

Abstract
Eukaryotic secretory proteins cross the endoplasmic reticulum (ER) membrane through a protein conducting channel contained within the Ribosome–Sec61Translocon Complex (RTC). Using a zinc-finger sequence as a folding switch, we show that cotranslational folding of a secretory passenger inhibits translocation in canine ER microsomes and in human cells. Folding occurs within a cytosolically inaccessible environment, after ER targeting but before translocation is initiated and is most effective when the folded domain is 15–54 residues beyond the signal sequence. Under these conditions, substrate is diverted into cytosol at the same stage of synthesis that unfolded substrate enters the ER lumen. Moreover, the translocation block is reversed by passenger unfolding even after cytosol emergence. These studies identify an enclosed compartment within the assembled RTC that allows a short span of nascent chain to reversibly abort translocation in a substrate-specific manner.
Keywords: Translocon, Sec61, SecY, Translocation, Protein secretion, Ribosome–translocon complex, Prolactin, endoplasmic reticulum, ER, zinc finger
Introduction
Most eukaryotic secretory proteins cross the endoplasmic reticulum (ER) membrane cotranslationally through the Sec61αβγ Protein Conducting Channel (PCC), which is contained within a large macromolecular machine referred to here as the Ribosome Sec61 Translocon Complex (RTC)1–4. Translocation is initiated during protein synthesis when a signal sequence emerges from the ribosome, binds the cytosolic Signal Recognition Particle (SRP), and targets the ribosome–nascent chain complex (RNC) to the ER membrane5. GTP hydrolysis by SRP and its cognate ER-bound receptor causes signal sequence release and promotes ribosome binding to cytosolic loops of Sec61α5,6, thereby shielding the nascent chain from the cytosol and aligning the ribosome exit tunnel with the PCC pore. Subsequent engagement of the signal sequence with Sec61α opens the axial pore of the PCC7 and establishes a continuous pathway for protein translocation that extends from the ribosome peptidyltransferase center to the ER lumen8–12. The signal sequence therefore plays two crucial roles in this process, membrane targeting and channel gating, which together ensure efficient substrate transport into the ER lumen13–15.
In contrast to signal sequences, less is known about how the passenger domain influences cotranslational translocation. While many signal sequences can transport non-cognate passengers16,17, translocation efficiency also depends on the cargo substrate14,17–20. For example, following ER targeting, residues in the passenger domain contact cytosolic loops of Sec61α, which extend into, and partially fill, the ribosome exit vestibule8,11. Within this environment, electrostatic interactions between the nascent polypeptide and RTC aid in orienting basic residues towards the cytosol, thereby dictating signal sequence topology and vectorial movement of the unfolded passenger domain through the narrow Sec61α pore10,21,22. These interactions take place within a restricted space that forms as the ribosome docks onto the translocon and prevents the elongating nascent chain from prematurely accessing the cytosolic compartment9,10,12,23.
A hallmark of cotranslational translocation is that protein movement through the Sec61 pore is temporally coupled to peptide synthesis. Indeed, potential passenger domains that fold prematurely in the cytosol prior to ER targeting inhibit translocation24–27. Ribosome binding to the ER can also delay folding of large passenger domains, indicating that the RTC itself plays an active role in maintaining the nascent polypeptide in an extended, translocation-competent state28. Consistent with this, most structural studies show the nascent polypeptide in an extended conformation as it passes through the ribosome–translocon junction, which in turn, provides limited space for folding8,11. These findings have established a general paradigm wherein tertiary folding is delayed during cotranslational translocation until the passenger domain exits the PCC into the ER lumen29,30.
On the other hand, a substantial region of nascent polypeptide (~50–60 residues) can accumulate within the assembled RTC after ER targeting and before translocation is initiated10. In addition, a short loop of polypeptide was recently shown by Cryo-electron microscopy to reside near the cytosolic surface of ribosome-bound bacterial SecY before passing through the pore31. Thus, nascent polypeptide structure at the ribosome–translocon junction and the functional implications of the junctional space remain poorly understood. Because the ribosome exit site serves as a folding vestibule for cytosolic domains and transmembrane segments32–34, we wondered whether it might also support folding of a nascent secretory passenger, and thereby influence translocation from within the interior of the RTC.
To test this hypothesis, we used a small, well-defined zinc finger domain whose tertiary structure can be precisely and reversibly controlled26,35. The C2H2-type Zn-fingers are 28–32 residue, autonomous, fast-folding domains that form highly stable tertiary structures in the presence of Zn+2 (ref 35,36) (Fig. 1a). Results show that tertiary passenger folding can indeed occur within a restricted space at the ribosome–translocon junction. Moreover, Zn-induced folding strongly inhibited translocation, diverting the normally secreted passenger into the cytosol, both in vitro and in mammalian cells and for chimeric as well as native proteins. Zn-finger placement 15–54 residues downstream of the signal sequence caused the greatest inhibition. Moreover, this translocation block was reversed when the passenger was unfolded during early but not late, stages of cytosolic exposure. These data demonstrate that functional mammalian RTCs contain a restricted compartment near the ribosome exit vestibule that permits structural properties of the nascent passenger domain to influence translocation outcome.
Figure 1. Zn-induced folding blocks cotranslational pPL translocation in vitro.

a) Ribbon diagram with peptide sequence (top) and space filling structure (bottom) of the S. cerevisiae ADR1a zinc finger domain (PDB: 2ADR)36. Note that the NMR structure shows the C11A variant indicated by parentheses. Zn+2 (red sphere) is coordinated by Cys5, Cys8, His21, and His26. b) Schematic showing location of the ADR1a zinc finger domain (black box) in pPL45-Zn relative to the pPL signal sequence (gray) and PL domain (white). Phosphorimage of in vitro synthesized [35S]Methionine-labeled pPL constructs translated in the presence and absence of CRMs and Zn+2 as indicated. Unprocessed and signal cleaved polypeptides are shown as filled and empty circles, respectively. Right panels show same products after proteinase K (PK) digestion to remove untranslocated protein. (Uncropped gel images are provided in Supplementary Fig. 2a.) c) Percent of Zn+2-mediated translocation inhibition at indicated Zn-finger insertion sites was calculated by dividing percentage of protected product in the presence of Zn+2 (0.5 mM) by that in the absence of Zn+2 (See also in supplementary Fig. 1). Results show mean and s.e.m. (n=3 independent experiments).
Results
Design of an inducibly folded secretory passenger
To control tertiary folding of a secretory passenger domain, we used the 29 residue ADR1a C2H2-type Zn-finger peptide from S. cerevisiae as an inducible folding switch26,35,36. This class of Zn-fingers comprises small, autonomously folding domains that coordinate a single Zn+2 ion between 2 cysteines and 2 histidines with picomolar affinity35,36. Folding is induced within seconds upon exposure to Zn+2 to form a highly stable tertiary β-strand and α-helical structure approximately 27 Å × 25 Å × 21 Å in size (Fig. 1a)36,37. Translation in the presence and absence of Zn+2 therefore provides an ideal method to induce cotranslational folding of otherwise identical polypeptides in a complex biological machine such as the RTC. This strategy enabled us to test whether Zn-induced folding occurred at the ribosome exit site on membrane-targeted ribosomes, whether folding occurred in the cytosol or a cytosolically inaccessible compartment, and whether folding influenced cotranslational translocation of the downstream passenger.
Zn-Induced folding blocks pPL translocation in vitro
The ADR1a Zn-finger was substituted for an equivalent number of residues in the well-studied secretory preprolactin (pPL) at multiple sites downstream of the signal sequence, and constructs were translated in rabbit reticulocyte lysate (RRL) with or without canine rough microsomes (CRMs) (Supplementary Fig. 1 and Fig. 1b). In the absence of Zn+2, both pPL and pPL45-Zn were translocated into the microsome lumen (>80% efficiency) (Fig. 1b). In contrast, translation in the presence of Zn+2 inhibited translocation only in those proteins that contained the Zn-finger sequence (Supplementary Fig. 1 and Fig. 1b, c). Although Zn-finger insertion inhibited translocation at all sites tested (from residues 32–170), maximal inhibition was observed for pPL45-Zn and pPL55-Zn where the Zn-finger occupies residues 45–74 and 55–84 respectively (Supplementary Fig. 1 and Fig. 1c). Thus, the optimal site for inhibition was located 15 to 54 residues beyond the C-terminus of the signal sequence, prompting us to use the pPL45-Zn construct in subsequent experiments.
