

Abstract
Rationale: Asymptomatic relatives of patients with familial interstitial pneumonia (FIP), the inherited form of idiopathic interstitial pneumonia, carry increased risk for developing interstitial lung disease.
Objectives: Studying these at-risk individuals provides a unique opportunity to investigate early stages of FIP pathogenesis and develop predictive models of disease onset.
Methods: Seventy-five asymptomatic first-degree relatives of FIP patients (mean age, 50.8 yr) underwent blood sampling and high-resolution chest computed tomography (HRCT) scanning in an ongoing cohort study; 72 consented to bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial biopsies. Twenty-seven healthy individuals were used as control subjects.
Measurements and Main Results: Eleven of 75 at-risk subjects (14%) had evidence of interstitial changes by HRCT, whereas 35.2% had abnormalities on transbronchial biopsies. No differences were noted in inflammatory cells in BAL between at-risk individuals and control subjects. At-risk subjects had increased herpesvirus DNA in cell-free BAL and evidence of herpesvirus antigen expression in alveolar epithelial cells (AECs), which correlated with expression of endoplasmic reticulum stress markers in AECs. Peripheral blood mononuclear cell and AEC telomere length were shorter in at-risk individuals than healthy control subjects. The minor allele frequency of the Muc5B rs35705950 promoter polymorphism was increased in at-risk subjects. Levels of several plasma biomarkers differed between at-risk subjects and control subjects, and correlated with abnormal HRCT scans.
Conclusions: Evidence of lung parenchymal remodeling and epithelial dysfunction was identified in asymptomatic individuals at risk for FIP. Together, these findings offer new insights into the early pathogenesis of idiopathic interstitial pneumonia and provide an ongoing opportunity to characterize presymptomatic abnormalities that predict progression to clinical disease.
Keywords: IPF, bronchoscopy, alveolar epithelial cell, telomere, biomarker
At a Glance Commentary
Scientific Knowledge on the Subject
The early events in pathogenesis of sporadic and familial forms of progressive pulmonary fibrosis are not well understood.
What This Study Adds to the Field
First-degree relatives of patients with familial interstitial pneumonia have a greatly increased risk of developing interstitial lung disease. In the asymptomatic stage, these at-risk individuals commonly have radiographic and histopathologic abnormalities, short telomeres in peripheral blood and alveolar epithelium, and abnormal plasma biomarker profiles. Use of these parameters holds promise in identifying individuals at high risk for developing interstitial lung disease in the presymptomatic stage.
Current evidence suggests that approximately 20% of idiopathic interstitial pneumonia (IIP) cases occur in families (1, 2), comprising the syndrome of familial interstitial pneumonia (FIP) (3, 4). First-degree relatives of FIP patients have a substantially greater likelihood of developing interstitial lung disease than the general population, potentially as high as 50% in families where the inheritance pattern is autosomal dominant (4, 5). Although more work is needed to fully understand the genetic predisposition to FIP, heterozygous mutations in surfactant proteins (SP) C (SFTPC) (6, 7) and A2 (SFTPA2) (8), along with three components of the telomerase complex (telomerase reverse transcriptase [TERT], the RNA component of telomerase [hTR] [9, 10], and dyskerin [DKC1] [11]), have been identified as the genetic cause of disease in 10–15% of FIP families. Additionally, common genetic variants, including a single nucleotide polymorphism in in the promoter for MUC5B, and environmental factors seem to impact risk for developing FIP (12). The ability to ascertain asymptomatic family members of FIP patients represents a unique opportunity to characterize a high-risk population in the presymptomatic stage and thereby identify factors that contribute to development of clinical disease.
Although there may be subtle radiographic features that distinguish FIP and sporadic idiopathic pulmonary fibrosis (IPF) (13), these diseases share important clinical and histopathologic features (3, 5, 14). Several groups have previously studied at-risk individuals in FIP families to gain insights into mechanisms that contribute to IIP, focused primarily on immune and inflammatory mechanisms (14, 15); however, few at-risk individuals have been followed over time to assess the natural history of disease development (16). Over the last 10–15 years, advances in understanding the genetics of IIP have aided in identifying pathways and mechanisms that contribute to the pathogenesis of FIP and sporadic IPF. The identification of SFTPC mutations as a cause of FIP led to the recognition that alveolar epithelial cell (AEC) dysfunction, including endoplasmic reticulum (ER) stress, is a common feature of FIP and sporadic IPF (17–19). In addition, discovery of mutations in the telomerase pathway and subsequent work demonstrated that short telomeres and telomere dysfunction are prominent in FIP and IPF (9, 10, 20, 21). More recently, identification of a common genetic variant in the promoter for MUC5B that correlates with risk for IPF has led to active investigation into the role of airway mucins in disease pathogenesis (22). As a result, we initiated a longitudinal cohort study of asymptomatic first-degree relatives of FIP patients to determine whether epithelial dysfunction is important in primary disease pathogenesis and whether we could identify factors that predict development of clinical disease. Here, we report initial phenotyping of 75 at-risk subjects from 41 FIP families who are part of this ongoing cohort study. Some of these data have been presented previously in abstract form (25–29).
Methods
For additional information, see the Methods section in the online supplement.
Subjects
First-degree relatives of patients with FIP were identified from the Vanderbilt FIP Registry and those ages 40–65 were eligible for screening. FIP was defined as the presence of IIP in two or more family members, including IPF in at least one affected individual. Based on present understanding of the genetic risk for FIP, we anticipate that at most 50% of these asymptomatic first-degree relatives will carry the relevant risk alleles in their family. These potential subjects were administered an interstitial lung disease questionnaire; those who had minimal to no dyspnea (dyspnea score ≤ 2) were invited to participate in our study; individuals with more significant dyspnea (dyspnea score ≥ 3) were ineligible and offered clinical evaluation. Eligible subjects were invited to undergo blood draw, a high-resolution computerized tomogram (HRCT) of the chest, and research bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial lung biopsies (TBLBx). Here, we report baseline data for the first 75 subjects (referred to hereafter as “at risk”) in this ongoing study.
