

Abstract
Objective:
To better define the genotype-phenotype correlations between the type of GBA (glucosidase, beta, acid) mutation, severe or mild, and the risk and age at onset (AAO), and potential mechanism of Parkinson disease (PD).
Methods:
We analyzed 1,000 patients of Ashkenazi-Jewish descent with PD for 7 founder GBA mutations, and conducted a meta-analysis of risk and AAO according to GBA genotype (severe or mild mutation). The meta-analysis included 11,453 patients with PD and 14,565 controls from worldwide populations. The statistical analysis was done with and without continuity correction (constant or empirical), considering biases that could potentially affect the results.
Results:
Among Ashkenazi-Jewish patients with PD, the odds ratios for PD were 2.2 and 10.3 for mild and severe GBA mutation carriers, respectively. The observed frequency of severe GBA mutation carriers among patients with PD was more than 4-fold than expected (4.4% vs 0.9%, respectively, p < 0.0001, Fisher exact test). In the different models of the meta-analysis, the odds ratios for PD ranged between 2.84 and 4.94 for mild GBA mutation carriers and 9.92 and 21.29 for severe GBA mutation carriers (p < 1 × 10−6 for all analyses). Pooled analysis demonstrated AAO of 53.1 (±11.2) and 58.1 (±10.6) years for severe and mild GBA mutation carriers, respectively (p = 4.3 × 10−5).
Conclusions:
These data demonstrate that mild and severe heterozygous GBA mutations differentially affect the risk and the AAO of PD. Our results have important implications for genetic counseling and clinical follow-up.
Mutations in GBA (glucosidase, beta, acid), encoding the lysosomal enzyme glucocerebrosidase, are important risk factors for Parkinson disease (PD) worldwide.1 Positive association between GBA mutations and PD was demonstrated in various populations, including Asians,2–6 Europeans,7–12 North Africans,13 North Americans14–16 and South Americans,17–20 but most frequently among Jews of Ashkenazi origin.21,22
When inherited from both parents, GBA mutations cause Gaucher disease (GD), a lysosomal storage disorder with 3 clinical types: nonneuropathic (type I), acute neuropathic (type II), and chronic neuropathic (type III). Accordingly, GBA mutations can be categorized as mild or severe: mild mutations are those that cause GD type I, and severe mutations are those that cause GD types II and III.23 Approximately 300 GBA mutations have been described in GD, many of which are also found in PD. We previously reported genotype-phenotype correlations between GBA mutation severity and PD in a cohort of 420 Ashkenazi patients with PD.22 Only a few case-control studies confirmed this observation,7,24 while most studies did not examine it. If this genotype-phenotype correlation is confirmed worldwide, it could be important for genetic counseling and clinical follow-up of GBA mutation carriers, and may also aid in understanding the pathophysiology of PD. The mechanism by which GBA mutations cause or increase the susceptibility for PD is not fully understood, and both gain- or loss-of-function mechanisms have been suggested.25
Herein, we examined whether severe and mild GBA mutations differentially affect PD susceptibility or age at onset (AAO) by studying the largest Ashkenazi PD cohort investigated to date and by conducting a meta-analysis of all relevant published case-control studies.
METHODS
Population.
The patient population in Tel Aviv included 1,000 consecutively recruited patients with PD, all unrelated, of full Ashkenazi-Jewish descent, who were examined at the Movement Disorders Unit at the Tel Aviv Sourasky Medical Center between July 2005 and August 2013. Details regarding their recruitment, diagnostic criteria, and interview procedure were previously described, including data for the first 420 patients recruited.22 In this cohort, 61% of the patients with PD are men, the average age at symptom onset is 60.1 ± 11.2 years, and the average age at enrollment is 67.4 ± 10.5 years. The control population included 3,805 individuals who have been previously described.22
Standard protocol approvals, registrations, and patient consents.
All participants provided informed consent before entering the study. The Institutional and National Supreme Helsinki (Institutional Review Board) Committees for Genetic Studies approved the study protocols and the informed consent.
