combination of a Compound having the Ability to Rearrange a Lysosomal Enzyme and Ambroxol and/or a derivative of Ambroxol” (International application no: PCT/EP2012/005363) was filed according to the findings presented in this manuscript.
Supplementary Material
References
- Golubev AM, Nagem RA, Brandão Neto JR, Neustroev KN, Eneyskaya EV, Kulminskaya AA.et al. (2004Crystal structure of alpha-galactosidase from Trichoderma reesei and its complex with galactose: implications for catalytic mechanism J Mol Biol 339413–422. [DOI] [PubMed] [Google Scholar]
- Moreland RJ, Jin X, Zhang XK, Decker RW, Albee KL, Lee KL.et al. (2005Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor J Biol Chem 2806780–6791. [DOI] [PubMed] [Google Scholar]
- Meikle PJ, Fuller M, Hopwood JJ. Mass spectrometry in the study of lysosomal storage disorders. Cell Mol Biol (Noisy-le-grand) 2003;49:769–777. [PubMed] [Google Scholar]
- Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U.et al. (2010Fabry disease: a review of current management strategies QJM 103641–659. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet C Semin Med Genet. 2012;160C:1–7. doi: 10.1002/ajmg.c.31324. [DOI] [PubMed] [Google Scholar]
- Güngör D, de Vries JM, Hop WC, Reuser AJ, van Doorn PA, van der Ploeg AT.et al. (2011Survival and associated factors in 268 adults with Pompe disease prior to treatment with enzyme replacement therapy Orphanet J Rare Dis 634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H.et al. (2006High incidence of later-onset fabry disease revealed by newborn screening Am J Hum Genet 7931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog A, Hartung R, Reuser AJ, Hermanns P, Runz H, Karabul N.et al. (2012A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations Orphanet J Rare Dis 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Giese AK, Markoff A, Grittner U, Kolodny E, Mascher H.et al. (2013Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease PLoS Genet 9e1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M, Alfalah M, Aerts JM, Naim HY, Zimmer KP. Impaired trafficking of mutants of lysosomal glucocerebrosidase in Gaucher's disease. Int J Biochem Cell Biol. 2005;37:2310–2320. doi: 10.1016/j.biocel.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Yam GH, Zuber C, Roth J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 2005;19:12–18. doi: 10.1096/fj.04-2375com. [DOI] [PubMed] [Google Scholar]
- Flanagan JJ, Rossi B, Tang K, Wu X, Mascioli K, Donaudy F.et al. (2009The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase Hum Mutat 301683–1692. [DOI] [PubMed] [Google Scholar]
- Coutinho MF, Prata MJ, Alves S. Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol Genet Metab. 2012;105:542–550. doi: 10.1016/j.ymgme.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Dietz HC. New therapeutic approaches to mendelian disorders. N Engl J Med. 2010;363:852–863. doi: 10.1056/NEJMra0907180. [DOI] [PubMed] [Google Scholar]
- Kemper AR, Hwu WL, Lloyd-Puryear M, Kishnani PS. Newborn screening for Pompe disease: synthesis of the evidence and development of screening recommendations. Pediatrics. 2007;120:e1327–e1334. doi: 10.1542/peds.2007-0388. [DOI] [PubMed] [Google Scholar]
- El Dib RP, Pastores GM. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2010;5:CD006663. doi: 10.1002/14651858.CD006663.pub2. [DOI] [PubMed] [Google Scholar]
- Wilcox WR, Linthorst GE, Germain DP, Feldt-Rasmussen U, Waldek S, Richards SM.et al. (2012Anti-α-galactosidase A antibody response to agalsidase beta treatment: data from the Fabry Registry Mol Genet Metab 105443–449. [DOI] [PubMed] [Google Scholar]
- Wang J, Lozier J, Johnson G, Kirshner S, Verthelyi D, Pariser A.et al. (2008Neutralizing antibodies to therapeutic enzymes: considerations for testing, prevention and treatment Nat Biotechnol 26901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TT, Banugaria SG, Case LE, Wenninger S, Schoser B, Kishnani PS. The impact of antibodies in late-onset Pompe disease: a case series and literature review. Mol Genet Metab. 2012;106:301–309. doi: 10.1016/j.ymgme.2012.04.027. [DOI] [PubMed] [Google Scholar]
- Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5:112–115. doi: 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- Asano N, Ishii S, Kizu H, Ikeda K, Yasuda K, Kato A.et al. (2000In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives Eur J Biochem 2674179–4186. [DOI] [PubMed] [Google Scholar]
- Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease. Proc Natl Acad Sci USA. 2002;99:15428–15433. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumiya T, Kroos MA, Vliet LV, Takeuchi H, Van der Ploeg AT, Reuser AJ. Chemical chaperones improve transport and enhance stability of mutant alpha-glucosidases in glycogen storage disease type II. Mol Genet Metab. 2007;90:49–57. doi: 10.1016/j.ymgme.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Parenti G, Zuppaldi A, Gabriela Pittis M, Rosaria Tuzzi M, Annunziata I, Meroni G.et al. (2007Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease Mol Ther 15508–514. [DOI] [PubMed] [Google Scholar]
- Andreotti G, Citro V, De Crescenzo A, Orlando P, Cammisa M, Correra A.et al. (2011Therapy of Fabry disease with pharmacological chaperones: from in silico predictions to in vitro tests Orphanet J Rare Dis 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Katz E, Della Valle MC, Mascioli K, Flanagan JJ, Castelli JP.et al. (2011A pharmacogenetic approach to identify mutant forms of α-galactosidase A that respond to a pharmacological chaperone for Fabry disease Hum Mutat 32965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin E, Do HV, Wu X, Flanagan J, Wustman B.2014Method to predict response to pharmacological chaperone treatment of diseases Amicus Therapeutics; USA; US Patent No 14/054,369. [Google Scholar]
- Maegawa GH, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D.et al. (2009Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease J Biol Chem 28423502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendikov-Bar I, Ron I, Filocamo M, Horowitz M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol Dis. 2011;46:4–10. doi: 10.1016/j.bcmd.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Sánchez-Ollé G, Duque J, Egido-Gabás M, Casas J, Lluch M, Chabás A.et al. (2009Promising results of the chaperone effect caused by imino sugars and aminocyclitol derivatives on mutant glucocerebrosidases causing Gaucher disease Blood Cells Mol Dis 42159–166. [DOI] [PubMed] [Google Scholar]
- Clark NE, Metcalf MC, Best D, Fleet GW, Garman SC. Pharmacological chaperones for human α-N-acetylgalactosaminidase. Proc Natl Acad Sci USA. 2012;109:17400–17405. doi: 10.1073/pnas.1203924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L.et al. (2008Chemical and biological approaches synergize to ameliorate protein-folding diseases Cell 134769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Agnello G, Sotolongo N, Segatori L. Ca2+ homeostasis modulation enhances the amenability of L444P glucosylcerebrosidase to proteostasis regulation in patient-derived fibroblasts. ACS Chem Biol. 2011;6:158–168. doi: 10.1021/cb100321m. [DOI] [PubMed] [Google Scholar]
- Zampieri S, Bembi B, Rosso N, Filocamo M, Dardis A. Treatment of Human Fibroblasts Carrying NPC1 Missense Mutations with MG132 Leads to an Improvement of Intracellular Cholesterol Trafficking. JIMD Rep. 2012;2:59–69. doi: 10.1007/8904_2011_49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti G, Guarracino MR, Cammisa M, Correra A, Cubellis MV. Prediction of the responsiveness to pharmacological chaperones: lysosomal human alpha-galactosidase, a case of study. Orphanet J Rare Dis. 2010;5:36. doi: 10.1186/1750-1172-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfella R, D'Amico M, Esposito K, Baldi A, Di Filippo C, Siniscalchi M.et al. (2006The ubiquitin-proteasome system and inflammatory activity in diabetic atherosclerotic plaques: effects of rosiglitazone treatment Diabetes 55622–632. [DOI] [PubMed] [Google Scholar]
