

Abstract
Pexa-Vec (pexastimogene devacirepvec, JX-594) is an oncolytic and immunotherapeutic vaccinia virus designed to destroy cancer cells through viral lysis and induction of granulocyte-macrophage colony-stimulating factor (GM-CSF)-driven tumor-specific immunity. Pexa-Vec has undergone phase 1 and 2 testing alone and in combination with other therapies in adult patients, via both intratumoral and intravenous administration routes. We sought to determine the safety of intratumoral administration in pediatric patients. In a dose-escalation study using either 106 or 107 plaque-forming units per kilogram, we performed one-time injections in up to three tumor sites in five pediatric patients and two injections in one patient. Ages at study entry ranged from 4 to 21 years, and their cancer diagnoses included neuroblastoma, hepatocellular carcinoma, and Ewing sarcoma. All toxicities were ≤ grade 3. The most common side effects were sinus fever and sinus tachycardia. All three patients at the higher dose developed asymptomatic grade 1 treatment-related skin pustules that resolved within 3–4 weeks. One patient showed imaging evidence suggestive of antitumor biological activity. The two patients tested for cellular immunoreactivity to vaccinia antigens showed strong responses. Overall, our study suggests Pexa-Vec is safe to administer to pediatric patients by intratumoral administration and could be studied further in this patient population.
Introduction
Over the past several decades, the use of multimodality therapy including chemotherapy, radiation, and surgery has resulted in significant improvements in outcomes for children with solid tumors and lymphomas.1 Progress has slowed in recent years, and there are still many patients with poor prognoses, especially those with recurrent or metastatic disease. Because of the many years of life remaining for those pediatric patients who are cured of their malignancy, minimization of toxicity and long-term morbidity are also key goals in the treatment of children with cancer. Thus, novel therapies are desperately needed.
The field of oncolytic virotherapy seeks to leverage the lytic lifecycle of viruses for tumoricidal activity, and their ability to be proinflammatory, for inducing antitumor immunity.2 Oncolytic viruses kill cancer cells through a novel mechanism of action (“oncolysis,” or virus replication–associated necrosis)3 and can be targeted to cancer cells with activated genetic pathways and/or loss of tumor suppressor function.4 In addition, oncolytic viruses can result in tumor cell death indirectly, including uninfected cells, through the recruitment of a tumor-specific adaptive immune response or by the expression of a therapeutic transgene such as a prodrug converting enzyme or proinflammatory cytokines.5,6,7 Finally, oncolytic viruses can also induce tumor necrosis through blockage of tumor-associated vasculature.8,9
Pexa-Vec (pexastimogene devacirepvec, JX-594) is a replication-competent vaccinia virus derived from the commonly used Wyeth vaccine strain (Dryvax; Wyeth Laboratories). Three genetic modifications are included in Pexa-Vec: (i) a thymidine kinase gene deletion to enable more selective replication in cancer cells, (ii) GM-CSF gene insertion under the control of the synthetic early-late promoter to induce a systemic antitumoral immune response, and (iii) lac-Z gene insertion under control of the p7.5 promoter.10,11,12 Numerous studies have demonstrated the safety of Pexa-Vec in rodent models, with side effects including treatment-related inappetence and reversible changes in hematology and clinical chemistry parameters. Reversible lymphoid depletion in the thymus and lymphoid hyperplasia with red pulp expansion in the spleen were also seen and considered to be physiological adaptations. Mild anemia was considered secondary to spleen enlargement.
Pexa-Vec has been administered successfully to over 300 adult patients by both intratumoral and intravenous routes. In a phase 1 trial, 14 patients with solid tumors were given intratumoral Pexa-Vec ranging from 108 to 3 × 109 plaque-forming units (pfu), with a dose of 1 × 109 pfu defined as the maximum tolerated dose (MTD) due to hyperbilirubinemia in two patients at the highest dose level.13 Expected toxicities included fever and flu-like symptoms. Interestingly, a secondary wave of viremia was detected, suggesting intratumoral replication and dissemination of Pexa-Vec to other tumor sites.
Given the safety and possible efficacy of Pexa-Vec in adult cancer patients, we sought to determine the safety of intratumoral Pexa-Vec injection in younger cancer patients. Because of the potential for systemic viremia and the expected range of body sizes, we chose to dose patients aged 2–21 years at study entry on a per-kilogram basis, beginning with a dose ~10–20-fold lower than the MTD in adults (calculated per kilogram). Whether lack of prior smallpox vaccination in children compared with adult populations (in which most of the patients born prior to the 1970s had likely been vaccinated) would increase the likelihood of benefit or pose increased risk for adverse events were also of interest.