Importantly, Zn+2 addition did not effect wild type pPL synthesis, processing (signal cleavage), or translocation (PK protection), thus supporting the conclusion that Zn+2-mediated translocation inhibition was indeed caused by induced folding of the Zn-finger domain. It should be noted that the free Zn+2 concentration in RRL is similar to that found in cells (i.e. in the sub-micromolar range) due to the presence of residual EGTA in the translation reaction (Kd (Zn+2) for EGTA = 7 × 10−9 at pH 7) (See methods).
Zn-Induced folding blocks translocation in vivo
We next tested whether the Zn-finger-induced translocation block might result from the slower rate of in vitro translation compared to that of intact cells (~0.5–1 aa/sec versus 5–7 aa/sec, respectively). 35S-methionine pulse-labeling revealed that wild type pPL was efficiently processed in HEK 293T cells in both the presence and absence of the Zn+2 chelator, N,N,N',N'-Tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) (Fig. 2a). In the presence of Zn+2, however, only 37 +/- 8% of pPL45-Zn underwent signal sequence cleavage, whereas translocation efficiency was restored to 93 +/- 3% following Zn+2 chelation (Fig 2a, b). These results are remarkably similar to those observed in vitro and confirm that the passenger-induced translocation block also occurred under physiological conditions and was not an artifact of in vitro translation kinetics, translocation, ER targeting, or translocon gating.
Figure 2. Zn-induced folding blocks translocation in cultured cells and in a native protein substrate.

a) Phosphorimage of [35S]Methionine-labeled wild type pPL and pPL45-Zn expressed in vitro (left panel, control) and in HEK 293T cells (right panel) grown under conditions of Zn+2 supplementation or chelation using TPEN as indicated. b) Quantification of experiments as in panel a shows percent pPL processing (signal cleavage) in the presence and absence of Zn+2. Results show mean and s.e.m. (n=4, independent experiments). c) Schematic of ZnF_788 1–218 construct (Uniprot entry Q6ZQV5) showing signal sequence (hatched), zinc finger sequences (black), and N-linked glycosylation sites (asterisks). d) In vitro translation of ZnF_788 1–218 in presence of CRMs and the NYT tripeptide as indicated. Singly and doubly glycosylated polypeptides are indicated by * and **, respectively. e) In vitro translation of ZnF_788 1–218 as in panel d, showing glycosylation is prevented by Zn+2 addition. f) Proteinase K (PK) protection of in vitro-translated ZnF_788 1–218 unprocessed and glycosylated bands in the absence and presence of Zn+2. Percent protection was calculated by dividing PK-treated band intensity by untreated band and normalizing to protection observed for wild type pPL signal cleaved protein. Results show mean and s.e.m. (n=4, independent experiments). (Uncropped gel images for panels a, d, e, and f are shown in Supplementary Fig. 2b, c, d and e.)
Substrate folding controls native protein translocation
Zn-finger domains are typically involved in nuclear DNA binding, and admittedly, represent a somewhat contrived substrate for cotranslational folding in the context of pPL. However, a survey of the Uniprot database identified a 615 residue human protein of unknown function (ZnF_788 (ID Q6ZQV5)) containing multiple Zn-finger motifs downstream of a weak, uncleaved N-terminal signal sequence (predicted by Signal 4.1. www.cbs.dtu.dk/services/SignalP)38. The first Zn-finger is located at residue 56, and two N-linked glycoslyation consensus sites are present at residues 67 and 161 (Fig. 2c). In vitro expression of the first 218 residues of ZnF_788 in the presence of CRMs generated a 27 kDa polypeptide and two N-linked glycosylated species migrating at 30 and 33 kDa (Fig 2d). Zn+2 addition prevented glycosylation at both sites (Fig 2e), and protease protection further confirmed that translocation of these glycosylated polypeptides was inhibited by Zn+2 (Fig. 2f). Thus, induced folding can effectively block cotranslational translocation of a native passenger domain.
pPL45-Zn constructs properly target to the ER
To rule out the trivial possibility that Zn-finger folding might simply interfere with membrane targeting, pPL45-Zn was expressed in the presence of Zn+2 from either full-length or truncated RNA transcripts, the latter of which lack a terminal stop codon and arrest translation at defined polypeptide lengths. This strategy allowed us to examine intact RNCs that contain a covalent peptidyl-tRNA bond (Fig. 3a). When translated in the absence of CRMs, truncated nascent chains as well as full-length pPL45-Zn remained in the supernatant after ultracentrifugation (Fig. 3a). In the presence of CRMs, however, nascent chains primarily pelleted with the membrane (Fig. 3a and Fig. 3b). Surprisingly, the majority of full-length pPL45-Zn remained uncleaved and was recovered in the supernatant (Fig. 3b). Zn-finger folding therefore not only blocked translocation without affecting membrane targeting, but also released the unprocessed nascent chain from the ER membrane after translation termination.
Figure 3. Zinc finger folding does not block ER targeting.

a) Phosphorimage of pPL45-Zn transcripts truncated at indicated codons translated in RRL in the presence of Zn+2 ± CRMs as indicated. Equivalent samples of input (i), supernatant (s) and membrane pellet (p) were examined by SDS-PAGE. Peptidyl-tRNA bands and spontaneously hydrolyzed polypeptides are indicated by empty and filled diamonds, respectively. (Uncropped gel images are provided in Supplementary Fig. 2f, g.) b) Percentage of cytosolic peptidyl-tRNA was calculated for each construct by dividing protein recovered in supernatant by the total input. Results show mean and s.d. (n=3 independent experiments).
Passenger folding occurs inside the membrane-bound RTC
How then does passenger folding block translocation after the RNC has docked onto the translocon? To address this question, we synthesized the pPL45-Zn 87-mer and wild type pPL 86-mer (control) in the presence of CRMs but without Zn+2 to capture the Zn-finger peptide in an unfolded state within the narrow confines of the ribosome exit tunnel (Fig. 4a)32,39,40. Zn+2 was then added (or not), and nascent chains were released from the ribosome by the aminoacyl-tRNA analog puromycin (Fig. 4b). Upon puromycin addition, wild type polypeptides translocated into the ER lumen irrespective of Zn+2, as evidenced by efficient signal sequence processing (Fig. 4b). As expected, no additional processing was observed if RNase was added after SDS denaturation. In the absence of Zn+2, the pPL45-Zn 87-mer behaved identically to wild type. However, translocation was inhibited by addition of Zn+2 prior to puromycin treatment.
Figure 4. Zn-induced folding blocks translocation at a post-ER targeting step.

a) Cartoon of pPL45-Zn 87-mer, showing approximate location of the zinc finger domain within the exit tunnel of translocon-bound ribosome. b) Protocol Schematic: pPL 86-mer and pPL45-Zn 87-mer were translated in RRL + CRMs in the absence of Zn+2. Cycloheximide was added ± Zn+2, and nascent chains were treated with puromycin or denatured in SDS followed by RNase digestion. Panels show phosphorimages of translation products treated as indicated. Migration of signal cleaved (translocated) and uncleaved polypeptide are designated by empty and filled circles, respectively. c) Schematic of pPL45-Zn showing location of cysteine residues. d, e) Puromycin-treated samples of pPL45-Zn prepared as in panel b in absence of Zn+2 (panel d) or presence of Zn+2 (panel e) were incubated with PEG-mal alone or after addition of 1% digitonin (dig) or 0.5M NaCl as indicated. Signal uncleaved and cleaved polypeptides are indicated by filled and empty circles, respectively. Arrows indicate ladder of peptides containing 1, 2, 3 or 4 PEG moieties. (Uncropped gel images for panels b, d and e are shown in Supplementary Fig. 2h, i, and j.)