BAL fluid and/or plasma from 27 healthy individuals undergoing bronchoscopy in a separate study were used as normal control subjects. These healthy control subjects had a mean age of 36.8 years (range, 21–58), 47% were female, and none were former smokers. DNA from 322 healthy subjects recruited through other studies was used as control subjects for MUC5B genotyping. IPF disease control subjects were identified from the Vanderbilt Pulmonary Fibrosis Registry, with DNA from 217 subjects available for genetic studies. Twelve of these subjects underwent research bronchoscopy through the COMET-IPF trial (30); these BALs were used as IPF disease control subjects. Mean age of these 12 subjects with IPF was 62.1; 91% were male and 83.3% were current or former smokers.
Survey Instrument
A modified version of the American Thoracic Society–Division of Lung Disease-78 respiratory questionnaire was used (31).
HRCT
At-risk subjects underwent a single prone HRCT scan without intravenous contrast using a customized protocol with review by an expert chest radiologist (J.A.W.). Subjects with extensive disease (>5% honeycombing) were referred for clinical interstitial lung disease evaluation.
Bronchoscopy
Flexible fiberoptic bronchoscopy with BAL and TBLBx was performed using standard procedures. Transbronchial biopsies were obtained from areas of HRCT abnormalities if present; with normal HRCT scans, biopsies were obtained from the right lower lobe.
Histopathology
Slides stained with hematoxylin and eosin were submitted for independent interpretation by two expert pulmonary pathologists (J.E.J., S.D.G.) who were masked to all demographic, clinical, and radiographic information.
Telomere Restriction Fragment Analysis
Telomere restriction fragment analysis was performed by Southern blot.
Tissue Telomere Measurements
Telomere length in type II AECs was measured by fluorescence in-situ hybridization using Cy3 fluorescence to mark telomeres relative to nuclear DNA (DAPI) (20, 32).
Immunohistochemistry
Paraffin-embedded lung tissue was sectioned (5 μm) and immunostaining performed using methods outlined previously (18, 33).
Muc5B Genotyping
MUC5B genotyping was performed using a competitive allele-specific polymerase chain reaction (PCR)–based Taqman single-nucleotide polymorphism genotyping assay (Applied Biosystems, Grand Island, NY) to detect the presence of rs35705950.
Herpesvirus Detection
DNA was isolated from 10 ml of concentrated BAL from each subject, and Epstein-Barr virus (EBV) and cytomegalovirus (CMV) copies were determined by quantitative PCR in comparison with a standard curve of 10-fold dilutions.
Biomarkers
Plasma biomarker levels were measured using a Luminex (Luminex Corp., Austin, TX) multiplex ELISA system (34, 35). Muc5B was measured in BAL supernatant by ELISA.
Statistical Analysis
The programming language R version 2.15.2 (36, 37) and GraphPad Prizm version 5.0 (GraphPad Software, San Diego, CA) were used to perform analysis of variance, Mann-Whitney U test, Pearson chi-square test, or Fisher exact test, mixed effects models and logistic regression as appropriate. P less than 0.05 was considered significant. Where noted, age, sex, and smoking adjusted analyses are presented.
Results
Subject enrollment is described in Figure 1. During the study period, 209 potential subjects were screened for eligibility and 75 first-degree relatives of FIP patients were enrolled. These 75 individuals had blood drawn and underwent HRCT (hereafter these subjects are considered “at risk”). Seventy-two at-risk subjects also consented to undergo bronchoscopy with BAL. One subject declined TBLBx, thus histopathology was available for 71 subjects. A subset of patients who underwent bronchoscopy had BAL cell populations analyzed by flow cytometry. Demographic characteristics of subjects are shown in Table 1. The mean age of at-risk subjects was 50.8 years and 64% were female. Twelve subjects reported a history of asthma. All subjects had a family history of FIP. As noted, healthy control subjects were younger and less likely to have smoked than at-risk subjects. In contrast, IPF disease control subjects were older, more likely male, and more likely to be current or former smokers.
Figure 1.

Schematic of enrollment and study population. AEC = alveolar epithelial cells; BAL = bronchoalveolar lavage; CMV = cytomegalovirus; EBV = Epstein-Barr virus; ER = endoplasmic reticulum; HRCT = high-resolution computed tomography; IHC = immunohistochemistry; qPCR = quantitative polymerase chain reaction; TBLBx = transbronchial lung biopsies; TRF = telomere restriction fragment.
Table 1.
Demographics of At-Risk Subjects
| n = 75 | |
|---|---|
| Age |
50.8 (7.7) |
| Female |
48 (64) |
| Ever smoker |
17 (22.6) |
| Family history of interstitial lung
disease |
75 (100) |
| Medical history |
|
| Emphysema |
1 (1.3) |
| Asthma |
11 (14.7) |
| Hypertension |
25 (33.3) |
| Pneumonia |
19 (25.2) |
| Radiation |
4 (5.3) |
| Chest injury |
2 (2.7) |
| Liver disease | 2 (2.7) |
Data are presented as mean (SD) or number (percentage).
