

Abstract
Objective:
We provide a nationwide population study of patients with congenital muscular dystrophy in Italy.
Methods:
Cases were ascertained from the databases in all the tertiary referral centers for pediatric neuromuscular disorders and from all the genetic diagnostic centers in which diagnostic tests for these forms are performed.
Results:
The study includes 336 patients with a point prevalence of 0.563 per 100,000. Mutations were identified in 220 of the 336 (65.5%). The cohort was subdivided into diagnostic categories based on the most recent classifications on congenital muscular dystrophies. The most common forms were those with α-dystroglycan glycosylation deficiency (40.18%) followed by those with laminin α2 deficiency (24.11%) and collagen VI deficiency (20.24%). The forms of congenital muscular dystrophy related to mutations in SEPN1 and LMNA were less frequent (6.25% and 5.95%, respectively).
Conclusions:
Our study provides for the first time comprehensive epidemiologic information and point prevalence figures for each of the major diagnostic categories on a large cohort of congenital muscular dystrophies. The study also reflects the diagnostic progress in this field with an accurate classification of the cases according to the most recent gene discoveries.
The term congenital muscular dystrophy (CMD) classically includes a group of genetically, clinically, and biochemically distinct entities sharing clinical and pathologic features such as early presentation of weakness and hypotonia and dystrophic features on muscle biopsy.1
The rapidly increasing identification of several genes responsible for different forms of CMD has dramatically expanded the spectrum of the known forms, allowing a better understanding of the individual forms and different underlying pathomechanisms.2
The incidence and prevalence of CMD in populations is not sufficiently known with only a few studies reporting incidence or point prevalence in relatively small regions.3–5 Furthermore, several new genes have only been described in the last few years and the subtyping and classification in those reports is therefore not updated.
In this study, we report the results of a detailed population study and the frequency of the various forms of CMD in all the patients with CMD referred to all the tertiary care centers for pediatric neuromuscular disorders in Italy.
METHODS
Our inclusion criteria were all patients with a diagnosis of CMD currently seen in the participating centers. The diagnosis of CMD was based on a combination of clinical and biopsy findings according to the recently proposed diagnostic criteria for CMD.6 This included clinical signs suggestive of CMD and/or dystrophic or myopathic features on muscle biopsy, with exclusion of other identifiable neuromuscular disorders. Clinical signs included weakness and hypotonia or contractures at birth or in the first months, but as in some CMD variants (e.g., Ullrich CMD), the onset can be delayed.6–8
All patients with a potential neuromuscular disorder, with weakness and/or increased creatine kinase (CK) serum levels, are referred to a tertiary care center for further testing. Because all the Italian tertiary care centers for pediatric and adult neuromuscular disorders participated in this study, we are therefore likely to have included all the patients who have undergone a diagnostic process for CMD. Cases were ascertained from the databases in each center, only including patients who were under current follow-up. To exclude that patients may have been referred by other physicians, we also checked all the molecular genetic centers performing analysis of the CMD genes. Other sources available for this study included the Collagen VI Database held at the University of Ferrara and the CMD Dystroglycan Database, a nationwide database used for our previous study9,10 and that is constantly updated.