We next used the membrane impermeable reagent polyethyleneglycol-maleimide 5,000 Da (PEG-mal)10 to determine whether passenger folding takes place in a cytosolically accessible compartment. PEG-mal covalently crosslinks aqueous, accessible cysteine residues and produces a readily detectible increase in molecular mass 41. Even though multiple cysteine residues were outside the ribosome exit tunnel in the pPL45-Zn 87-mer (Fig. 4c), these nascent chains remained inaccessible to cytosolic PEG-mal after ribosome release both in the presence and absence of Zn+2 (Fig. 4d,e). In the absence of Zn+2, most pPL45-Zn 87-mer underwent signal cleavage and translocated into the ER lumen as demonstrated by variable pegylation of cysteines after membrane permeabilization (Fig. 4d). In the presence of Zn+2, however, most of the pPL45-Zn 87-mer remained unprocessed, and its cysteine residues became accessible to cytosolic PEG-mal only after the ribosome–translocon junction was disrupted by high salt10 (Fig. 4e). Zn-induced folding therefore trapped the 87-mer in a shielded environment within the assembled RTC, presumably at or near the ribosome–translocon junction, thereby preventing it from accessing either the cytosol or ER lumen even after the peptidyl-tRNA bond was cleaved. Because there was ample time after ER targeting for the pore to open, as shown for the wild type pPL 86-mer, the translocation block appears to result from rapid, Zn-induced folding as the nascent chain exited the ribosome but before it passed through the open translocon pore.
These data identify a mechanism for folding-induced translocation block that differs from N-terminus folding in the cytosol, as in the case of signal anchor sequences26. Our results also contrast with models where the growing nascent chain is predicted to pass through a gap beneath the ribosome and subsequently fold in the cytosol before it has a chance to move through the translocon pore8,11,29. Rather, the cargo substrate is sequestered and folded within the assembled RTC after targeting, thereby mechanically blocking translocation from within the complex itself.
A shielded RTC compartment controls substrate localization
If cargo folding blocks translocation from within the RTC, then at some point during synthesis the folded passenger domain must be diverted from its lumenal destination into the cytosol. Previous in vitro studies have indicated that pPL signal sequence cleavage first occurs at a nascent chain length between 132 and 169 residues42. This is also the case for pPL45-Zn. In the absence of Zn+2, pPL45-Zn signal sequence cleavage was first observed at a nascent chain length of 136 aa (~50% cleavage), and processing efficiency increased at longer truncations (Fig. 5). Because the signal sequence is 30 residues long, approximately 100 residues of the passenger domain was therefore synthesized before the nascent chain accessed signal peptidase in the ER lumen. In each instance, addition of Zn+2 blocked processing, indicating that passenger folding also inhibits translocation of ribosome-bound truncated nascent chain intermediates.
Figure 5. Passenger folding inhibits translocation of ribosome-bound nascent chains.

Phosphorimage of truncated translocation intermediates synthesized ± CRMs and ± Zn+2. Peptidyl-tRNA and released polypeptide are shown by empty and filled diamonds, respectively. Arrows show signal sequence cleaved peptidyl-tRNA polypeptides, observed for truncations 136–187 only in the absence of Zn+2. (Uncropped gel images are provided in Supplementary Fig. 2k.)
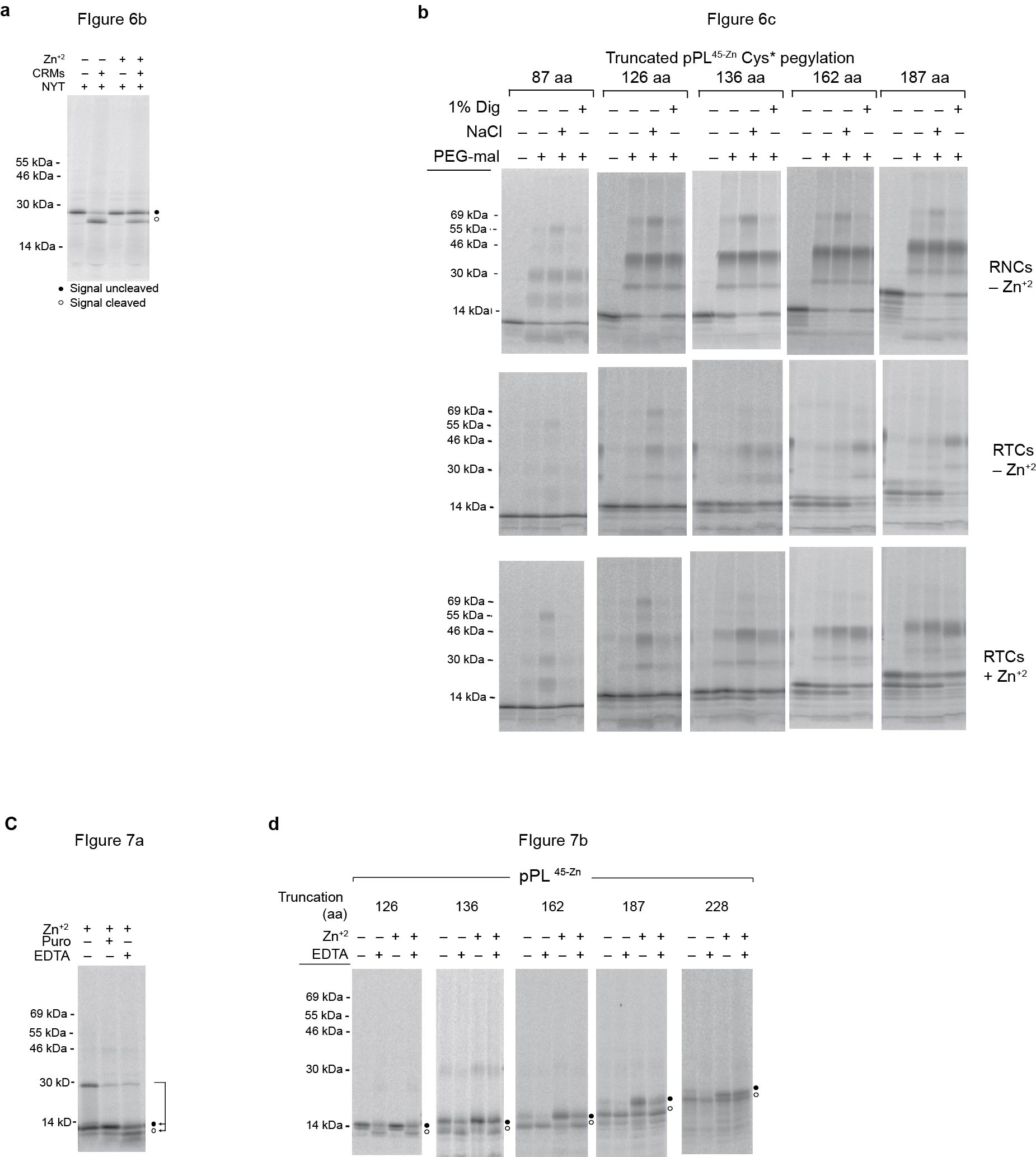
To determine the stage of synthesis at which the folded passenger gains access to cytosol, pegylation was performed at progressive nascent chain lengths. To simplify the pegylation pattern, Cys25, Cys34, and Cys87 were converted to serine, generating pPL45-Zn Cys*, leaving four cysteine residues (Cys41, Cys49, Cys52, Cys55) within or adjacent to the Zn-finger (Fig. 6a). These changes had no effect on signal sequence activity or Zn-induced translocation block (Fig. 6b).
Figure 6. Zinc finger folding directs the nascent chain into cytosol at 162aa.