Of the 75 at-risk subjects who underwent HRCT, 11 (14.7%) had evidence of interstitial abnormalities consistent with early interstitial lung disease (Table 2). Among abnormal HRCT scans with evidence of interstitial changes, interlobular reticular opacities were present in all 11 subjects and irregular septal thickening was found in 9 of 11 subjects. Examples of these findings are shown in Figure 2. After examination by two experienced lung pathologists, 25 of 71 (35.2%) transbronchial biopsies were considered abnormal with such features as interstitial fibrosis, inflammation, granulomas, and respiratory bronchiolitis (Table 3). Examples of abnormal transbronchial biopsies are shown in Figure 3. In five individuals, both HRCT and biopsy were considered abnormal.
Table 2.
High-Resolution Computed Tomography Findings in At-Risk Subjects
| n = 75 | |
|---|---|
| Consistent with early interstitial lung disease | 11 (14.7) |
| Type of interstitial abnormalities | |
| Intralobular reticular opacities | 11 (14.7) |
| Irregular intralobular septal thickening | 9 (12) |
| Ground glass opacities | 3 (4) |
| Traction bronchiectasis | 1 (1.3) |
| Traction bronchiolectasis | 1 (1.3) |
| Honeycombing | 1 (1.3) |
Data are expressed as number (percentage).
Figure 2.

High-resolution computed tomography abnormalities in at-risk subjects. Axial and coronal images of three different individuals demonstrating representative radiographic abnormalities are shown by arrows. (A and B) Intralobular reticular opacities. (C and D) Irregular intralobular septal thickening. (E and F) Traction bronchiolectasis.
Table 3.
Transbronchial Biopsy Histologic Findings
| n = 71 | |
|---|---|
| Normal | 45 (63.4) |
| Abnormal | 26 (36.6) |
| Interstitial fibrosis | 12 (16.9) |
| Peribronchiolar fibrosis | 15 (21.1) |
| Chronic inflammation | 10 (14.1) |
| Respiratory bronchiolitis | 2 (2.8) |
| Giant cells/granulomas | 6 (8.5) |
Data are expressed as number (percentage). Multiple abnormalities could be identified within a single biopsy.
Figure 3.

Transbronchial biopsy histology from at-risk subjects. Abnormal features identified on transbronchial biopsy specimens include (A) coarse interstitial fibrosis associated with normal tissue, hyperplastic epithelial cells and (B) peribronchiolar metaplasia with perivascular inflammation. (C) Association between high-resolution computed tomography (HRCT) and transbronchial biopsies among 71 subjects. Original magnification (A) 40×, and (B) 200×.
Previous studies reported alterations in BAL inflammatory cell populations in family members of FIP patients (14, 15). In our cohort, no differences were detected in total or differential BAL cell counts in at-risk subjects compared with normal control subjects (Table 4). In addition, no differences in lymphocyte subsets were observed in BAL cells compared with normal control subjects or IPF patients (CD4, CD25/FoxP3) (see Table E2 in the online supplement). Similar to previous reports (14, 15), there were increased neutrophils and eosinophils in BAL from IPF subjects. Inflammatory cell populations were similar among at-risk subjects with and without radiographic or histologic abnormalities.
Table 4.
Bronchoalveolar Lavage Characteristics
| Normal (n = 23) | At-Risk (n = 75) | Idiopathic Pulmonary Fibrosis (n = 12) | P Value | |
|---|---|---|---|---|
| Cells (104)/ml | 6.7 (2.3) | 8.3 (6.1) | 19.8 (23.1) | 0.006 |
| Differential | ||||
| Macrophages | 90 (8.0) | 91 (7.8) | 76 (22.2) | 0.002 |
| Lymphocytes | 8 (8.0) | 7 (6.5) | 15 (19.9) | 0.116 |
| Neutrophils | 2 (1.6) | 1 (1.2) | 7 (8.5) | 0.004 |
| Eosinophils | 0 (0.2) | 0 (2.5) | 3 (2.5) | <0.001 |
Data expressed as mean (SD). Data were compared by analysis of variance. Pairwise comparison of normal versus at-risk was not significant.
Prior studies have detected herpesviruses including EBV and CMV by immunohistochemical staining and PCR from whole lung tissue in patients with IPF (38, 39). To evaluate these herpesviruses in the lungs, we used quantitative PCR from cell-free BAL fluid to determine viral load in at-risk patients, normal control subjects, and IPF patients. Although inflammatory cell populations were similar among groups, at-risk and IPF subjects had elevated herpesvirus load in BAL fluid. IPF subjects had significantly increased EBV DNA (median, 161 [interquartile range, 131–492] copies/ml) (Figure 4A) and CMV DNA (3,620 [318–5,290] copies/ml) (Figure 4B) compared with normal control subjects in BAL fluid (P < 0.001). At-risk subjects also had higher copy numbers of EBV (25.3 [15–49] copies/ml) and CMV (78 [33–371] copies/ml) compared with normal control subjects (P < 0.001). In addition, EBV (Figure 4C) and CMV (Figure 4D) antigens were also frequently detectable by immunohistochemistry in type II AECs from transbronchial biopsies of at-risk subjects, which is uncommon in normal lungs (18). In IPF lungs, we previously reported that ER stress markers were commonly identified in AECs and were associated with herpesvirus antigens (18). In 62% of at-risk subjects, ER stress markers Bip (Figure 4E) or XBP1 (Figure 4F) were identified in AECs by immunohistochemistry and were significantly associated with presence of herpesvirus antigens (Figure 4G).
Figure 4.