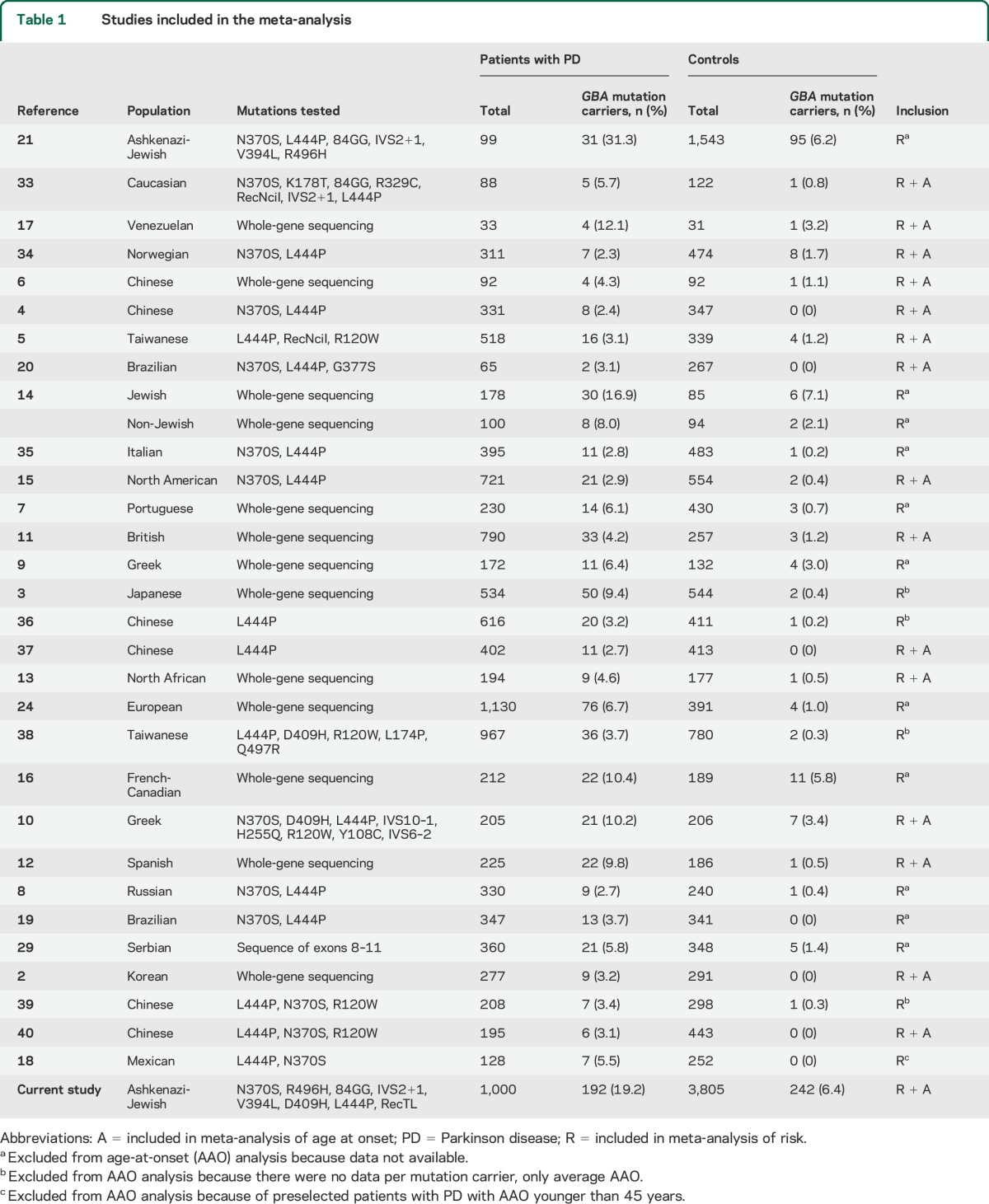
Selection of studies for meta-analysis.
To identify all studies that analyzed GBA mutations in PD populations, we searched PubMed using all combinations of the following search terms: “GBA,” “glucocerebrosidase,” “Parkinson,” “Parkinson's,” and “parkinsonism.” The search was performed in May 2014. All studies that reported the genotypes of GBA mutations in patients with PD and controls (n = 31 including the current study; table 1) were included in the analysis of risk, and all studies that reported the AAO of the different GBA mutation carriers were included in the analysis of AAO (n = 16 including the current study; table 1). Studies were excluded from the analysis of AAO for one of the following reasons: (1) there were no data on the mutations or AAO (11 studies were excluded based on this criterion; table 1); (2) there were no data on AAO per carrier (4 studies were excluded based on this criterion; table 1); or (3) if early-onset cases were preselected (one study was excluded, defined as AAO <45 years18).