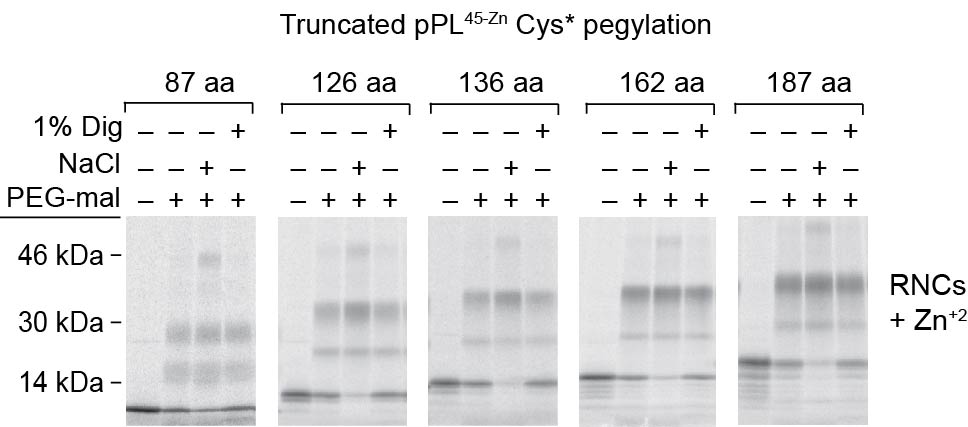
a) Schematic of pPL45-Zn Cys* showing approximate location of cysteines (empty circles) at residues 41, 49, 52 and 55 and N-linked glycosylation site (asterisk). C-terminal cysteines are not present in truncations examined. b) Phosphorimage of full length pPL45-Zn Cys* translated in the presence of NYT tripeptide ± CRMs, ± Zn+2. c) Phosphorimages of pPL45-Zn Cys* truncated at indicated residues and translated in the absence (upper panels) or presence of CRMs ± Zn+2 (middle and bottom panels). Pegylation of RNCs or RTCs was performed in the presence and absence of 1% digitonin (dig) or 0.5 M NaCl as indicated. Processed and unprocessed polypeptides are indicated by empty and filled circles, respectively. d) Percent pegylation of pPL45-Zn Cys* free RNCs and membrane-bound RTCs generated in absence of Zn+2 was calculated as described in Methods and plotted as a function of nascent chain length. e) Percent pegylation of pPL45-Zn Cys* calculated as in panel d but for RTCs generated in absence of Zn+2 ± 1% digitonin or 0.5 M NaCl. f) Percent pegylation of pPL45-Zn Cys* was calculated as in panel e but for RTCs translated in the presence of Zn+2. g) Percent pegylation of unprocessed (signal uncleaved) pPL45-Zn Cys* generated in the presence of Zn+2. Values reflect disappearance of unprocessed nascent chain following addition of PEG-mal. All results show mean and s.e.m. (n=3, independent experiments). (Uncropped gel images for panels b and c are shown in Supplementary Fig. 4a, b.)
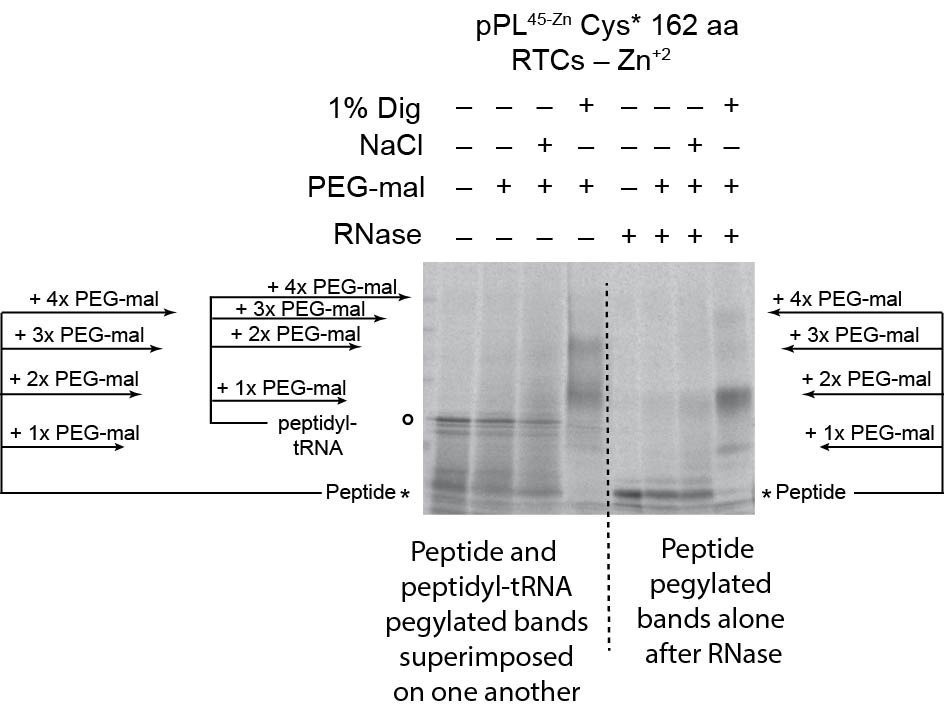
When translation was carried out in the absence of CRMs, PEG-mal readily modified RNCs containing truncated pPL45-Zn Cys* constructs from 87 to 187 aa in length, with doubly pegylated species predominating (Fig. 6c, d). Similar results were observed in both the presence and absence of Zn+2 (Supplementary Fig. 3 and Fig. 6d). For these experiments, samples were digested with RNase prior to SDS-PAGE to better quantify pegylation efficiency and avoid overlap between pegylated nascent chains and peptidyl-tRNA species (Supplementary Fig. 5). When CRMs were present in the translation reaction, pegylation was decreased in the absence of Zn+2 for all truncations examined, again showing that the ribosome–translocon junction prevents the nascent chain from prematurely accessing cytosol (Fig 6c, d). Consistent with this, high salt treatment increased pegylation efficiency for truncations 126, and 136 (Fig. 6c, e), although to a level less than that observed for free RNCs (Fig. 6c). In the absence of Zn+2, nascent chains also began to translocate into the ER lumen at a length of 136 aa, as shown by pegylation of the processed band following membrane solubilization (Fig. 6c). Under these conditions, translocation efficiency further increased as the nascent chain was extended to 162 and 187 residues (Fig 6c, e).
Addition of Zn+2, resulted in a very different outcome. Nascent chains 87 to 136 residues in length were still shielded from cytosolic PEG-mal in intact RTCs; unprocessed polypeptides remained partially sensitive to high salt, and processed, lumenal polypeptides were pegylated following digitonin solubilization, (Fig. 6c, f). At a length of 162 aa, however, most nascent chains failed to undergo signal cleavage (Fig. 6c), and these unprocessed polypeptides gained access to the cytosol as evidenced by increased pegylation efficiency (Fig. 6c, f, g). Similar results were observed for the 187 truncation, indicating that the critical decision to direct the nascent polypeptide into the lumen or cytosol was made as the nascent chain length was extended from 136 to 162 residues. Thus Zn-finger folding arrested the passenger domain within a shielded compartment in the RTC before the nascent chain passed beneath the ribosome and into the cytosol.
Passenger-induced translocation block is reversible
Passenger domain folding disrupts translocation in a unique way because the RTC is primed for lumenal delivery by the signal sequence but faces a mechanical block that eventually releases the substrate into the cytosol when translation is terminated (Fig. 3). This raises an interesting question of whether the translocon remains in a translocation-competent state after the substrate has gained access to the cytosol. To address this question, we took advantage of the ability of ethylenediaminetetraacetic acid (EDTA) to rapidly and reversibly unfold Zn-finger domains43. In addition to stimulating peptidyl-tRNA bond cleavage and dissociation of small and large ribosomal subunits by Mg+2 chelation44, EDTA chelates free Zn+2 and decreases the Zn+2 off-time of folded Zn-finger domains from 1–3 hours to less than 1 second (~106 decrease in off rate) via a ternary “catalytic” intermediate37,43. We therefore compared the effects of puromycin and EDTA on the pPL45-Zn 87-mer. Both treatments induced peptidyl-tRNA bond cleavage (Fig. 7a). However, puromycin release failed to initiate translocation due Zn-finger folding (also shown in Fig. 3). In contrast, signal sequence processing increased ~400% (from <10% to 41 ± 6%) when folding was prevented by EDTA (Fig. 7a,d). Similar results were observed for the pPL45-Zn 127 and 136-mer, in which the Zn-finger had emerged from the ribosome but not yet entered the ER lumen or cytosol (Fig. 7b–d). EDTA therefore not only prevented folding as the Zn-finger exited the ribosome (Fig. 7a), but also re-initiated translocation after the Zn-finger had likely folded within in the RTC (Fig. 7b). Remarkably, EDTA treatment also increased translocation efficiency nearly 200% for the pPL45-Zn 162-mer (Fig. 7c,d) and by 30% for the 187-mer. The extent of reversibility decreased at longer lengths (187 and 228 aa), coincident with an increase in basal processing. Taken together, these data show that the folding-induced translocation block maintains the RTC in a translocation-competent state and can be reversed when the passenger is unfolded even after gaining access to the cytosol (Fig. 7e).
Figure 7. Passenger-induced translocation block is reversible upon unfolding.