Herpesviruses are detectable in lungs from at-risk subjects. DNA was isolated from cell-free bronchoalveolar lavage (BAL) supernatant from normal control subjects, subjects at-risk for familial interstitial pneumonia, and idiopathic pulmonary fibrosis (IPF) control subjects. Copies of Epstein-Barr virus (EBV) (A) and cytomegalovirus (CMV) (B) were quantified by quantitative polymerase chain reaction (bar represents median and interquartile range). EBV (C) and CMV (D) antigens were also detected by immunostaining in type II alveolar epithelial cells in transbronchial biopsy specimens from at-risk subjects. (E) Bip and (F) XBP1 were identified in alveolar epithelial cells in transbronchial biopsies from at-risk subjects. (G) Quantification of transbronchial biopsy specimens from at-risk subjects stained for endoplasmic reticulum (ER) stress markers Bip and XBP1 and herpesvirus antigens from EBV and CMV. *P < 0.001 at-risk versus normal control subjects. **P < 0.001 IPF versus normal control subjects.
Recently, a genome-wide linkage study identified a common polymorphism (rs35705950) in the promoter of the gene encoding for Mucin 5B (MUC5B) that was associated with increased risk of sporadic IPF and FIP (22). To determine whether this polymorphism was enriched among at-risk subjects, we genotyped at-risk subjects, 217 IPF patients from the Vanderbilt Pulmonary Fibrosis Registry, and 339 healthy control subjects for rs35705950. Minor allele frequency was 8.7 and 35.0% in control subjects and IPF subjects, respectively, similar to previous reports (Figure 5A) (22, 40, 41). Among at-risk subjects, minor allele frequency was intermediate at 17.4%. The MUC5B promoter polymorphism has been associated with increased Muc5B expression (22). We measured Muc5B levels in BAL fluid and found Muc5B was higher in at-risk subjects compared with healthy control subjects who had undergone bronchoscopy; levels in IPF patients were higher than at-risk subjects (Figure 5B). Among at-risk subjects, there seemed to be a dose-response relationship of Muc5B protein in BAL fluid and copies of the minor allele (Figure 5C).
Figure 5.
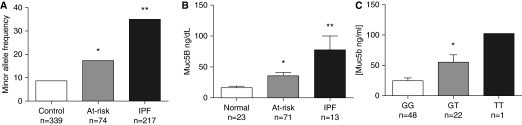
Muc5B promoter polymorphism rs35705950 is found with increased frequency in at-risk subjects. (A) At-risk subjects along with 339 healthy control subjects and 217 idiopathic pulmonary fibrosis (IPF) patients were genotyped for the Muc5B rs35705950 promoter polymorphism. Minor allele frequency is increased in at-risk subjects compared with control subjects. (B) Muc5B level in bronchoalveolar lavage supernatant was measured by ELISA in normal control subjects, at-risk subjects, and IPF control subjects. (C) Muc5B levels in homozygote major allele carriers (GG), heterozygote minor allele carriers (GT), and homozygote minor allele carriers (TT). *P < 0.05 at-risk versus normal control subjects. **P < 0.05 IPF versus normal control subjects.
Short telomeres have been associated with sporadic IPF and FIP (9, 10, 20, 21); however, a temporal relationship with disease has not been established. To determine if telomere shortening precedes development of clinically evident disease, we measured peripheral blood mononuclear cell (PBMC) telomere length in at-risk subjects. Twenty-seven of 75 at-risk subjects (36%) had short telomeres (<10th percentile for age) in PBMCs (Figure 6A), strikingly similar to the proportion previously reported among sporadic IPF patients (20, 21). A prior report identified short telomeres specifically in type II AECs in IPF lungs (20); therefore, we sought to determine whether at-risk subjects also had short telomeres in AECs. Using a dual-fluorescence approach to measure telomere length by fluorescence in-situ hybridization in type II AECs from lung tissue (Figures 6C–6E), we found that telomere length in type II AECs from at-risk subjects was significantly shorter than type II AECs from normal lungs. Consistent with this prior report (20), telomere length in type II AECs from IPF patients was uniformly short (20) (Figure 6D). Surprisingly, type II AEC telomere length did not correlate with PBMC telomere length in the at-risk population (Figure 6E).
Figure 6.

At-risk subjects have short telomeres in peripheral blood and type II alveolar epithelial cells. (A) DNA was isolated from peripheral blood mononuclear cells (PBMCs) of at-risk subjects and telomere restriction fragment (TRF) was measured by Southern blot. TRF was then plotted against age in comparison with a population of normal control subjects. Telomeres less than the 10th percentile for age were considered abnormally short. (B and C) Telomere length in type II alveolar epithelial cells (AECs) was measured using a dual-fluorescence approach wherein type II AECs were identified by costaining for pro–surfactant protein C, and telomeric DNA was quantified relative to total nuclear DNA (DAPI) by relative fluorescence in (B) control and (C) at-risk subjects. Green = pro–surfactant protein C; blue = DAPI (nuclear DNA); red within nucleus = telomeres. (D) Quantification of telomere fluorescence intensity relative to nuclear DNA in type II AECs. *P < 0.001, **P < 0.005. (E) PBMC telomere length was plotted versus AEC telomere length and a linear regression was performed to determine the relationship between PBMC and AEC telomere length. IPF = idiopathic pulmonary fibrosis.
Finally, we sought to determine whether at-risk subjects had alterations in plasma biomarker profiles. Several protein biomarkers including matrix metalloproteinase 7 (MMP7), SP-D, and other chemokines have shown promise as diagnostic or prognostic biomarkers in IPF patients (30, 42–48). We were interested to determine whether plasma biomarkers could discriminate at-risk subjects from healthy control subjects, a finding that could be used to develop a prognostic model. We measured plasma levels of 19 proposed biomarkers using a multiplex ELISA platform in at-risk and control subjects. After controlling for age and sex, and correcting for multiple comparisons, levels of 11 biomarkers including previously validated markers SP-D, MMP7, TIMP2, and ET-1 were statistically different between at-risk subjects and control subjects (Figure 7; see Table E1).