Table 1.
Studies included in the meta-analysis
Classification of mutations in meta-analysis.
Mutations were defined as mild or severe according to a previously published classification that inferred the definition of the mutation as mild or severe based on the resulting GD. Mutations that caused the nonneuropathic type I GD were classified as mild, and mutations that caused the neuropathic types II and III were classified as severe.23 Because this classification was published in 2005, we searched for new information regarding mutations that were found in the studies analyzed here and were classified as unknown in the reference.23 The p.I260T mutation (described in a patient with PD in reference 12) is now classified as severe based on a report of a patient with type II GD with this mutation,26 the p.S271G mutation (described in a patient with PD in reference 2) is now classified as mild based on a report of a patient with type I GD with this mutation,27 and the p.R277C mutation (described in a patient with PD in reference 2) is now classified as mild based on a report of a patient with type I GD with this mutation.28 The classification of all the mutations is detailed in table e-1 on the Neurology® Web site at Neurology.org.
Genotyping in the Tel Aviv sample.
DNA was extracted from white blood cells by using a standard salting-out protocol, and the genotyping of founder GBA mutations and the LRRK2 p.G2019S mutation was performed as previously described.22 In brief, patients and controls were tested for the 84GG, IVS2+1, p.N370S, p.L444P, p.V394L, p.R496H, and 370Rec (previously referred to as RecTL23) GBA mutations using PRONTO Gaucher kits (Pronto Diagnostics, Rehovot, Israel). The 3,805 controls were not tested for the p.R496H mutation, because it was not recommended by the Israeli Society of Medical Geneticists to be included in the GBA mutation screening panel. The LRRK2 p.G2019S mutation (rs34637584) was also detected using TaqMan assay ID C_63498123_10 in the StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA).
Statistical analysis.
Analysis of 1,000 Ashkenazi-Jewish patients with PD.
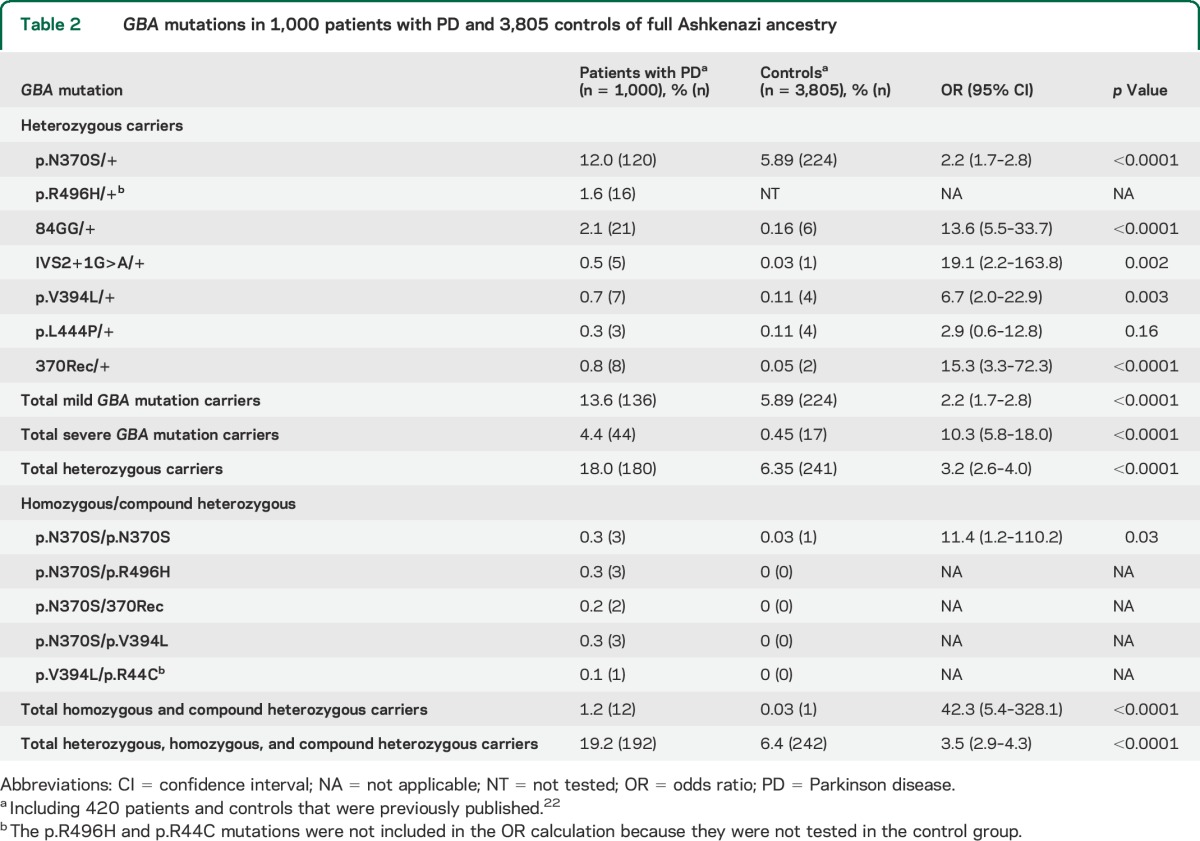