Results
Demographics
Six patients were enrolled to the study from November 2010 to February 2012. All were evaluable. A summary of patient characteristics is shown in Table 1. Ages ranged from 4 to 21 years at first enrollment. Prior vaccination with vaccinia was negative in four and unknown in two patients. Diagnoses included hepatocellular carcinoma (three patients), neuroblastoma (two patients), and Ewing sarcoma (one patient). All had failed multiple prior local and systemic therapies; both neuroblastoma patients had previously undergone high-dose chemotherapy with autologous stem cell rescue. The total Pexa-Vec dose to be administered for each patient (column 8, Table 1) was based on the dose level (column 1, Table 1) and calculated based on total body weight. The dose was diluted into a total volume (column 8, Table 1) based on the target tumor volume(s) and was injected into two different tumor sites in patient 01, into three different tumor sites in patient 03, and into a single tumor in the other patients. Patient 05 received a second injection into the same tumor site 6 weeks after the first injection. Thus, there were a total of 10 tumor injections on seven occasions in six patients. Total injection volumes into which each total virus dose (either 106 or 107 pfu/kg) was diluted were individually determined to be 50% of the calculated volume of the specific tumor site(s) to be injected and ranged from 0.1 to 120 ml (average 19.7 ml; median: 2.0 ml). Ultrasound guidance was used exclusively for six injections, CT guidance for three injections, and both for one injection. A multipronged needle was used for three injections (patients 02, 04, and 06); all others were with a straight needle.
Table 1. Patient demographics, treatment, and radiographic response to Pexa-Vec.
Adverse events
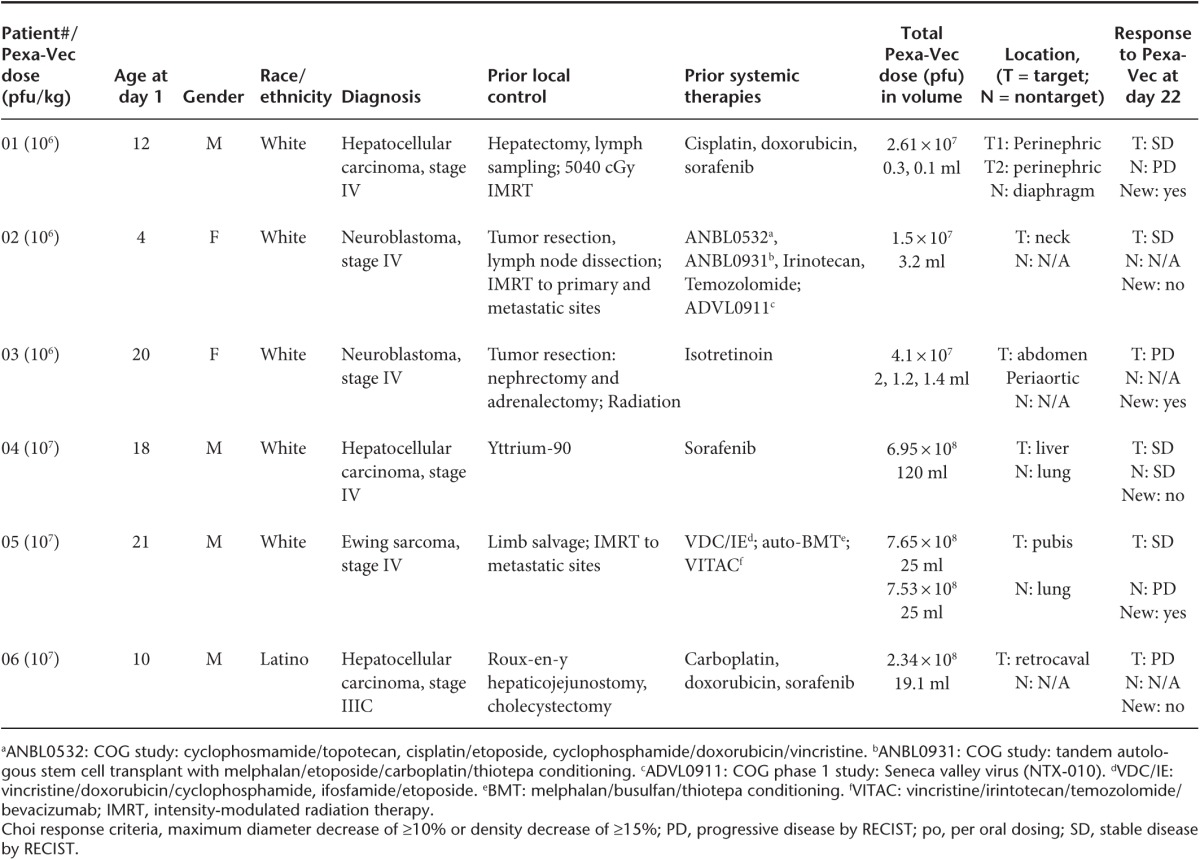