Figure 7.

Plasma biomarkers differ between at-risk subjects and healthy control subjects. Plasma levels of (A) matrix metalloproteinase 7, (B) surfactant protein-D, (C) ET-1, and (D) TIMP2 in 67 at-risk and 27 healthy control subjects. Data were log transformed to normalize distribution for analysis. Boxes indicate 25th–75th percentiles; whiskers depict minimum to maximum values. At-risk and control subjects were compared using a mixed-effects model controlling for age and sex. *P < 0.001 is considered significant after Bonferroni correction for multiple comparisons.
We then performed exploratory analyses of known or potential risk factors for IPF to determine whether they predicted HRCT abnormalities among these asymptomatic at-risk subjects (Table 5). In univariate analyses, age, smoking history, short AEC telomeres, and several plasma biomarkers were significant predictors of HRCT abnormalities; controlling for age, sex, and smoking history did not significantly alter these findings.
Table 5.
Univariate Predictors of HRCT Abnormalities
| Estimate | Log Odds Ratio | 95% CI | P Value | |
|---|---|---|---|---|
| PBMC telomere percent | 0.42 | −0.53 to 1.38 | 0.375 | |
| AEC telomere | −0.68 | −1.13 to −0.23 | 0.005 | |
| EBV | 0.33 | −0.81 to 1.47 | 0.56 | |
| CMV | −0.13 | −1.56 to 1.29 | 0.853 | |
| Muc5B quant | 0.07 | −0.48 to 0.62 | 0.804 | |
| BAL cells | 0.32 | −0.08 to 0.72 | 0.11 | |
| Macrophages | −0.05 | −0.27 to 0.16 | 0.623 | |
| Neutrophils | 0.01 | −0.6 to 1.11 | 0.986 | |
| Lymphocytes | 0.13 | −0.76 to 1.01 | 0.767 | |
| SP-D | 0.61 | 0.07 to 1.15 | 0.029 | |
| MMP7 | 0.65 | 0.14 to 1.16 | 0.015 | |
| ET1 | 0.27 | −0.17 to 0.71 | 0.218 | |
| TIMP2 | −0.22 | −0.51 to 0.08 | 0.141 | |
| Age | 0.186 | 0.06 to 0.32 | 0.005 | |
| Sex | 0.631 | −0.98 to 2.24 | 0.444 | |
| Ever smoker | 1.94 | 0.15 to 3.74 | 0.034 |
Definition of abbreviations: AEC = alveolar epithelial cells; BAL = bronchoalveolar lavage; CI = confidence interval; CMV = cytomegalovirus; EBV = Epstein-Barr virus; HRCT = high-resolution computed tomography; MMP = matrix metalloproteinase; PBMC = peripheral blood mononuclear cell; SP-D = surfactant protein-D.
Discussion
Human studies attempting to gain insights into primary pathologic mechanisms in IPF have been limited by the fact that IPF patients are rarely recognized until disease is advanced. Capturing events early in the disease course has proved challenging. In this prospective cohort study, we report the baseline characterizations of a group of subjects at high risk for the development of pulmonary fibrosis. Over time, following this cohort will offer a unique opportunity to characterize the natural history of FIP, identify diagnostic and prognostic biomarkers, and identify which mechanisms contribute to early FIP pathogenesis. Remarkably, in many of these asymptomatic individuals, there is evidence of dysfunction in pathways linked to FIP pathogenesis, including ER stress, telomere shortening in type II AECs, increased Muc5B levels, and elevated plasma biomarkers of lung epithelial injury. Strikingly, these abnormalities are found in some subjects without evidence of disease by HRCT or TBLBx, suggesting that abnormalities in these pathways may precede the development of clinically detectable lung fibrosis, a requisite for a causative relationship. Over the coming years, as at-risk subjects develop symptomatic disease we anticipate that there will be greater clarity as to which abnormalities are predictive of disease and play a role in disease pathogenesis.
As was anticipated based on prior studies (5, 14), we found that a substantial proportion of at-risk subjects have subtle radiographic abnormalities on HRCT, most commonly reticular changes and septal thickening. Furthermore, more than one-third of these asymptomatic at-risk subjects had abnormal TBLBx. Given the limited sensitivity of this approach in patients with advanced disease (49–51), this finding likely underestimates the true breadth of abnormal pathology in this cohort. Surprisingly, relatively few subjects had both radiographic and pathologic changes, suggesting that current radiographic and biopsy approaches are likely both of limited sensitivity for early disease. Although it is not clear that these radiographic or histologic abnormalities will ultimately develop into symptomatic FIP, the possibility is intriguing. If the presence of these changes proves to be predictive of disease development, this would provide the strongest evidence to date that there is a long but identifiable presymptomatic period during which targeted therapies could be used to prevent progression of FIP and IPF.
One surprising finding in our study is that inflammatory cell populations in BAL fluid were similar in at-risk and normal control subjects, which contrasts to a prior report (15). There were also no differences in T-cell subsets, including regulatory T cells, which were previously reported to be decreased in IPF patients (52). Cumulatively, our data provide further evidence that chronic inflammation is not the principal mediator of epithelial injury and dysfunction in the early stages of FIP and IPF.