Differences in continuous variables were tested using analysis of variance, Mann–Whitney, or Kruskal-Wallis tests, and χ2 or Fisher exact test was used for comparison of categorical variables. To test for any deviation from Hardy-Weinberg equilibrium among patients with PD and controls, a goodness-of-fit test with 1 degree of freedom was applied. Odds ratios (ORs) and 95% confidence intervals (CIs) were determined using an online calculator (DJR Hutchon Calculator). There are 2 mild founder mutations in the Ashkenazi population: p.R496H and p.N370S. Because the p.R496H mutation was not tested among the young Ashkenazi controls, the calculated OR of mild mutation carriers in the Ashkenazi population refers only to p.N370S mutation carriers. For the analysis of risk and AAO, carriers of the LRRK2 p.G2019S mutation were excluded. SPSS software version 17 (SPSS Inc., Chicago, IL) was used for all other data analyses.
Meta-analysis.
Cochran-Mantel-Haenszel test was used for pooling study outcomes, and the Tarone test was used for determining heterogeneity. The analysis was conducted using the “metafor” package in R. Because some of the studies included in the meta-analysis had no (zero) severe or mild GBA mutation carriers in either patients with PD or controls, not allowing for calculation of ORs, the meta-analysis was done both with and without a continuity correction. AAO of severe or mild GBA mutation carriers were pooled together, and Student t test was used for statistical analysis. To avoid bias regarding the analysis of AAO, studies that included only patients with early-onset PD were excluded. In addition, all individuals with available data on mutations other than GBA, which may affect the AAO of PD, such as LRRK2, Parkin, and PINK1 mutations, were excluded.
RESULTS
PD risk in mild vs severe GBA mutation carriers among 1,000 patients of Ashkenazi origin.
Table 2 details the frequencies of GBA mutations that were identified among 1,000 patients with PD and 3,805 controls of full Ashkenazi-Jewish origin. The OR for PD among mild GBA mutation carriers was 2.2 (95% CI 1.7–2.8, including only the p.N370S mutation, because the p.R496H mutation was not tested in controls; see the methods section), compared with 10.3 (95% CI 5.8–18.0) among severe GBA mutation carriers. To examine whether these ORs were significantly different, the expected vs observed frequency of severe GBA mutations among patients was analyzed. Based on the frequencies of mild GBA mutations, which are 2.04 more frequent in patients than in controls, the expected frequency of carriers of a severe GBA mutation among 1,000 patients with PD was 0.92%, while the observed frequency was significantly higher, at 4.4% (p < 0.0001, Fisher exact test).
Table 2.
GBA mutations in 1,000 patients with PD and 3,805 controls of full Ashkenazi ancestry
Differential effects of severe vs mild GBA mutations: A meta-analysis.