There were no deaths during the observation period of this study. The only grade 4 event was thrombocytopenia in a single patient in the lower dose cohort and was deemed not related to Pexa-Vec treatment (preexisting due to prior chemotherapy). A summary of total related and unrelated treatment-emergent adverse events is provided in Table 2. The most common adverse reaction was fever, which occurred in six of the seven (86%) injection episodes, beginning 6–10 hours following the injection and resolving by 24 hours. The only grade 3 fevers occurred in two patients in the higher dose cohort. Other common events were sinus tachycardia (likely due to fever) and flu-like symptoms including nausea, vomiting, chills, back pain, and headache. Interestingly, all three patients given the higher dose developed pustular lesions consistent with vaccinia-related pustules on skin (Figure 1) and, in one case, lips, which developed within a few days to a week after Pexa-Vec treatment. Due to their widespread nature and the timing of their appearance, they were assumed to be Pexa-Vec–induced pox lesions and were not cultured. Patients remained in modified contact isolation (see Materials and Methods) during clinic visits until pustules resolved. The pustules resolved after 3–4 weeks in two of the patients (unknown duration in the third), consistent with onset and resolution of pustules related to vaccinia vaccination. The pustules were not bothersome to the patients and did not leave any scars (according to the treating physicians, though follow-up photos were not taken). The patient who received a second injection only developed pustules after the first but not after the second injection, presumably due to the development of antivaccinia immunity.
Table 2. Adverse events grade 1–3 occurring more than once or grade 4/5.
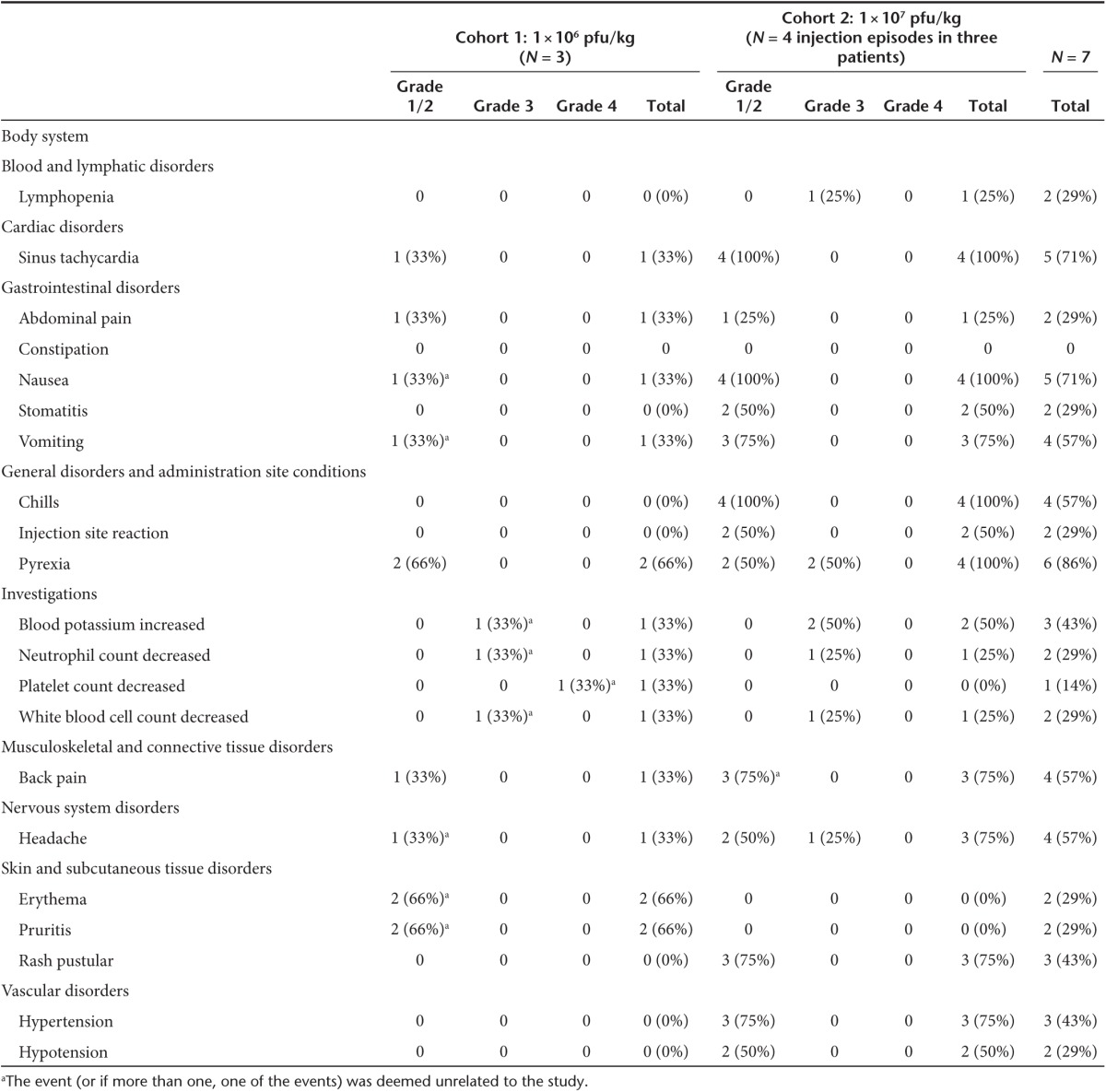
Figure 1.