A potential role of viral infection in IPF has been suspected for decades, but evidence supporting an important role for herpesviruses in disease initiation or progression has been limited. There are several postulated mechanisms by which reactivation of latent herpesviruses might contribute to lung fibrosis (23, 24). Although it has been recognized by multiple groups that herpesvirus antigens can be detected in IPF lungs by histology and PCR (18, 38, 39), these studies could not differentiate latent from active infection. Surprisingly, we found IPF patients had increased copy numbers of EBV and CMV DNA in cell-free BAL supernatant, which may reflect ongoing viral replication. Interestingly, at-risk subjects had levels of EBV and CMV DNA that were scattered between normal control subjects and IPF patients with suggestion of a dichotomous pattern. Several at-risk subjects had very elevated levels similar to those detected in IPF patients, suggesting that enhanced herpesvirus replication in the lung may be a source of ongoing epithelial cell injury and precede clinically evident disease. Although a recent study suggested viral infection is not commonly detectable in patients with acute exacerbations of IPF (53), further investigation is needed to clarify the role herpesviruses play as cofactors in chronic fibrotic remodeling in the lung. In addition, as we have previously shown in IPF lungs (18), markers of ER stress are common in AECs from at-risk subjects and are associated with the presence of herpesvirus antigens, suggesting herpesvirus infection may be an important source of AEC ER stress in early IPF pathogenesis. Further studies are needed to clarify the mechanisms through which herpesviruses and ER stress contribute to lung fibrosis.
Telomere dysfunction represents another potential factor contributing to AEC injury and dysfunction. IPF subjects had marked telomere shortening in type II AECs in the lung, suggesting that specific loss of telomeres in the lung might be a key step in IPF pathogenesis. Nearly half of at-risk subjects, including subjects with histologically normal biopsies, had AEC telomeres shorter than those found in any normal control lung. Notably, we found short telomeres (<10th percentile for age) in approximately 35% of at-risk subjects. In contrast to a previous study of IPF patients (20), there was no correlation between AEC telomere length and PBMC telomere length among at-risk individuals. This discordance may be caused by differences in the study populations (established disease vs. asymptomatic individuals), or other factors, such as smoking history or environmental exposures. Although these measurements used different methodologies, this finding suggests that AEC telomere shortening is very common in IPF and only a subset of patients also have short telomeres in peripheral blood. Furthermore, telomere shortening in AECs may represent an important marker of early disease. Additional study is necessary to better understand mechanisms of telomere shortening in the lung and how this contributes to the development of lung fibrosis.
The MUC5B polymorphism rs35705950 minor allele has been detected at increased frequency in multiple independent IPF cohorts (22, 40, 41), and has also been associated with interstitial lung abnormalities in the Framingham cohort (54). We found similar allele frequencies in a separate cohort of IPF and control subjects to those reported in multiple large cohorts (22, 40, 41). At-risk subjects carried the minor allele at an intermediate frequency, which fits with the increased risk of disease in our cohort.
An area of particular interest within this cohort is the development of diagnostic and predictive biomarkers of FIP and IPF. We were surprised to find that numerous plasma biomarkers, including several previously associated with IPF including MMP7 and SP-D were found to differ between at-risk and healthy control subjects. Furthermore, several of these biomarkers were also associated with HRCT abnormalities among at-risk subjects. Demographic differences between at-risk and control subjects (including age and smoking history) represent potential confounders; however, controlling for these potential confounders did not significantly alter these findings. Further validation of these biomarkers is required to determine if this approach may hold promise in identifying presymptomatic disease.
There are several limitations of this study. Although there is increased disease risk across our cohort, we do not know which individual subjects carry risk alleles or which subjects will ultimately develop symptomatic FIP. As a result, the greatest insights from the present study will be evident in the future when an analysis can be performed comparing those with known genetic risk alleles or known FIP with those who do not carry risk alleles or do not develop disease. We performed exploratory analyses comparing subjects with HRCT abnormalities (who we suspect are most likely to develop clinical disease) with those with normal HRCTs; however, given the small number of subjects, these analyses are likely underpowered to detect any but very large effects, thus we consider these findings to be exploratory and hypothesis generating. With additional subject recruitment, more robust analyses will be possible. Additionally, as some subjects progress to overt disease over time, we anticipate the opportunity to develop models to identify a group of factors associated with disease risk.
In summary, this report represents the most extensive histologic, radiographic, and molecular evaluation of a population at high risk for developing FIP and IPF performed to date. We identified subtle radiographic and/or histopathologic abnormalities in more than 25% of subjects, and found that at-risk subjects harbor many traits related to epithelial cell dysfunction, which is linked to advanced FIP and IPF. We anticipate that, over time, following this cohort will offer an unprecedented view of the natural history of FIP and IPF and mechanisms of early disease pathogenesis.
Conclusions
By studying a cohort of individuals at high risk of developing FIP and IPF, and performing extensive radiographic, histologic, and molecular characterization of these asymptomatic individuals, we identified a variety of abnormalities that have the potential to represent the earliest phases in IPF pathogenesis. At-risk individuals frequently have radiographic and histopathologic abnormalities, evidence of telomere dysfunction, and alterations of plasma biomarker profiles. Over time, we anticipate gaining additional insights into the predictive utility of these abnormalities, and better characterizing the mechanisms that contribute to the morbidity and mortality of IPF.
Acknowledgments
Acknowledgment
The authors thank the patients and families who contributed data and samples during the past two decades and who have made this work possible.