a) Phosphorimage of pPL45-Zn 87-mer translated in presence of Zn+2 following EDTA or puromycin treatment. Signal cleaved and uncleaved peptides are indicated by empty and filled circles, respectively. b) Experimental protocol for EDTA-induced translocation initiation. c) Phosphorimages of pPL45-Zn truncated at residues indicated and expressed in vitro ± Zn+2 and ± EDTA d) Graph showing percent of polypeptide recovered that underwent signal cleavage for each truncation, before and after EDTA treatment. Results show mean and s.e.m.. (n=3 independent experiments). e) Diagram of model in which peptide accumulation at the ribosome–translocon junction leads to Zn-induced folding of the passenger domain, which mechanically blocks translocation from within the RTC. This block can be reversed upon unfolding prior to and after the nascent chain gains access to cytosol, but the efficiency of reversal decreases as more polypeptide enters the cytosol. Ultimately the passenger is released either into the cytosol in uncleaved form, or into the lumen as the processed domain. (Uncropped gel images for panels a and c are shown in Supplementary Fig. 4 c, d.)
Discussion
In this study we used a small, well-defined peptide, whose tertiary structure can be precisely controlled, to show that cotranslational folding of a nascent passenger domain can directly inhibit translocation across the ER membrane from within the assembled RTC. By comparing otherwise identical folded versus unfolded substrates, we identified a region 15–54 residues C-terminal to the signal sequence that is particularly sensitive to translocation inhibition. Results revealed several unexpected findings with implications for both translocon function and protein localization. First, tertiary folding of a secretory passenger can occur after ER targeting but before translocation is initiated, as the nascent polypeptide accumulates near the ribosome–translocon junction. Second, folding takes place within a restricted environment that prevents the nascent polypeptide from prematurely accessing cytosolic contents. Third, the folded domain appears to induce a mechanical block within the RTC that diverts the passenger into the cytosol at the same stage of synthesis that an unfolded passenger would normally translocate into the ER lumen. Fourth, despite this block, the translocon remains in a translocation-competent state, and translocation can be initiated if the passenger is unfolded even after accessing the cytosol. Fifth, with continued synthesis, the block becomes irreversible, and the folded passenger is released into the cytosol when translation terminates. Finally, this translocation block is observed both in vitro and in mammalian cells and for both native as well as engineered substrates. Protein secretion therefore results from a complex interplay between a functional signal sequence and structural properties of its cargo.
A central paradigm of cotranslational translocation is that SRP-mediated ER targeting controls the timing and location of substrate folding. When an N-terminal signal sequence emerges from a cytosolic ribosome and binds SRP, translation is slowed until the RNC docks onto the ER membrane5,6,45. Translation resumes following SRP release, allowing the signal sequence to insert into Sec61α and open the PCC pore7,31,46–48. Strong signal sequences such as pPL thus ensure that the elongating nascent chain contacts the PCC before sufficient polypeptide emerges into the cytosol to stably fold45. Peptide domains that fold prior to membrane targeting prevent translocation24–26, and ribosome binding to Sec61 can actually delay tertiary folding of large passenger domains28. These findings, together with Cryo-electron microscopy structural studies showing that cytosolic loops of Sec61 partially fill the ribosome vestibule8,11, have suggested the RTC actively maintains nascent chain in an extended conformation until it reaches its destination in the ER lumen.
Results of this study support an alternate model, wherein the nascent chain does not pass immediately from the ribosome through the PCC after ER targeting10, but rather accumulates transiently within the RTC in a cytosolically inaccessible space of sufficient size to accommodate a rigid domain with dimensions 27 Å × 25 Å × 21 Å35. Importantly, our model differs from that of Cheng et al. who inserted a rapidly folding ubiquitin domain downstream of a signal anchor sequence29 and concluded that slow translocon gating, rather than substrate folding, was responsible for diverting the passenger into the cytosol30. This raises the question as to how the decision is ultimately made to direct peptide movement into different cellular compartments42. In our system, ribosome-attached pPL nascent chains begin to translocate at a chain length of 136 aa, and become fully committed at a length of 162 aa, quite late after ER targeting. Interestingly, this is the same stage of synthesis that the passenger domain begins to emerge from beneath the ribosome and into the cytosol when translocation is blocked by the folded passenger. Thus one factor that may contribute to localization is the amount of peptide that can be accommodated within the RTC.
It is not yet evident from current electron density maps precisely where in the RTC tertiary passenger folding might take place8,11. However, given the narrow dimensions of the ribosome exit tunnel and PCC pore, it likely occurs near the ribosome exit site. One possibility is that assembly of Sec6αβγ, together with various translocon- associated proteins in native ER membranes, creates a vestibule of sufficient size to accommodate the folded Zn-finger that is not evident in current structural studies containing only the ribosome and PCC2,10,12. Alternatively, the mode of ribosome binding during initial targeting and passenger accumulation (studied here), may differ from the commitment phase of ongoing translocation captured in structural studies8,11. Potential dynamic changes at the ribosome–translocon junction associated with targeting and translocation could also potentially explain this discrepancy4,9,10,49. Consistent with this latter possibility, Park et al. recently reported a stage of translocation in which a short peptide loop resided near the cytosolic interface of ribosome-bound bacterial SecY before translocation31. Thus, similar to its reported role for cytosolic subdomains and transmembrane segments32,34, the ribosome exit site may also provide a folding vestibule for small secreted domains that reduces translocation efficiency, even after ribosome docking.
Tertiary passenger folding therefore provides a unique structural feature that can prevent translocation in the absence of other known translocation modifiers (i.e. hydrophobic transmembrane (stop transfer) segment50–52, a positively charged cluster53, or a pause transfer sequence54). In certain respects, passenger folding is functionally analogous to pause transfer sequences such as those found in apolipoprotein-B54 and other translocated proteins55 (i.e. prion protein). Pause transfers are short hydrophilic sequences that transiently terminate translocation by disrupting the ribosome–translocon junction and exposing the nascent chain to the cytosol while allowing translocation of the resulting cytosolic loop to resume after a limited period of synthesis. In the case of Apolipoprotein-B, pausing allows lipid transfer during translocation56, and targets Apolipoprotein-B to ER-associated degradation when insufficient lipid is available57, or when the microsomal triglyceride transfer protein is absent or inhibited58. Despite their discovery nearly 25 years ago, the mechanism by which a sequence element can reversibly block translocation from within the RTC has remained a mystery. Our study demonstrates tertiary folding within a localized region of the nascent secretory passenger may provide one such method.
In summary, we have shown that translocation efficiency of a signal sequence can be coupled to tertiary structural properties of its downstream passenger domain. This relationship was recapitulated in mammalian cells grown at physiologic temperature, indicating that it was not caused by the reduced in vitro kinetics of translation, folding, ER targeting, translocon gating, or translocation. These findings raise the possibility that cotranslational folding might act more generally to impact protein translocation in cells. Toward this goal, we identified an endogenous, Zn-finger-containing protein, ZnF_788, whose signal sequence activity was also modified by zinc availability. Thus it may not be unreasonable to speculate that ZnF_788 translocation could be affected by free cellular Zn+2 levels which exhibit wide variations in response to cell stress and cytosolic redox changes 59. While Zn-finger domains are common in cytosolic and nuclear proteins, analysis of the SCOP and CATH structural databases revealed that they are rare in the secretory proteome(Supplementary Fig. 6). Surprisingly, however, small structurally defined (sub)domains are nearly twice as common within the first 100 residues following a signal sequence as are domains of similar size within the first 100 residues of cytosolic and nuclear proteins (Supplementary Fig. 6). As expected, approximately 70% of these secretory domains contain disulfide bonds, which would stabilize folding only after ER entry or perhaps under conditions of oxidative stress. These findings raise the possibility that folding kinetics or stability may confer unique translocation properties on secretory cargo that are coupled to their cognate signal sequences15. However, further work is needed to determine the extent to which protein domains less stable than the Zn-finger examined here may impact general translocation processes.