To further determine whether the effects of severe vs mild GBA mutations on risk and AAO of PD are a common phenomenon worldwide, we conducted a meta-analysis that included all studies published until May 2014. We found 31 peer-reviewed publications with data on GBA mutation types, including our current study, with a total of 11,453 patients with PD and 14,565 controls, and 16 studies with data on AAO per individual with a GBA mutation.
When including studies that identified severe and mild GBA mutation carriers among both patients and controls, the pooled ORs for PD in mild and severe GBA mutation carrier groups were 2.84 (95% CI 2.34–3.45; 11 studies were included; figure e-1A) and 10.28 (95% CI 6.95–15.20; 14 studies were included; figure e-1B), respectively (p < 1 × 10−20 for both). Using a constant continuity correction of 0.5 for studies with no (zero) severe or mild GBA mutation carriers among either patients or controls, the ORs for PD in mild and severe GBA mutation carrier groups were 3.07 (95% CI 2.53–3.71) and 15.49 (95% CI 10.50–22.86), respectively (p < 1 × 10−20 for both; figures e-1C and e-1D). Using empirical continuity correction based on case/control ratio and estimated prior OR (see methods), the ORs for PD among mild and severe GBA mutation carriers were 3.01 (95% CI 2.50–3.63) and 14.59 (95% CI 10.00–21.31), respectively (p < 1 × 10−20 for both; figure 1).
Figure 1. Meta-analysis of severe and mild GBA mutations and Parkinson disease risk.

(A) Forest plot of studies with data on mild GBA mutations, using an empirical continuity correction (see methods) for studies with zero cases with mild GBA mutations. The analysis included data from 31 studies with a total of 11,453 cases and 14,565 controls. The p value for heterogeneity was 0.62. (B) Forest plot of studies with data on severe GBA mutations, using an empirical continuity correction (see methods) for studies with zero cases with severe GBA mutations. The analysis included data from 31 studies with a total of 11,453 cases and 14,565 controls. The p value for heterogeneity was 0.94. FE = fixed effect; Ref = reference number.
The same analysis was conducted excluding our current data for 1,000 Ashkenazi patients and 3,805 controls to avoid the possibility of bias of the results (figure e-2, A–F). When excluding studies with no (zero) severe or mild GBA mutation carriers in either patients or controls, the pooled ORs for PD among mild and severe GBA mutation carriers were 3.78 (95% CI 2.62–5.46) and 9.92 (95% CI 6.05–16.25), respectively (p < 1 × 10−13 for both; figures e-2A and e-2B). Using a constant continuity correction of 0.5, the ORs for PD among mild and severe GBA mutation carriers were 4.47 (95% CI 3.14–6.35) and 17.03 (95% CI 10.49–27.64), respectively (p < 1 × 10−18 for both; figures e-2C and e-2D). Using empirical continuity correction, the ORs for PD among mild and severe GBA mutation carriers were 4.25 (95% CI 3.03–5.95) and 15.67 (95% CI 9.84–24.94), respectively (p < 1 × 10−19 for both; figures e-2E and e-2F).
Because 12 studies used a whole GBA gene sequencing approach instead of only analyzing specific mutations, it was possible to conduct a meta-analysis for these studies separately (table 1, figure e-3, A–F). When excluding studies with no severe or mild GBA mutation carriers in either patients or controls, the pooled ORs for PD among mild and severe GBA mutation carriers were 4.69 (95% CI 2.44–9.01, p < 1 × 10−6; figure e-3A) and 12.2 (95% CI 4.92–30.24, p < 1 × 10−11; figure e-3B), respectively. Using a constant continuity correction of 0.5, the ORs for PD among mild and severe GBA mutation carriers were 4.94 (95% CI 2.72–8.98, p < 1 × 10−8; figure e-3C) and 21.29 (95% CI 8.65–52.42, p < 1 × 10−20; figure e-3D), respectively. Using empirical continuity correction, the ORs for PD among mild and severe GBA mutation carriers were 4.73 (95% CI 2.65–8.43, p < 1 × 10−8; figure e-3E) and 19.34 (95% CI 8.19–45.63, p < 1 × 10−20; figure e-3F), respectively.