Footnotes
Supported by HL085317 (T.S.B.); HL092870 (T.S.B.); HL85406 (W.E.L.); HL105479 (W.E.L.); HL94296 (J.A.K.); HL109118 (E.S.W.); HL 119,503 (L.R.Y.); HL 87,738 (P.F.C. and A.L.D.); HL088263 (L.B.W.); National Institutes of Health NCRR UL1 RR024975; American Thoracic Society/Boehringer Ingelheim Pharmaceuticals, Inc. Career Development Award in Idiopathic Pulmonary Fibrosis (J.A.K.); and the Department of Veterans Affairs (T.S.B. and W.E.L.). The flow cytometry core is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
Author Contributions: J.A.K. designed studies, performed experiments, analyzed and interpreted data, and wrote and revised the manuscript. J.M.P. designed studies, performed experiments, interpreted data, and wrote and revised the manuscript. D.F.Z. and P.F.C. designed studies, performed experiments, and revised the manuscript. C.M., E.T.G., A.L.D., D.B.M., V.V.P., O.B.R., D.-S.C., M.E.M., B.R.J., L.A.G., F.B.M., J.A.W., J.F.S., L.B.W., J.W.L., P.P.M., R.Z., E.S.W., J.D.K., J.E.J., and S.D.G. performed experiments and revised the manuscript. L.C. analyzed data and revised the manuscript. L.H.L., L.R.Y., M.P.S., J.A.P., J.D.C., and J.E.L. designed studies and revised the manuscript. W.E.L. and T.S.B. designed studies, interpreted data, and wrote and revised the manuscript.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201406-1162OC on November 12, 2014
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.García-Sancho C, Buendía-Roldán I, Fernández-Plata MR, Navarro C, Pérez-Padilla R, Vargas MH, Loyd JE, Selman M. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med. 2011;105:1902–1907. doi: 10.1016/j.rmed.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 2.Loyd JE. Pulmonary fibrosis in families. Am J Respir Cell Mol Biol. 2003;29(3) Suppl:S47–S50. [PubMed] [Google Scholar]
- 3.Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech. 2013;6:9–17. doi: 10.1242/dmm.010736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawson WE, Loyd JE, Degryse AL. Genetics in pulmonary fibrosis—familial cases provide clues to the pathogenesis of idiopathic pulmonary fibrosis. Am J Med Sci. 2011;341:439–443. doi: 10.1097/MAJ.0b013e31821a9d7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips JA, III, Sporn TA, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005;172:1146–1152. doi: 10.1164/rccm.200408-1104OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nogee LM, Dunbar AE, III, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 7.Thomas AQ, Lane K, Phillips J, III, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84:52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, III, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 10.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kropski JA, Mitchell DB, Markin C, Polosukhin VV, Choi L, Johnson JE, Lawson WE, Phillips JA, III, Cogan JD, Blackwell TS, et al. A novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest. 2014;146:e1–e7. doi: 10.1378/chest.13-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee HY, Seo JB, Steele MP, Schwarz MI, Brown KK, Loyd JE, Talbert JL, Schwartz DA, Lynch DA. High-resolution CT scan findings in familial interstitial pneumonia do not conform to those of idiopathic interstitial pneumonia. Chest. 2012;142:1577–1583. doi: 10.1378/chest.11-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosas IO, Ren P, Avila NA, Chow CK, Franks TJ, Travis WD, McCoy JP, Jr, May RM, Wu HP, Nguyen DM, et al. Early interstitial lung disease in familial pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:698–705. doi: 10.1164/rccm.200702-254OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bitterman PB, Rennard SI, Keogh BA, Wewers MD, Adelberg S, Crystal RG. Familial idiopathic pulmonary fibrosis. Evidence of lung inflammation in unaffected family members. N Engl J Med. 1986;314:1343–1347. doi: 10.1056/NEJM198605223142103. [DOI] [PubMed] [Google Scholar]
- 16.El-Chemaly S, Ziegler SG, Calado RT, Wilson KA, Wu HP, Haughey M, Peterson NR, Young NS, Gahl WA, Moss J, et al. Natural history of pulmonary fibrosis in two subjects with the same telomerase mutation. Chest. 2011;139:1203–1209. doi: 10.1378/chest.10-2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 19.Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol. 2005;32:521–530. doi: 10.1165/rcmb.2005-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, Rosenblatt RL, Garcia CK. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178:729–737. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kropski JA, Lawson WE, Blackwell TS. Right place, right time: the evolving role of herpesvirus infection as a “second hit” in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L441–L444. doi: 10.1152/ajplung.00335.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naik PK, Moore BB. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev Respir Med. 2010;4:759–771. doi: 10.1586/ers.10.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritchett JM, Avula S, Solus JF, Zoz D, Crossno PF, Markin C, Johnson JE, Worrell JA, Choi L, Rickman OB, et al. A MUC5B promoter polymorphism is associated with increased risk of IPF but not ALI or lung cancer. Am J Respir Crit Care Med. 2013;185:A5164. [Google Scholar]
- 26.Pritchett JM, Cheng D-S, Zoz DS, Crossno PF, Markin C, Johnson JE, Worrell JA, Choi L, Loyd JE, Lawson WE, et al. Elevated levels of human herpesviruses are detectable by QPCR in cell-free BAL of IPF patients and asymptomatic individuals at-risk for familial interstitial pneumonia. Am J Respir Crit Care Med. 2013;187:A3370. [Google Scholar]
- 27.Zoz DF, Crossno PF, Degryse AL, Markin C, Polosukhin VV, Gleaves LA, McMahon FB, Hemingway KR, Rickman OB, Worrell JA, et al. Radiographic and histologic abnormalities are frequently seen in asymptomatic individuals at risk for familial interstitial pneumonia. Am J Respir Crit Care Med. 2011;183:A5297. [Google Scholar]