Examples of skin pustules developed by subject 05. The pustules first began appearing at day 5 postinjection and slowly faded over the course of a few weeks. They were asymptomatic as they were nonpruritic and did not leave scars. He and the other two subjects who developed similar lesions were kept in contact isolation during hospital visits until the lesions had crusted over.
Clinical and radiographic responses
Prior to enrollment in the study, all patients had progressive disease refractory to prior therapy. Repeat imaging using either computed tomography or magnetic resonance imaging was obtained on day 22 and compared to the same modality at baseline (within 14 days prior to injection). Four of the six patients had stable disease and two had progressive disease in the injected target lesion (summarized in Table 1). Uninjected lesions, when present, progressed in all patients except patient 04 whose lung nodules were stable at day 22. Of the four with stable disease at the injection site, two had either progression of other noninjected lesions or the development of new lesions at other sites and were taken off study. The other two chose to pursue other treatment options rather than receive subsequent injections. Following the 28-day observation period, all patients underwent other types of cancer therapy or palliative care per their treating oncologist, and all have subsequently died of their underlying disease.
Despite the lack of objective response by Response Evaluation Criteria in Solid Tumors (RECIST) measurements, there was evidence of biological activity by other imaging characteristics in patient 05, including necrotic changes in the injected tumor on magnetic resonance imaging and a decrease in standard uptake value on positron emission tomography on day 22 following the first injection (Figure 2).
Figure 2.

Radiographic evaluation of the injected lesion at the pubis bone in subject 05. Magnetic resonance imaging showed a necrotic change within the lesion following the injection with some clearing and diminishment in the PET signal. Based on these findings suggesting possible efficacy, the subject underwent a second injection as allowed by the protocol, but his disease progressed elsewhere, and he sought other therapy.
Immunologic response
To measure the immune response to oncolytic virotherapy, we measured IFN-γ production in response to five vaccinia proteins using enzyme-linked immunospot (ELISPOT) assays on one patient from each dose level. Neither patient had measurable immunity prior to treatment, and both developed robust responses to some or all proteins tested at each of three postinjection time points (Figure 3). These data demonstrated immunocompetence in these patients despite both their prior immunosuppressive chemotherapy and the presumed immunosuppressive microenvironment that is characteristic of many solid tumors.
Figure 3.
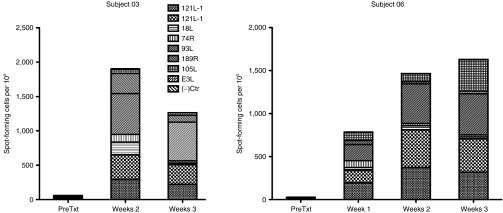
Assessment of immunologic response to vaccinia antigens in two patients following Pexa-Vec injection. No responses to vaccinia antigens could be detected prior to Pexa-Vec injection. Both subjects showed robust responses to most of the vaccinia-derived peptides tested in this ELISPOT assay that were largely maintained during the three-week testing period.
Discussion
We present the first report of intratumoral injection of an oncolytic vaccinia virus in children and adolescents. No serious adverse events attributed to Pexa-Vec or the study procedures were reported in these patients. We also found some immunological effects of the injections and in one case, evidence of antitumor activity.