Calculations of p values for data heterogeneity were performed for all 3 meta-analyses presented above: all studies included, all studies excluding the Tel Aviv Ashkenazi cohort, and only studies in which the entire GBA gene had been sequenced. Because the best p values were obtained for the empirical continuity correction model (p = 0.62–0.95), this model is thought to be most accurately estimating the ORs for mild and severe GBA mutation carriers. It is important to emphasize, however, that both with or without continuity correction, the effects remained the same, demonstrating differential effects of severe and mild GBA mutations.
To analyze the effects of severe vs mild GBA mutation on AAO, we pooled the results from 16 studies that included data on AAO of specific GBA mutation carriers (table 1). The AAO was 53.1 (±11.2) years among severe GBA mutation carriers (n = 166), and 58.1 (±10.6) years among mild GBA mutation carriers (n = 162, p = 4.3 × 10−5). After excluding our current study, the AAO was 52.0 (±11.5) years among severe GBA mutation carriers (n = 122), and 56.1 (±10.6) years among mild GBA mutation carriers (n = 40, p < 0.05). Of note, in both analyses, with and without the current study, the AAO of severe GBA mutation carriers was 4 to 5 years younger than the AAO of mild GBA mutation carriers. In our population alone, although not statistically significant, the AAO were 56.2 ± 9.9 and 58.5 ± 10.6 years among severe and mild GBA mutation carriers, respectively, which is comparable to our previous report from 420 patients.22
DISCUSSION
The meta-analysis study presented here included data from a large variety of populations around the world, including from North, Central, and South America, Western and Eastern Europe, Asia, North Africa, and Ashkenazi Jews (table 1). While a previous meta-analysis examined whether GBA mutations are associated with PD, it did not determine the role of severe vs mild mutations in PD risk and onset.1 In the current study, we demonstrated that there is a clear, significant differential effect of severe vs mild GBA mutations on the risk and AAO of PD, not only in Ashkenazi patients, where founder GBA mutations are common (approximately 20%), but also worldwide. Carriers of severe GBA mutations have about 3- to 4-fold higher risk and about 5 years younger AAO than carriers of mild GBA mutations. These results were demonstrated in all the models used for the analysis, with and without continuity correction.
Additional published studies that support our findings were not included in our meta-analysis of AAO because they did not contain per-individual AAO information19,29: in 347 Brazilian patients with PD, 5 patients with the mild GBA mutation p.N370S had an average AAO of 54.6 years, and 8 patients with the severe GBA mutation p.L444P had an average AAO of 47.0 years.19 In a Serbian population of 360 patients with PD, the average AAO for mild GBA mutation carriers (n = 7) was 56.2 years, and 45.1 years among severe GBA mutation carriers (n = 10).29 It is possible that other disease phenotypes may also be differentially presented among severe vs mild GBA mutation carriers. Because recent studies reported that carriers of GBA mutations may be at higher risk of cognitive impairment,30 it could be of interest to study large cohorts of patients to determine whether this phenotype may be associated with severe vs mild GBA genotype as well. While in the current study there were no available data on the subtypes of PD (tremor-dominant or akinetic-rigid), it would be of interest to examine these phenotypes and their association with GBA genotypes.
Based on the meta-analysis results, the estimated risk of PD (OR) among mild GBA mutation carriers ranged from 3.0 to 4.7, and from 14.6 to 19.3 among severe GBA mutation carriers. These estimates suggest that known healthy carriers of severe GBA mutations should undergo genetic counseling regarding the increased risk of PD. In addition, they suggest that the approach described for the LRRK2 p.G2019S mutation carriers31 might be adopted, i.e., a closer clinical follow-up, to identify early symptoms of PD among carriers of severe GBA mutations. Such follow-up will be particularly important when preventive treatment for PD becomes available.
Although the role of GBA mutations as risk factors for PD is clearly established, the mechanism underlying GBA-associated PD is still not clear. Several suggestions have been made, including loss-of-function mechanisms or toxic gain-of-function mechanisms.25 Our findings may support the loss-of-function hypothesis. This could be further exemplified by the 2 founder null mutations that have been identified in the Ashkenazi population, 84GG and IVS2+1. These 2 mutations are found in 26 of 1,000 patients (2.6%) compared with only 7 of 3,805 controls (0.19%), with ORs of 13.6 and 19.1, respectively. These null mutations result in significantly reduced production of the glucocerebrosidase protein, and a possible loss-of-function mechanism was already suggested32: depletion of glucocerebrosidase results in the accumulation of α-synuclein, subsequently leading to inhibition of trafficking of glucocerebrosidase into the lysosome, thus creating a pathogenic positive feedback loop.32 However, more studies are required to identify the specific mechanism by which GBA mutations cause α-synuclein accumulation and predispose to PD.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Prof. Yoav Ben-Shlomo for statistical advice in the early stages of this study.