- 28.Zoz DF, Crossno PF, Degryse AL, Markin C, Polosukhin VV, Gleaves LA, Worrell JA, Johnson JE, Loyd JE, Lawson WE, et al. Evaluation of asymptomatic relatives of patients with familial interstitial pneumonia for evidence of subclinical fibrotic remodeling in the lung. Am J Respir Crit Care Med. 2010;181:A6269. [Google Scholar]
- 29.Kropski JA, Pritchett JM, Zoz DF, Crossno PF, Markin C, Garnett ET, Mitchell DB, Polosukhin VV, Choi L, Cheng D-S, et al. Evidence for early lung epithelial injury and dysfunction in individuals at-risk for familial interstitial pneumonia. Am J Respir Crit Care Med. 2014;189:A3921. [Google Scholar]
- 30.Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA, Fry CD, White ES, Sisson TH, Tayob N, et al. COMET Investigators. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;303:L1046–L1056. doi: 10.1152/ajplung.00139.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferris BG. Epidemiology Standardization Project (American Thoracic Society) Am Rev Respir Dis. 1978;118:1–120. [PubMed] [Google Scholar]
- 32.Meeker AK, Gage WR, Hicks JL, Simon I, Coffman JR, Platz EA, March GE, De Marzo AM. Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol. 2002;160:1259–1268. doi: 10.1016/S0002-9440(10)62553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawson WE, Polosukhin VV, Zoia O, Stathopoulos GT, Han W, Plieth D, Loyd JE, Neilson EG, Blackwell TS. Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:899–907. doi: 10.1164/rccm.200311-1535OC. [DOI] [PubMed] [Google Scholar]
- 34.McDonald EA, Cheng L, Jarilla B, Sagliba MJ, Gonzal A, Amoylen AJ, Olveda R, Acosta L, Baylink D, White ES, et al. Maternal infection with Schistosoma japonicum induces a profibrotic response in neonates. Infect Immun. 2014;82:350–355. doi: 10.1128/IAI.01060-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonald EA, Friedman JF, Sharma S, Acosta L, Pond-Tor S, Cheng L, White ES, Kurtis JD. Schistosoma japonicum soluble egg antigens attenuate invasion in a first trimester human placental trophoblast model. PLoS Negl Trop Dis. 2013;7:e2253. doi: 10.1371/journal.pntd.0002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dean CB, Nielsen JD. Generalized linear mixed models: a review and some extensions. Lifetime Data Anal. 2007;13:497–512. doi: 10.1007/s10985-007-9065-x. [DOI] [PubMed] [Google Scholar]
- 37.R Development Core Team. Vienna, Austria: R Foundation for Statistical Computing: 2005. R: A language and environment for statistical computing. [Google Scholar]
- 38.Egan JJ, Stewart JP, Hasleton PS, Arrand JR, Carroll KB, Woodcock AA. Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis. Thorax. 1995;50:1234–1239. doi: 10.1136/thx.50.12.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang YW, Johnson JE, Browning PJ, Cruz-Gervis RA, Davis A, Graham BS, Brigham KL, Oates JA, Jr, Loyd JE, Stecenko AA. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41:2633–2640. doi: 10.1128/JCM.41.6.2633-2640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, Airo P, Matucci-Cerinic M, Wallaert B, Israel-Biet D, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8:e70621. doi: 10.1371/journal.pone.0070621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–1577. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vuga LJ, Tedrow JR, Pandit KV, Tan J, Kass DJ, Xue J, Chandra D, Leader JK, Gibson KF, Kaminski N, et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;189:966–974. doi: 10.1164/rccm.201309-1592OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. 2013;143:1422–1429. doi: 10.1378/chest.11-2735. [DOI] [PubMed] [Google Scholar]
- 44.Samukawa T, Hamada T, Uto H, Yanagi M, Tsukuya G, Nosaki T, Maeda M, Hirano T, Tsubouchi H, Inoue H. The elevation of serum napsin A in idiopathic pulmonary fibrosis, compared with KL-6, surfactant protein-A and surfactant protein-D. BMC Pulm Med. 2012;12:55. doi: 10.1186/1471-2466-12-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, Li K, Choi J, Vuga LJ, Lindell KO, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;185:67–76. doi: 10.1164/rccm.201101-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K, Hiwada K, Kohno N. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med. 2002;165:378–381. doi: 10.1164/ajrccm.165.3.2107134. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi H, Fujishima T, Koba H, Murakami S, Kurokawa K, Shibuya Y, Shiratori M, Kuroki Y, Abe S. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med. 2000;162:1109–1114. doi: 10.1164/ajrccm.162.3.9910080. [DOI] [PubMed] [Google Scholar]
- 48.Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008;5:e93. doi: 10.1371/journal.pmed.0050093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shim HS, Park MS, Park IK. Histopathologic findings of transbronchial biopsy in usual interstitial pneumonia. Pathol Int. 2010;60:373–377. doi: 10.1111/j.1440-1827.2010.02528.x. [DOI] [PubMed] [Google Scholar]
- 50.Berbescu EA, Katzenstein AL, Snow JL, Zisman DA. Transbronchial biopsy in usual interstitial pneumonia. Chest. 2006;129:1126–1131. doi: 10.1378/chest.129.5.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomassetti S, Cavazza A, Colby TV, Ryu JH, Nanni O, Scarpi E, Tantalocco P, Buccioli M, Dubini A, Piciucchi S, et al. Transbronchial biopsy is useful in predicting UIP pattern. Respir Res. 2012;13:96. doi: 10.1186/1465-9921-13-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wootton SC, Kim DS, Kondoh Y, Chen E, Lee JS, Song JW, Huh JW, Taniguchi H, Chiu C, Boushey H, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:1698–1702. doi: 10.1164/rccm.201010-1752OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, Nishino M, Araki T, Zazueta OE, Kurugol S, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. 2013;368:2192–2200. doi: 10.1056/NEJMoa1216076. [DOI] [PMC free article] [PubMed] [Google Scholar]