Based on this small number of patients, our data suggest that intratumoral injection of Pexa-Vec at the doses tested is relatively safe in young children, causing transient flu-like symptoms and pustules. The MTD of intratumoral Pexa-Vec within the liver in adults is 1 × 109 pfu.14 For the purposes of this study in children, we chose to administer a weight-based dose, beginning with 1 × 106 pfu/kg, with a log de-escalation (dose level −1) if needed due to toxicity and a log escalation to 1 × 107 pfu/kg for dose level 2. Although the original study was designed to allow dose escalation to 3 × 107 pfu/kg, the study was amended to eliminate the top planned dose cohort due to the high costs of continuing the study. The highest dose level was thus a conservative equivalent of the adult MTD, as it would be equivalent on a per-kg basis to the adult MTD for a large person (100 kg). We observed similar expected adverse reactions as noted in adult studies, primarily flu-like symptoms that were low grade and transient. Interestingly, we observed skin and mucosal vaccinia-related skin pustules in all three patients on the higher dose, whereas in adults, the incidence of skin pustules among all studies has been ~12%. Although the numbers are small, it is possible that children and adolescents are at higher risk for developing skin pustules than adults. It is possible that the lack of prior exposure to vaccinia vaccination in children puts them at increased risk for disseminated viremia than an adult population some of whom had prior exposure to the vaccine. In adult studies, however, the development of pustular skin lesions has not correlated with prior lack of vaccinia vaccination (unpublished).
In adult trials, virtually all patients treated with Pexa-Vec have experienced mild-to-moderate flu-like symptoms, which included fever, chills, anorexia, aches/pain, fatigue, headache, and/or nausea. Acute, mild-to-moderate hypotension had an onset within 1–2 hours following treatment and was then observed to peak in severity between 8 and 12 hours; hypotension continued intermittently throughout the first 24 hours following Pexa-Vec treatment in some patients. Acute, moderate-to-severe fever has been observed within 4–6 hours posttreatment and had a typical duration of 18–24 hours. The severity of fever and hypotension appeared to be dose related. Other mild-to-moderate flu-like symptoms had a typical duration of 1–3 days. Our experience in this small cohort of pediatric patients was consistent with these prior experiences in adults.
Our study was not designed to test efficacy, but the stable disease observed in the majority of patients and the changes on magnetic resonance imaging and fluorodeoxyglucose-positron emission tomography (FDG-PET) we observed in some patients suggest that Pexa-Vec treatment may have provided some biological activity. It is likely, however, in many of our patients that the natural history of tumor growth may have been too slow to detect a change in the rate of growth given the short time frame (3 weeks) of the observation period. In other Pexa-Vec studies, patients developed antitumor immunity as measured by the presence of complement-dependent antitumor antibodies in serum, and tumor shrinkage in some cases developed over the course of several months following the injection including in noninjected lesions, consistent with an immunologic component to efficacy.15,16 We did not observe any changes in noninjected lesions that might have resulted from an immunologic effect possibly in part due to the relatively short time frame that the patients were observed in the study prior to seeking other treatments.
A randomized phase 2 dose-ranging study of Pexa-Vec in 30 patients with advanced liver cancer demonstrated an improvement in survival in patients receiving intratumoural injections of high-dose Pexa-Vec (1 × 109 pfu) versus low-dose Pexa-Vec (1 × 108 pfu), demonstrating a dose threshold for systemic antitumor activity.17 Four of 28 evaluable patients exhibited responses by modified RECIST criteria for hepatocellular carcinoma. A phase 2b clinical trial in hepatocellular carcinoma patients who failed sorafenib therapy (n = 120) was recently completed and did not achieve the primary endpoint of prolonging overall survival in Pexa-Vec–treated patients when comparing to patients treated with best supportive care in this last-line, treatment refractory, poor prognosis patient population.
In adult trials, after successful intratumoral dose escalation, an intravenous dose-escalation trial in advanced solid tumors was initiated to evaluate the tolerability of intravenous Pexa-Vec.18 No dose-limiting toxicities were observed and a maximum feasible dose of 3 × 107 pfu/kg was defined. Notably, intravenous Pexa-Vec was associated with dose-dependent delivery to multiple solid tumor types (including colorectal cancer, lung cancer, pancreatic cancer, and mesothelioma) and resulted in antitumor activity at high doses. Whether intravenous dosing is safe in young patients has not yet been tested.
In conclusion, our data support the continued evaluation of Pexa-Vec in children and adolescent cancer patient, which could include further dose escalation. Although the majority of our patients had stable disease at the injection site, most showed growth of uninjected lesions or the development of new lesions. Thus, systemic administration would be an attractive option for patients with metastatic disease, and future study designs should also evaluate combinations of oncolytic vaccinia with other anticancer therapies as multimodal approaches are hallmarks of successful cancer treatment strategies.