ONLINE METHODS
Plasmid construction
Coding sequences of pPL derivatives were cloned between NcoI and PstI restriction sites downstream of the 5’ UTR from Xenopus globin as in the previously described plasmid pSP-BPI60,61 using an pSP64T vector (Promega, Madison, WI). The ADR1a zinc-finger domain DNA sequence (AAG CCA TAT CCA TGC GGC CTC TGT AAT AGA TGC TTC ACC AGA AGA GAC CTG CTG ATC AGA CAC GCT CAG AAG ATC CAC AGC GGT AAC) was derived from the corresponding amino acid sequence described by Denzer et al.26, noting that cited NMR structures used a C11A variant36. Briefly, the Zn-finger sequence was used to replace an identical number of codons in pPL with the resulting plasmid designated by the first residue in pPL that was replaced. For pPL79-Zn, the Zn-finger containing product was subcloned into pSP-BPI using an EcoRI restriction site generated by converting codons 43 and 44 to GAA TCC. Other ADR1a-containing constructs were generated by PCR overlap extension without additional codon changes. pPL45-Zn-Cys* was generated by converting pPL residues Cys25, Cys34 and Cys87 to serine (codons TCC, TCT and TCC, respectively) using the parent plasmid pPL45-Zn. For mammalian cell expression, pPL and pPL45-Zn coding sequences were cut from pSP64 and ligated into Hind III and BamHI sites in pcDNA3 (Life Techologies, Grand Island, NY) using standard molecular biology techniques. The full length cDNA for ZnF_788 was obtained from the “full-length long Japan” clone collection from the Japanese Biological Resource Center (http://www.nbrc.nite.go.jp/e/hflcdna-e.html) (ID FLJ46867)62. The 5’ UTR and codons 1–218 of ZnF_788 were amplified by PCR and subcloned into pSP64-BPI NcoI and EcoRI cloning sites, which introduced a classical Kozak sequence and inserted an additional aspartate after the initiation methionine. All site-directed mutagenesis was performed using the PCR overlap extension technique63, and all PCR-amplified and cloned regions were verified by DNA sequencing.
In vitro transcription and translation
cDNA templates for in vitro translation were generated by PCR amplification using a sense oligonucleotide complementary to BP 2744–2763 in pSP64T and antisense oligonucleotides complimentary the pPL coding sequence. Truncation sites correspond to the last (3’) codon of pPL included in the PCR fragment which was converted to a Val codon (GTG) to stabilize the peptidyl-tRNA bond. RNA transcripts were synthesized using SP6 polymerase for 60 min at 40°C under conditions identical to those described elsewhere64. Translation was carried out in vitro at 24°C for 60 min as described64,65 in a reaction containing 20% vol/vol crude RNA transcript, 0.05 µCi/µl EasyLabel [35S]Methionine (Perkin Elmer, Waltham, MA), and 40% vol/vol rabbit reticulocyte lysate prepared as described64,65. Prior to use, RRL was digested for 10 min by addition of 1 mM CaCl2, followed by 0.15 U/µl S7 micrococcal nuclease (Roche Diagnostics, Indianapolis, IN), and digestion was terminated by addition of 2.0 mM EGTA (final concentrations are given). Where indicated ZnCl2 solution in 6 mM HCl was added to translation reactions at final concentration of 0.5 mM unless otherwise noted. Since RRL contains ~1mM excess EGTA, the free [Zn+2] is estimated to be in the sub-micromolar range similar to that found in cells (Kd (Zn+2) for EGTA~7 × 10−9 at pH 7)66. Where indicated, rough ER microsomes prepared from canine pancreas64 were added prior to the start of translation at a final concentration of 5.0 OD280. For pPL45-Zn Cys* and elsewhere where indicated, a tripeptide inhibitor of oligosaccharyltransferase (NYT) was added at 50 µM concentration prior to the start of translation to prevent N-linked glycosylation. All samples were analyzed by SDS-PAGE on 10–18% gradient gels and imaged on KODAK imaging screens using a BioRad personalFx phosphorimager and Quantity One software (BioRad, Hercules, CA).
Proteaseinase K Protection Assay
Translation reactions were incubated with 0.1 mg/ml proteinase K (EMD Millipore, Darmstadt, Germany) for 1 hr at 4°C. Protease digestion was stopped by addition of phenylmethylsulfonylfluoride (1 mM final concentration) followed by immediate immersion in 10 volumes of 98°C 1% SDS (wt/vol), 0.1M Tris-HCl, pH 6.8.
ER Membrane Targeting
Translation reactions were cooled to 4° C and diluted two-fold in Buffer A (50 mM HEPES-KOH), pH 7.5, 100 mM KOAc, 1 mM dithiotrhreitol, and 5 mM MgOAc2) and then layered onto 400 µl of 0.6 M sucrose in Buffer A and centrifuged at 187,000 × g for 10 min (TLA 120.1 rotor). Equivalent volumes of input, supernatant, and pellet fractions were collected and analyzed by SDS-PAGE.
PEG-mal cysteine crosslinking
Pelleted microsomes were resuspended in buffer A containing 0.1 M sucrose and 50 µM dithiothreitol and polyethyleneglycol-maleimide 5,000 Da (Fluka, St. Louis, MO) was added to a final concentration of 2 mM. Samples were incubated at 4°C for 1 hr and quenched with 200 mM DTT for 1 hr. Where indicated, 0.5 M NaCl or 1% digitonin (wt/vol) (EMD Millipore, Darmstadt, Germany) was added prior to PEG-mal. Samples were added directly to SDS-sample buffer (Figure 3) or solubilized with 4 volumes of 0.1% SDS (wt/vol) in PBS, and treated with RNase A (0.04µg/µl) (Roche Diagnostics, Indianapolis, IN), prior to SDS-PAGE and phosphorimaging. Pegylation efficiency was determined using Quantity One software by dividing the net sum of pegylated band intensities (volume) by the net sum of pegylated and unpegylated protein bands. In Figure 6, pegylation efficiency of unprocessed pPL polypeptides was calculated by:
% pegylation = 1 − (UPeg/U)
where UPeg is the unprocessed band after incubation in PEG-mal and U is the unprocessed band in the absence of PEG-mal.
HEK 293T metabolic labeling and immunoprecipitation
HEK 293T cells were grown on 6-well plates in 5% CO2 at 37°C and DMEM high glucose containing 10% fetal calf serum supplemented with standard concentrations of penicillin/streptomycin, L-glutamate, and sodium pyruvate. 3 µg pcDNA-pPL or pcDNA-pPL45-Zn were transfected onto cells using calcium phosphate precipitation. The next day, cells were incubated for 30 minutes in cysteine and methionine-free DMEM containing either 25 µM ZnCl2 or 50 µM N,N,N',N'-Tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) (Sigma-Aldrich, St. Louis, MO) and then supplemented with 50 µCi/ml EasyLabel ([35S]Methionine) (Perkin Elmer, Waltham, MA) for an additional 30 min. Cells were washed and lysed in Buffer B (0.1% SDS (wt/vol), 1% TX-100 (vol/vol) containing 100 mM Tris pH 8.0, 100 mM NaCl and 5 mM EDTA). 1 µl anti-preprolactin antiserum (MP Biomedicals, Solon, OH) and 5 µL protein A Affi-Gel (BioRad, Hercules, CA) were added, and samples were rotated overnight at 4°C, washed four times with Buffer B, once with Buffer B lacking detergent, and eluted in SDS-loading buffer. Samples were analyzed by SDS-PAGE and phosphorimaging as above.
Post-translational translocation assays
In vitro translation reactions were arrested after 45 minutes by addition of 20 µg/ml cycloheximide. Where indicated, 0.5 mM ZnCl2 and 1 mM puromycin (EMD Millipore, Darmstadt, Germany) were added, and samples were further incubated for 30 minutes at 24°C. Membranes were pelleted for 10 minutes at 187,000 × g through 0.5 M sucrose in Buffer A (recovery of nascent chain was 80–90% (data not shown)) and analyzed by SDS-PAGE. For Figure 7, membranes were pelleted and resuspended in Buffer A containing 0.1M sucrose, 1 mM ATP, 1 mM GTP, 500 mM KOAc, 5 mM dithiothreitol and protease inhibitors (4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride, leupeptin, and pepstatin). Nascent chain was released from the ribosome by 1 mM puromycin or 25 mM EDTA for 30 minutes at room temperature. Reactions were terminated with 4 volumes of 0.1% SDS in PBS, and peptidyl-tRNA was digested with RNase A (0.04µg/µl) prior to SDS-PAGE.