- Zheng W, Padia J, Urban DJ, Jadhav A, Goker-Alpan O, Simeonov A.et al. (2007Three classes of glucocerebrosidase inhibitors identified by quantitative high-throughput screening are chaperone leads for Gaucher disease Proc Natl Acad Sci USA 10413192–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropak MB, Blanchard JE, Withers SG, Brown ED, Mahuran D. High-throughput screening for human lysosomal beta-N-Acetyl hexosaminidase inhibitors acting as pharmacological chaperones. Chem Biol. 2007;14:153–164. doi: 10.1016/j.chembiol.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman RL, D'aquino JA, Ringe D, Petsko GA. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry. 2009;48:4816–4827. doi: 10.1021/bi9002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini RJ, Hou X, Parkington H, Coleman H, Klaver DW, Vincent AJ.et al. (2011TRPM8 and Nav1.8 sodium channels are required for transthyretin-induced calcium influx in growth cones of small-diameter TrkA-positive sensory neurons Mol Neurodegener 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser T. Ambroxol: a CNS drug. CNS Neurosci Ther. 2008;14:17–24. doi: 10.1111/j.1527-3458.2007.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekierska A, De Baets G, Reumers J, Gallardo R, Rudyak S, Broersen K.et al. (2012α-Galactosidase aggregation is a determinant of pharmacological chaperone efficacy on Fabry disease mutants J Biol Chem 28728386–28397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A.et al. (2014Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells Brain 137Pt 51481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto C, Ferrara MC, Meli M, Acampora E, Avolio V, Rosa M.et al. (2012Pharmacological enhancement of α-glucosidase by the allosteric chaperone N-acetylcysteine Mol Ther 202201–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii S, Chang HH, Kawasaki K, Yasuda K, Wu HL, Garman SC.et al. (2007Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin Biochem J 406285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilroy GE, Zhang X, Floyd ZE. PPAR-gamma AF-2 domain functions as a component of a ubiquitin-dependent degradation signal. Obesity (Silver Spring) 2009;17:665–673. doi: 10.1038/oby.2008.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Moreau F, Chadee K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat Commun. 2012;3:1300. doi: 10.1038/ncomms2270. [DOI] [PubMed] [Google Scholar]
- Ron I, Rapaport D, Horowitz M. Interaction between parkin and mutant glucocerebrosidase variants: a possible link between Parkinson disease and Gaucher disease. Hum Mol Genet. 2010;19:3771–3781. doi: 10.1093/hmg/ddq292. [DOI] [PubMed] [Google Scholar]
- Maor G, Filocamo M, Horowitz M. ITCH regulates degradation of mutant glucocerebrosidase: implications to Gaucher disease. Hum Mol Genet. 2013;22:1316–1327. doi: 10.1093/hmg/dds535. [DOI] [PubMed] [Google Scholar]
- Steet RA, Chung S, Wustman B, Powe A, Do H, Kornfeld SA. The iminosugar isofagomine increases the activity of N370S mutant acid beta-glucosidase in Gaucher fibroblasts by several mechanisms. Proc Natl Acad Sci USA. 2006;103:13813–13818. doi: 10.1073/pnas.0605928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Soska R, Lun Y, Feng J, Frascella M, Young B.et al. (2010The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease Mol Ther 1823–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Flanagan JJ, Feng J, Soska R, Frascella M, Pellegrino LJ.et al. (2012The pharmacological chaperone AT2220 increases recombinant human acid α-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease PLoS One 7e40776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto C, Pisani A, Rosa M, Acampora E, Avolio V, Tuzzi MR.et al. (2012Synergy between the pharmacological chaperone 1-deoxygalactonojirimycin and the human recombinant alpha-galactosidase A in cultured fibroblasts from patients with Fabry disease J Inherit Metab Dis 35513–520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.