Materials and Methods
Clinical study approvals, consents, and registration. The trial proposal underwent public review and discussion by the National Institute of Health Recombinant Advisory Committee on 1 December 2009 (http://osp.od.nih.gov/sites/default/files/RAC_Minutes_12-09.pdf). It was registered with clinicaltrials.gov (NCT01169584) and received approval from the Cincinnati Children's Hospital Institutional and the Texas Children's Hospital Review Boards. The Declaration of Helsinki protocols were followed; informed consent was obtained from all patients aged 18 years or older and from the parents or legal guardians of patients younger than 18 years. Child assent was provided when appropriate and in accordance with individual institutional policies.
The study had two primary aims: (i) to determine the maximally tolerated dose and/or maximum feasible dose of Pexa-Vec administered by intratumoral injection in pediatric patients and (ii) to determine the safety/toxicity of Pexa-Vec administered by intratumoral injection in this patient population. Secondary aims included: (i) to determine the Pexa-Vec pharmacokinetics and pharmacodynamics over time following intratumoral injection and (ii) to determine the immune response to Pexa-Vec following intratumoral injection.
Eligibility. Patients were greater than 1 year of age and less than 22 years of age with recurrent or refractory non–central nervous system solid tumors with no known curative pathway. Patients with lymphomas were excluded at the request of the National Institute of Health Recombinant DNA Advisory Committee due to their inherit immunodeficiency. Patients were required to have a tumor deemed safe to inject by interventional radiology, with a minimum size of 1 cm in at least one dimension. Patients were also required to have a Lansky performance score of ≥50. To confirm lack of profound immunosuppression, patients were required to have a CD4 count ≥200/mm3. Due to the invasive injection procedure, coagulation status requirements included international normalized ratio < 1.5, platelet count greater than 75,000/ml, and hemoglobin ≥9 g/dl, the latter two with or without transfusions. No antiplatelet, anticoagulation, or antiviral medications were allowed within 7 days prior to Pexa-Vec injection except low-dose heparin needed to maintain venous catheter patency. Organ function requirements included: absolute neutrophil count greater than 750/ml; adequate renal function (serum creatinine ≤1.8 times the upper limit of normal for age or a creatinine clearance or radioisotope glomerular filtration rate (GFR) ≥ 70 ml/minute/1.73 m2), and adequate hepatic function (total bilirubin, alanine aminotransferase, and aspartate aminotransferase less than 2.5 times the upper limit of normal for age). Patients must have recovered from the acute toxic effects of prior therapy, including a 3-week interval since the last myelosuppressive therapy. Live virus immunizations were not permitted within 30 days of enrollment or during the on-study treatment period. Patients known to be at highest risk from vaccinia immunization were excluded, including those with a known congenital immunodeficiency, HIV, severe eczema requiring systemic therapy, pregnancy, or nursing mothers. Patients requiring ongoing immunosuppression including high-dose steroids were also excluded from enrollment.
Study design. The dose escalation followed standard three plus three design. Briefly, three patients were enrolled at each of two dose levels, either 106 or 107 pfu/kg of body weight. Because the weights of the patients varied significantly, each patient received widely different total doses. If any enrolled patient at risk for a dose-limiting toxicity (DLT) experienced a DLT, three additional patients were to be enrolled at that level. The MTD was defined to be the maximum dose at which fewer than one-third of patients experience DLT during cycle 1 of therapy, inclusive of the expansion cohort. Descriptions and grading scales for adverse event reporting are found in the revised NCI Common Terminology Criteria for Adverse Events version 3.0. A DLT was defined as any grade 3 or 4 nonhematological toxicity possibly, probably, or definitely attributable to protocol therapy with the exclusion of grade 3 headache, nausea, and vomiting, grade 3 hypotension, grade 3 or 4 fever of less than 72 hours in duration, grade 3 infection, grade 3 supplement-responsive electrolyte disturbance, and grade 3 tumor pain. Hematologic DLTs were defined as grade 4 neutropenia for more than 7 days, a platelet count less than 25,000/mm3 on 2 separate days within a 7-day period, and prolonged myelosuppression.
In consultation with hospital infection control, we adopted a modified contact isolation procedure for patients following Pexa-Vec injection. Healthcare providers wore gown, gloves, mask, and eye shield, due to risk of transmission through inadvertent touching of mouth, nose, and eyes. Individuals were advised by training and placards to remove their gloves first, prior to removing the eye shield, again to minimize the risk of transmission of glove-resident virus via contact with mucosal membranes. Blood work was sent to the lab with a note identifying the study and reminding lab technicians to not handle the specimen if pregnant or immunocompromised. Following patient discharge, all disposable items from the patient's room were discarded into biohazard bags, bedding and linens were placed in transport bags by trained nursing staff for decontamination, and all surfaces were wiped with bleach wipes.