Structural database analysis
Reviewed entries of human proteins (20,266 total) from the Uniprot database [23161681] were parsed into soluble secretory (1,905) and soluble cytosolic and nuclear proteins (8,485) based on their cellular location and signal-peptide annotations. Proteins with annotated transmembrane segments were excluded. The search strings were “organism:9606 AND reviewed: yes AND annotation:(type: location Cytoplasm OR Nucleus) NOT annotation: (type:transmem) NOT annotation:(type:signal)” for cytosolic and nuclear proteins and “organism:9606 AND reviewed: yes AND annotation:(type: signal) NOT annotation: (type:transmem)” for secretory proteins.
To determine whether proteins in the two cohorts contained predicted structured domains in their N-terminal regions, Uniprot identifiers were mapped to the Superfamily [19036790] and Gene3D [11875040] databases. These databases annotate domains within proteins sequences using hidden Markov models based on the structural classification databases SCOP [14681400] and CATH [23203873]. From these annotations the percentage of proteins with domains within the first 100 residues downstream of the predicted signal sequence cleavage (secretory proteins) or within 100 residues following the N-terminal methionine (cytosolic and nuclear proteins) was calculated.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank V. Hilser, P. Devaraneni, and members of the Skach laboratory for valuable discussion. This work was supported by US National Institutes of Health Grants GM53457 (W.S.), DK51818 (W.S), F32 GM083568 (B.C.), T32 HL083808 (B.C.) and by the Cystic Fibrosis Foundation Therapeutics (W.S.)
Abbreviations
- aa
amino acid
- CRMs
canine pancreas rough microsomes
- EDTA
ethylenediaminetetraacetic acid
- ER
endoplasmic reticulum
- PCC
protein conducting channel
- PEG-mal
polyethyleneglycol maleimide-5000 Da
- PK
proteinase K
- pPL
preprolactin
- RNC
ribosome–nascent chain complex
- RRL
rabbit reticulocyte lysate
- RTC
ribosome–Sec61 translocon complex
- SRP
signal recognition particle
Footnotes
Authors Contributions
B. Conti conceived the project, designed and executed experiments, analyzed results, and assisted in writing the manuscript. J. Elferich and U. Shinde carried out the bioinformatics analysis and assisted in writing the manuscript. Z. Yang, designed and carried out molecular biology experiments. W. Skach designed experiments, analyzed results, and assisted in writing the manuscript.
Competing Financial Interests
The authors have no other competing financial interests.
References
- 1.Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu Rev Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- 2.Shao S, Hegde RS. Membrane protein insertion at the endoplasmic reticulum. Annu Rev Cell Dev Biol. 2011;27:25–56. doi: 10.1146/annurev-cellbio-092910-154125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skach WR. Cellular mechanisms of membrane protein folding. Nat Struct Mol Biol. 2009;16:606–612. doi: 10.1038/nsmb.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson AE. The structural and functional coupling of two molecular machines, the ribosome and the translocon. J Cell Biol. 2009;185:765–767. doi: 10.1083/jcb.200902014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song W, Raden D, Mandon EC, Gilmore R. Role of Sec61alpha in the regulated transfer of the ribosome-nascent chain complex from the signal recognition particle to the translocation channel. Cell. 2000;100:333–343. doi: 10.1016/s0092-8674(00)80669-8. [DOI] [PubMed] [Google Scholar]
- 6.Holtkamp W, et al. Dynamic switch of the signal recognition particle from scanning to targeting. Nat Struct Mol Biol. 2012;19:1332–1337. doi: 10.1038/nsmb.2421. [DOI] [PubMed] [Google Scholar]
- 7.Van den Berg B, et al. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 8.Becker T, et al. Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science. 2009;326:1369–1373. doi: 10.1126/science.1178535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crowley KS, Liao S, Worrell VE, Reinhart GD, Johnson AE. Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell. 1994;78:461–471. doi: 10.1016/0092-8674(94)90424-3. [DOI] [PubMed] [Google Scholar]
- 10.Devaraneni PK, et al. Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex. Cell. 2011;146:134–147. doi: 10.1016/j.cell.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frauenfeld J, et al. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat Struct Mol Biol. 2011;18:614–621. doi: 10.1038/nsmb.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson AE, van Waes MA. The translocon: a dynamic gateway at the ER membrane. Annu Rev Cell Dev Biol. 1999;15:799–842. doi: 10.1146/annurev.cellbio.15.1.799. [DOI] [PubMed] [Google Scholar]
- 13.Jungnickel B, Rapoport TA. A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell. 1995;82:261–270. doi: 10.1016/0092-8674(95)90313-5. [DOI] [PubMed] [Google Scholar]
- 14.Kim SJ, Mitra D, Salerno JR, Hegde RS. Signal sequences control gating of the protein translocation channel in a substrate-specific manner. Dev Cell. 2002;2:207–217. doi: 10.1016/s1534-5807(01)00120-4. [DOI] [PubMed] [Google Scholar]
- 15.Rutkowski DT, Lingappa VR, Hegde RS. Substrate-specific regulation of the ribosome-translocon junction by N-terminal signal sequences. Proc Natl Acad Sci U S A. 2001;98:7823–7828. doi: 10.1073/pnas.141125098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lingappa VR, Chaidez J, Yost CS, Hedgpeth J. Determinants for protein localization: beta-lactamase signal sequence directs globin across microsomal membranes. Proc Natl Acad Sci U S A. 1984;81:456–460. doi: 10.1073/pnas.81.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levine CG, Mitra D, Sharma A, Smith CL, Hegde RS. The efficiency of protein compartmentalization into the secretory pathway. Mol Biol Cell. 2005;16:279–291. doi: 10.1091/mbc.E04-06-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrews DW, Perara E, Lesser C, Lingappa VR. Sequences beyond the cleavage site influence signal peptide function. J Biol Chem. 1988;263:15791–15798. [PubMed] [Google Scholar]
- 19.Kim SJ, Rahbar R, Hegde RS. Combinatorial control of prion protein biogenesis by the signal sequence and transmembrane domain. J Biol Chem. 2001;276:26132–26140. doi: 10.1074/jbc.M101638200. [DOI] [PubMed] [Google Scholar]
- 20.Hegde RS, Kang SW. The concept of translocational regulation. J Cell Biol. 2008;182:225–232. doi: 10.1083/jcb.200804157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goder V, Junne T, Spiess M. Sec61p contributes to signal sequence orientation according to the positive-inside rule. Mol Biol Cell. 2004;15:1470–1478. doi: 10.1091/mbc.E03-08-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Heijne G. Analysis of the distribution of charged residues in the N-terminal region of signal sequences: implications for protein export in prokaryotic and eukaryotic cells. Embo J. 1984;3:2315–2318. doi: 10.1002/j.1460-2075.1984.tb02132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crowley KS, Reinhart GD, Johnson AE. The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell. 1993;73:1101–1115. doi: 10.1016/0092-8674(93)90640-c. [DOI] [PubMed] [Google Scholar]
- 24.Arkowitz RA, Joly JC, Wickner W. Translocation can drive the unfolding of a preprotein domain. EMBO J. 1993;12:243–253. doi: 10.1002/j.1460-2075.1993.tb05650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonardi F, et al. Probing the SecYEG translocation pore size with preproteins conjugated with sizable rigid spherical molecules. Proc Natl Acad Sci U S A. 2011;108:7775–7780. doi: 10.1073/pnas.1101705108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denzer AJ, Nabholz CE, Spiess M. Transmembrane orientation of signal-anchor proteins is affected by the folding state but not the size of the N-terminal domain. Embo J. 1995;14:6311–6317. doi: 10.1002/j.1460-2075.1995.tb00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perara E, Rothman RE, Lingappa VR. Uncoupling translocation from translation: implications for transport of proteins across membranes. Science. 1986;232:348–352. doi: 10.1126/science.3961485. [DOI] [PubMed] [Google Scholar]