GLOSSARY
- AAO
age at onset
- CI
confidence interval
- GBA
glucosidase, beta, acid
- GD
Gaucher disease
- OR
odds ratio
- PD
Parkinson disease
Footnotes
Editorial, page 866
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Ziv Gan-Or: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, statistical analysis, study supervision. Idan Amshalom: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis. Laura L. Kilarski: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Anat Bar-Shira: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Mali Gana-Weisz: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Anat Mirelman: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data, study supervision. Karen Marder: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Susan Bressman: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, obtaining funding. Nir Giladi: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, obtaining funding. Avi Orr-Urtreger: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, study supervision, obtaining funding.
STUDY FUNDING
This work was supported by Tel Aviv Sourasky Medical Center Grant of Excellence, by the Kahn Foundation, by the Chief Scientist of the Israeli Ministry of Health (grant 3-4893), by the Legacy Heritage Biomedical Science Partnership Program of the Israel Science Foundation (grant 1922/08), and by The Michael J. Fox Foundation for Parkinson's Research.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi JM, Kim WC, Lyoo CH, et al. Association of mutations in the glucocerebrosidase gene with Parkinson disease in a Korean population. Neurosci Lett 2012;514:12–15. [DOI] [PubMed] [Google Scholar]
- 3.Mitsui J, Mizuta I, Toyoda A, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009;66:571–576. [DOI] [PubMed] [Google Scholar]
- 4.Tan EK, Tong J, Fook-Chong S, et al. Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch Neurol 2007;64:1056–1058. [DOI] [PubMed] [Google Scholar]
- 5.Wu YR, Chen CM, Chao CY, et al. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J Neurol Neurosurg Psychiatry 2007;78:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziegler SG, Eblan MJ, Gutti U, et al. Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol Genet Metab 2007;91:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bras J, Paisan-Ruiz C, Guerreiro R, et al. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol Aging 2009;30:1515–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emelyanov A, Boukina T, Yakimovskii A, et al. Glucocerebrosidase gene mutations are associated with Parkinson's disease in Russia. Mov Disord 2012;27:158–159. [DOI] [PubMed] [Google Scholar]
- 9.Kalinderi K, Bostantjopoulou S, Paisan-Ruiz C, Katsarou Z, Hardy J, Fidani L. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci Lett 2009;452:87–89. [DOI] [PubMed] [Google Scholar]
- 10.Moraitou M, Hadjigeorgiou G, Monopolis I, et al. Beta-glucocerebrosidase gene mutations in two cohorts of Greek patients with sporadic Parkinson's disease. Mol Genet Metab 2011;104:149–152. [DOI] [PubMed] [Google Scholar]
- 11.Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain 2009;132:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seto-Salvia N, Pagonabarraga J, Houlden H, et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson's disease course. Mov Disord 2012;27:393–399. [DOI] [PubMed] [Google Scholar]
- 13.Lesage S, Condroyer C, Hecham N, et al. Mutations in the glucocerebrosidase gene confer a risk for Parkinson disease in North Africa. Neurology 2011;76:301–303. [DOI] [PubMed] [Google Scholar]
- 14.Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007;69:1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mata IF, Samii A, Schneer SH, et al. Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol 2008;65:379–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noreau A, Riviere JB, Diab S, et al. Glucocerebrosidase mutations in a French-Canadian Parkinson's disease cohort. Can J Neurol Sci 2011;38:772–773. [DOI] [PubMed] [Google Scholar]