Manufacturing and preparation of Pexa-Vec. Pexa-Vec is a Wyeth strain vaccinia modified by insertion of the human GM-CSF and Lac-Z genes into the vaccinia TK gene region under control of the synthetic early-late promoter and p7·5 promoter, respectively. Pexa-Vec was provided and distributed by Jennerex Biotherapeutics (now SillaJen Biotherapeutics, Seoul, South Korea). Clinical trial material lots used in this study were manufactured according to Good Manufacturing Practice guidelines (n = 2). Pexa-Vec was grown in adherent mammalian cells and purified through sucrose-gradient centrifugation or by tangential flow filtration. In vitro and in vivo comparability testing demonstrated equivalence of the two lots. Final product quality control tests included assays for sterility and endotoxin, DNA, protein, pfu, and genome concentration in the clinical trial material; functional assays included potency and GM-CSF production. Clinical trial material was formulated in either phosphate-buffered saline with 10% v/v glycerol (pH 7.1) or 30 mmol/l Tris with 10% (w/v) sucrose (pH 7.7). Vials remained at −80 °C prior to use. Pexa-Vec was diluted in 0.9% bicarbonate-buffered normal saline. The volume of Pexa-Vec solution to be injected was proportional to the volume of the tumor to be injected (50% of the tumor volume, not exceeding 120 ml per tumor).
Study drug and treatment plan. The starting dose was chosen as 1 × 106 pfu/kg, with a log de-escalation (dose level −1) if needed due to toxicity and a log escalation to 1 × 107 pfu/kg for dose level 2. Patients were admitted the day prior to injection and started on intravenous hydration to mitigate virus-mediated hypotension. The following day, patients were premedicated with acetaminophen, and Pexa-Vec was administered within a 6-hour period after thaw/dilution in the interventional radiology suite with the patient under general anesthesia. Pexa-Vec was given through a straight needle into an area of the tumor thought to be most viable based on CT, magnetic resonance imaging, and PET scans, or using the Quadra-Fuse needle (Rex Medical, Radnor, PA) when tumors were large enough to accommodate the projected tines. Based on the discretion of the investigator, Pexa-Vec could be distributed in up to three different tumor sites. Patients remained inpatient for observation at least overnight or longer depending on clinical status. Patients were seen in the outpatient clinic on days 4 and 8, then weekly through day 29 and every 6 weeks thereafter for 6 months. Repeat imaging studies were performed at day 22 to assess response. For patients who derived clinical benefit, up to three retreatments were allowed at the discretion of the sponsor.
Vaccinia virus–specific T-cell analysis. Blood was drawn from subjects 03 and 06, before and after treatment with Pexa-Vec. Peripheral blood mononuclear cells were isolated by centrifugation on Lymphoprep (Axis Shield, Oslo, Norway). Whole peripheral blood mononuclear cells were pulsed directly with overlapping peptide libraries (pepmixes) comprising peptides of 15 amino acids in length overlapping by 11 amino acids and spanning the entire protein sequences of seven vaccinia antigens, A10L, D8L, B22R, H3L, C7L, G5R (JPT Technologies, Berlin, Germany), and E3L (Genemed Synthesis, San Antonio, TX). Stimulated cells were cultured in human T-cell medium (RPMI 1640 (Hyclone, Waltham, MA) supplemented with 45% Click's medium (Irvine Scientific, Santa Ana, CA), 2 mmol/l GlutaMAX TM-I (Invitrogen, Carlsbad, CA), and 5% Human AB Serum (Valley Biomedical, Winchester, VA)), in the presence of 1,000 U/ml IL-4 (R&D Systems, Minneapolis, MN) and 10 ng/ml IL-7 (PeproTech, Rocky Hill, NJ), as previously described.19 We harvested cells on day 9 and tested for vaccinia virus specificity using ELISPOT assay.
We used ELISPOT analysis as a semiquantitative measure of antigen-specific effector T cells. Briefly, we seeded 2.5 × 104 to 105 T cells in triplicate with individual pepmixes at 0.1 μg per peptide per well. We used a pepmix of a testis cancer antigen, NY-ESO1, at 0.1 μg per peptide per well and phytohemagluttinin at 2 μl (1 mg/ml) as negative and positive controls, respectively. After 18 hours of incubation, we developed the plates and sent them to Zellnet Consulting, New Jersey, for quantification. Spot forming cell counts and input cell numbers were plotted, and a linear regression was calculated after excluding plateau data points. We expressed the frequency of T cells specific to each antigen as specific spot forming cell per input cell numbers.
Statistical analysis. The study sample size was set to assess safety issues. The likelihood of dose escalation, given varying true DLTs in the treated population, was calculated as per routine in phase 1 dose-escalation trials. Expected sample size was 6–12 patients for the two dose cohorts.