- 28.Kowarik M, Kung S, Martoglio B, Helenius A. Protein folding during cotranslational translocation in the endoplasmic reticulum. Mol Cell. 2002;10:769–778. doi: 10.1016/s1097-2765(02)00685-8. [DOI] [PubMed] [Google Scholar]
- 29.Cheng Z, Gilmore R. Slow translocon gating causes cytosolic exposure of transmembrane and lumenal domains during membrane protein integration. Nat Struct Mol Biol. 2006;13:930–936. doi: 10.1038/nsmb1146. [DOI] [PubMed] [Google Scholar]
- 30.Mandon EC, Trueman SF, Gilmore R. Translocation of proteins through the Sec61 and SecYEG channels. Curr Opin Cell Biol. 2009;21:501–507. doi: 10.1016/j.ceb.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park E, et al. Structure of the SecY channel during initiation of protein translocation. Nature advance online publication. 2013 doi: 10.1038/nature12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosolapov A, Deutsch C. Tertiary interactions within the ribosomal exit tunnel. Nat Struct Mol Biol. 2009;16:405–411. doi: 10.1038/nsmb.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Brien EP, Hsu S-TD, Christodoulou J, Vendruscolo M, Dobson CM. Transient Tertiary Structure Formation within the Ribosome Exit Port. Journal of the American Chemical Society. 2010;132:16928–16937. doi: 10.1021/ja106530y. [DOI] [PubMed] [Google Scholar]
- 34.Tu L, Khanna P, Deutsch C. Transmembrane Segments Form Tertiary Hairpins in the Folding Vestibule of the Ribosome. J Mol Biol. 2013 doi: 10.1016/j.jmb.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parraga G, et al. Zinc-dependent structure of a single-finger domain of yeast ADR1. Science. 1988;241:1489–1492. doi: 10.1126/science.3047872. [DOI] [PubMed] [Google Scholar]
- 36.Bowers PM, Schaufler LE, Klevit RE. A folding transition and novel zinc finger accessory domain in the transcription factor ADR1. Nat Struct Biol. 1999;6:478–485. doi: 10.1038/8283. [DOI] [PubMed] [Google Scholar]
- 37.Buchsbaum JC, Berg JM. Kinetics of metal binding by a zinc finger peptide. Inorganica Chimica Acta. 2000;297:217–219. [Google Scholar]
- 38.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 39.Lu J, Deutsch C. Folding zones inside the ribosomal exit tunnel. Nat Struct Mol Biol. 2005;12:1123–1129. doi: 10.1038/nsmb1021. [DOI] [PubMed] [Google Scholar]
- 40.Voss NR, Gerstein M, Steitz TA, Moore PB. The geometry of the ribosomal polypeptide exit tunnel. J Mol Biol. 2006;360:893–906. doi: 10.1016/j.jmb.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 41.Lu J, Deutsch C. Secondary structure formation of a transmembrane segment in Kv channels. Biochemistry. 2005;44:8230–8243. doi: 10.1021/bi050372q. [DOI] [PubMed] [Google Scholar]
- 42.Mothes W, Prehn S, Rapoport TA. Systematic probing of the environment of a translocating secretory protein during translocation through the ER membrane. Embo J. 1994;13:3973–3982. doi: 10.1002/j.1460-2075.1994.tb06713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz U, Kiefer M, Tholey A, Adolph HW. On the competition for available zinc. J Biol Chem. 2005;280:3197–3207. doi: 10.1074/jbc.M409425200. [DOI] [PubMed] [Google Scholar]
- 44.Connolly T, Collins P, Gilmore R. Access of proteinase K to partially translocated nascent polypeptides in intact and detergent-solubilized membranes. J Cell Biol. 1989;108:299–307. doi: 10.1083/jcb.108.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walter P, Blobel G. Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol. 1981;91:557–561. doi: 10.1083/jcb.91.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clemons WM, Jr, Menetret JF, Akey CW, Rapoport TA. Structural insight into the protein translocation channel. Curr Opin Struct Biol. 2004;14:390–396. doi: 10.1016/j.sbi.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 47.du Plessis DJ, Berrelkamp G, Nouwen N, Driessen AJ. The lateral gate of SecYEG opens during protein translocation. J Biol Chem. 2009;284:15805–15814. doi: 10.1074/jbc.M901855200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lycklama a Nijeholt JA, Wu ZC, Driessen AJ. Conformational dynamics of the plug domain of the SecYEG protein-conducting channel. J Biol Chem. 2011;286:43881–43890. doi: 10.1074/jbc.M111.297507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hegde RS, Lingappa VR. Sequence-specific alteration of the ribosome-membrane junction exposes nascent secretory proteins to the cytosol. Cell. 1996;85:217–228. doi: 10.1016/s0092-8674(00)81098-3. [DOI] [PubMed] [Google Scholar]
- 50.Liao S, Lin J, Do H, Johnson AE. Both lumenal and cytosolic gating of the aqueous ER translocon pore are regulated from inside the ribosome during membrane protein integration. Cell. 1997;90:31–41. doi: 10.1016/s0092-8674(00)80311-6. [DOI] [PubMed] [Google Scholar]
- 51.Sadlish H, Pitonzo D, Johnson AE, Skach WR. Sequential triage of transmembrane segments by Sec61alpha during biogenesis of a native multispanning membrane protein. Nat Struct Mol Biol. 2005;12:870–878. doi: 10.1038/nsmb994. [DOI] [PubMed] [Google Scholar]
- 52.Yost CS, Hedgpeth J, Lingappa VR. A stop transfer sequence confers predictable transmembrane orientation to a previously secreted protein in cell-free systems. Cell. 1983;34:759–766. doi: 10.1016/0092-8674(83)90532-9. [DOI] [PubMed] [Google Scholar]
- 53.Fujita H, Yamagishi M, Kida Y, Sakaguchi M. Positive charges on the translocating polypeptide chain arrest movement through the translocon. J Cell Sci. 2011;124:4184–4193. doi: 10.1242/jcs.086850. [DOI] [PubMed] [Google Scholar]
- 54.Chuck SL, Lingappa VR. Pause transfer: a topogenic sequence in apolipoprotein B mediates stopping and restarting of translocation. Cell. 1992;68:9–21. doi: 10.1016/0092-8674(92)90202-n. [DOI] [PubMed] [Google Scholar]
- 55.Nakahara DH, Lingappa VR, Chuck SL. Translocational pausing is a common step in the biogenesis of unconventional integral membrane and secretory proteins. J Biol Chem. 1994;269:7617–7622. [PubMed] [Google Scholar]
- 56.Mitchell DM, et al. Apoprotein B100 has a prolonged interaction with the translocon during which its lipidation and translocation change from dependence on the microsomal triglyceride transfer protein to independence. Proc Natl Acad Sci U S A. 1998;95:14733–14738. doi: 10.1073/pnas.95.25.14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fisher EA, et al. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J Biol Chem. 1997;272:20427–20434. doi: 10.1074/jbc.272.33.20427. [DOI] [PubMed] [Google Scholar]
- 58.Cuchel M, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–156. doi: 10.1056/NEJMoa061189. [DOI] [PubMed] [Google Scholar]
- 59.Fukada T, Yamasaki S, Nishida K, Murakami M, Hirano T. Zinc homeostasis and signaling in health and diseases: Zinc signaling. J Biol Inorg Chem. 2011;16:1123–1134. doi: 10.1007/s00775-011-0797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
References for Online Methods
- 60.Skach WR, Calayag MC, Lingappa VR. Evidence for an alternate model of human P-glycoprotein structure and biogenesis. J Biol Chem. 1993;268:6903–6908. [PubMed] [Google Scholar]
- 61.Skach WR, Lingappa VR. Amino-terminal assembly of human P-glycoprotein at the endoplasmic reticulum is directed by cooperative actions of two internal sequences. J Biol Chem. 1993;268:23552–23561. [PubMed] [Google Scholar]
- 62.Ota T, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- 63.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 64.Carlson E, Bays N, David L, Skach WR. Reticulocyte lysate as a model system to study endoplasmic reticulum membrane protein degradation. Methods Mol Biol. 2005;301:185–205. doi: 10.1385/1-59259-895-1:185. [DOI] [PubMed] [Google Scholar]
- 65.Matsumura Y, Rooney L, Skach WR. In vitro methods for CFTR biogenesis. Methods Mol Biol. 741:233–253. doi: 10.1007/978-1-61779-117-8_16. [DOI] [PubMed] [Google Scholar]
- 66.Lee B-C, Chu TK, Dill KA, Zuckermann RN. Biomimetic Nanostructures: Creating a High-Affinity Zinc-Binding Site in a Folded Nonbiological Polymer. Journal of the American Chemical Society. 2008;130:8847–8855. doi: 10.1021/ja802125x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.