- 17.Eblan MJ, Nguyen J, Ziegler SG, et al. Glucocerebrosidase mutations are also found in subjects with early-onset parkinsonism from Venezuela. Mov Disord 2006;21:282–283. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Del Rincon Mde L, Monroy Jaramillo N, Suarez Martinez AI, et al. The L444P GBA mutation is associated with early-onset Parkinson's disease in Mexican Mestizos. Clin Genet 2013;84:386–387. [DOI] [PubMed] [Google Scholar]
- 19.Guimaraes Bde C, Pereira AC, Rodrigues Fda C, et al. Glucocerebrosidase N370S and L444P mutations as risk factors for Parkinson's disease in Brazilian patients. Parkinsonism Relat Disord 2012;18:688–689. [DOI] [PubMed] [Google Scholar]
- 20.Spitz M, Rozenberg R, Pereira Lda V, Reis Barbosa E. Association between Parkinson's disease and glucocerebrosidase mutations in Brazil. Parkinsonism Relat Disord 2008;14:58–62. [DOI] [PubMed] [Google Scholar]
- 21.Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 2004;351:1972–1977. [DOI] [PubMed] [Google Scholar]
- 22.Gan-Or Z, Giladi N, Rozovski U, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008;70:2277–2283. [DOI] [PubMed] [Google Scholar]
- 23.Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis 2005;35:355–364. [DOI] [PubMed] [Google Scholar]
- 24.Lesage S, Anheim M, Condroyer C, et al. Large-scale screening of the Gaucher's disease-related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet 2011;20:202–210. [DOI] [PubMed] [Google Scholar]
- 25.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012;11:986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chabas A, Gort L, Diaz-Font A, et al. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol Dis 2005;35:253–258. [DOI] [PubMed] [Google Scholar]
- 27.Sunwoo MK, Kim SM, Lee S, Lee PH. Parkinsonism associated with glucocerebrosidase mutation. J Clin Neurol 2011;7:99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JY, Lee BH, Kim GH, et al. Clinical and genetic characteristics of Gaucher disease according to phenotypic subgroups. Korean J Pediatr 2012;55:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar KR, Ramirez A, Gobel A, et al. Glucocerebrosidase mutations in a Serbian Parkinson's disease population. Eur J Neurol 2013;20:402–405. [DOI] [PubMed] [Google Scholar]
- 30.Alcalay RN, Caccappolo E, Mejia-Santana H, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD Study. Neurology 2012;78:1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giladi N, Mirelman A, Thaler A, Bar-Shira A, Gurevich T, Orr-Urtreger A. Fighting the risk of developing Parkinson's disease: clinical counseling for first degree relatives of patients with Parkinson's disease. J Neurol Sci 2011;310:17–20. [DOI] [PubMed] [Google Scholar]
- 32.Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sato C, Morgan A, Lang AE, et al. Analysis of the glucocerebrosidase gene in Parkinson's disease. Mov Disord 2005;20:367–370. [DOI] [PubMed] [Google Scholar]
- 34.Toft M, Pielsticker L, Ross OA, Aasly JO, Farrer MJ. Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian population. Neurology 2006;66:415–417. [DOI] [PubMed] [Google Scholar]
- 35.De Marco EV, Annesi G, Tarantino P, et al. Glucocerebrosidase gene mutations are associated with Parkinson's disease in southern Italy. Mov Disord 2008;23:460–463. [DOI] [PubMed] [Google Scholar]
- 36.Mao XY, Burgunder JM, Zhang ZJ, et al. Association between GBA L444P mutation and sporadic Parkinson's disease from mainland China. Neurosci Lett 2010;469:256–259. [DOI] [PubMed] [Google Scholar]
- 37.Sun QY, Guo JF, Wang L, et al. Glucocerebrosidase gene L444P mutation is a risk factor for Parkinson's disease in Chinese population. Mov Disord 2010;25:1005–1011. [DOI] [PubMed] [Google Scholar]
- 38.Huang CL, Wu-Chou YH, Lai SC, et al. Contribution of glucocerebrosidase mutation in a large cohort of sporadic Parkinson's disease in Taiwan. Eur J Neurol 2011;18:1227–1232. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Liu L, Xiong J, et al. Glucocerebrosidase L444P mutation confers genetic risk for Parkinson's disease in central China. Behav Brain Funct 2012;8:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Bao QQ, Zhuang XS, et al. Association of common variants in the glucocerebrosidase gene with high susceptibility to Parkinson's disease among Chinese. Chin J Physiol 2012;55:398–404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.