Acknowledgments
We thank members of the Office for Clinical and Translational Research at Cincinnati Children's Hospital Medical Center (CCHMC) including Sheri Uber, Susan Radtke, Marianne Brunner, Rubina Dosani, and Jeanie Bailey, as well as the Division of Oncology clinical research team including Rebecca Turner, Kristina Kruskamp, Stacey Crane, Beth Stockman, Chris Kramer, and Renee Richardson. We thank members of the CCHMC Vector Core for virus dose preparations including Johannes Vanderloo, Diana Nordling, Scott Cross, Amy Terwilliger, and Anne Kaiser. We thank the CCHMC infectious disease and infection control teams for oversight including David Bernstein, Beverly Connelly, and Michael Cloughessy. We thank Denise Lagory in the CCHMC investigative pharmacy and other members of the CCHMC interventional radiology department including Kamlesh Krukreja, Manish Patel, and Neil Johnson. We thank David Kerr, J. Andrea McCart, Peter Forsyth, and John Crowley for serving on the Data Safety Monitoring Board and reviewing the safety/toxicity data. This study was supported by a grant from Solving Kids Cancer, Pierce Phillips Charity, The Catherine Elizabeth Blair Memorial Foundation, and Make Some Noise: Cure Kids Cancer Foundation.
C.J.B., A.M., J.M.B., and D.H.K. are employees of SillaJen, Inc. SillaJen, Inc. holds the license for Pexa-Vec.
References
- Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O'Leary M.et al. (2010Outcomes for children and adolescents with cancer: challenges for the twenty-first century J Clin Oncol 282625–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur B, Chiocca EA, Cripe TP. Oncolytic HSV-1 virotherapy: clinical experience and opportunities for progress. Curr Pharm Biotechnol. 2012;13:1842–1851. doi: 10.2174/138920112800958814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S.et al. (2003VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents Cancer Cell 4263–275. [DOI] [PubMed] [Google Scholar]
- Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JS, Kim JW, Ahmed AU, Lesniak MS. Therapeutic cell carriers: a potential road to cure glioma. Expert Rev Neurother. 2014;14:651–660. doi: 10.1586/14737175.2014.917964. [DOI] [PubMed] [Google Scholar]
- Shulak L, Beljanski V, Chiang C, Dutta SM, Van Grevenynghe J, Belgnaoui SM.et al. (2014Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy J Virol 882927–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA.et al. (2007Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow Mol Ther 151686–1693. [DOI] [PubMed] [Google Scholar]
- Spurrell E, Gangeswaran R, Wang P, Cao F, Gao D, Feng B.et al. (2014STAT1 interaction with E3-14.7K in monocytes affects the efficacy of oncolytic adenovirus J Virol 882291–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima F, Esaki S, Luo C, Kamakura M, Kimura H, Nishiyama Y. Oncolytic viral therapy with a combination of HF10, a herpes simplex virus type 1 variant and granulocyte-macrophage colony-stimulating factor for murine ovarian cancer. Int J Cancer. 2014;134:2865–2877. doi: 10.1002/ijc.28631. [DOI] [PubMed] [Google Scholar]
- Blackham AU, Northrup SA, Willingham M, Sirintrapun J, Russell GB, Lyles DS.et al. (2014Molecular determinants of susceptibility to oncolytic vesicular stomatitis virus in pancreatic adenocarcinoma J Surg Res 187412–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C.et al. (2012The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers Mol Ther 20749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurayoshi K, Ozono E, Iwanaga R, Bradford AP, Komori H, Ohtani K. Cancer cell specific cytotoxic gene expression mediated by ARF tumor suppressor promoter constructs. Biochem Biophys Res Commun. 2014;450:240–246. doi: 10.1016/j.bbrc.2014.05.102. [DOI] [PubMed] [Google Scholar]
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC.et al. (2008Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial Lancet Oncol 9533–542. [DOI] [PubMed] [Google Scholar]
- Kim MK, Breitbach CJ, Moon A, Heo J, Lee YK, Cho M.et al. (2013Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans Sci Transl Med 5185ra63. [DOI] [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M.et al. (2013Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer Nat Med 19329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workenhe ST, Mossman KL. Rewiring cancer cell death to enhance oncolytic viro-immunotherapy. Oncoimmunology. 2013;2:e27138. doi: 10.4161/onci.27138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ.et al. (2011Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans Nature 47799–102. [DOI] [PubMed] [Google Scholar]
- Gerdemann U, Keirnan JM, Katari UL, Yanagisawa R, Christin AS, Huye LE.et al. (2012Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections Mol Ther 201622–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]