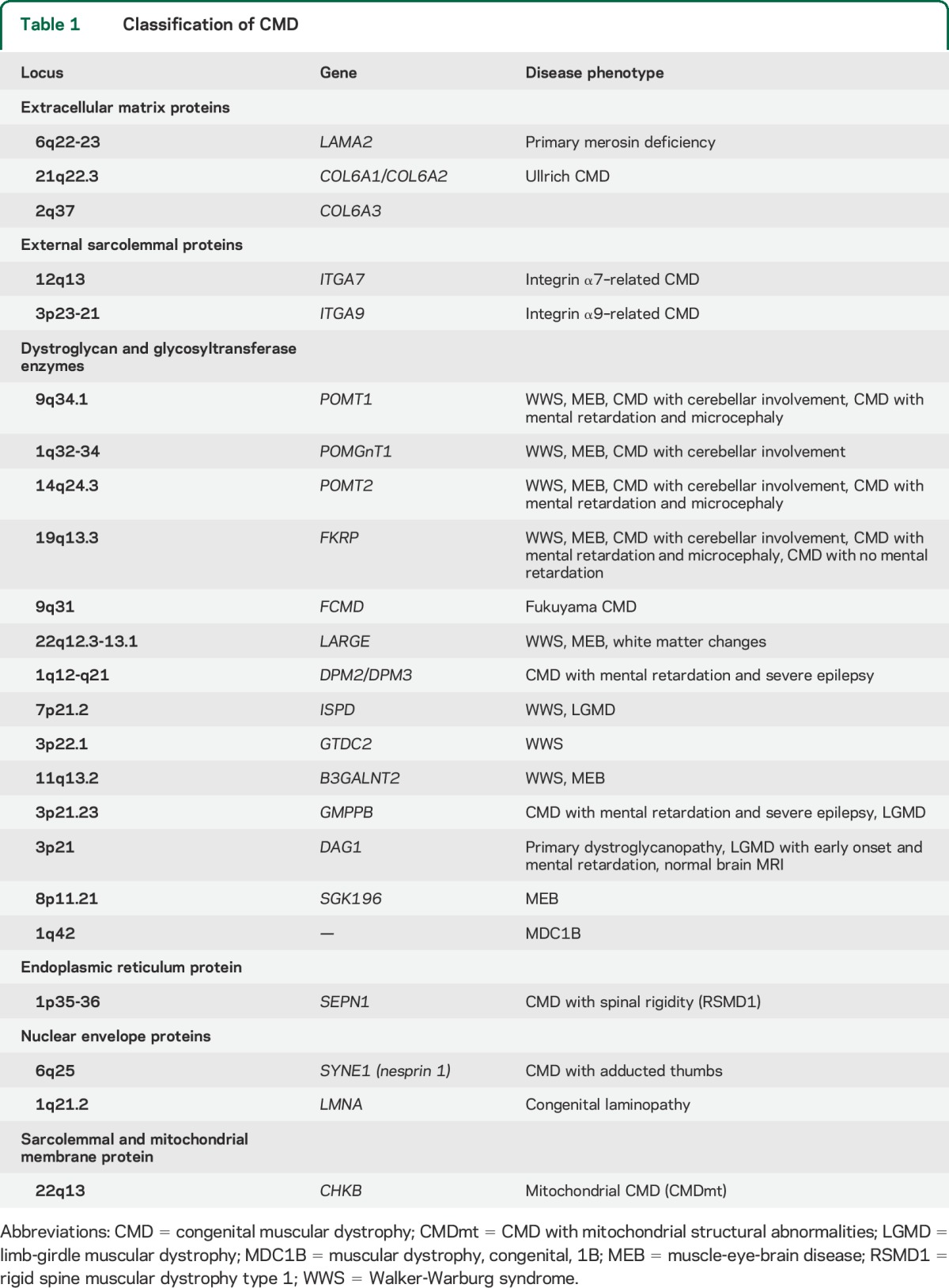
Disease groups were established using the most recent CMD classifications2 (table 1):
Forms of CMD due to mutations in genes encoding for structural proteins of the basal lamina or extracellular matrix or receptors for extracellular matrix proteins. This category includes mutations in the collagen 6 genes, laminin α2 (merosin), and integrin α7 and α9.
Forms secondary to genes encoding for putative or demonstrated glycosyltransferases, which affect the glycosylation of α-dystroglycan (αDG). This includes Fukuyama CMD, muscle-eye-brain disease (MEB), and Walker-Warburg syndrome, and other phenotypes.
Forms due to mutations in genes encoding for nuclear envelope proteins (LMNA and nesprin).
Forms due to abnormalities of proteins with, so far, unknown function localized in the endoplasmic reticulum, which include the form of CMD with rigid spine syndrome secondary to mutations in SEPN1.
Table 1.
Classification of CMD
For each group, and for the whole cohort, point prevalence was established. The prevalence day was January 31, 2013. The 95% confidence interval was measured using the Wilson score interval with continuity correction.
Standard protocol approvals, registrations, and patient consents.
The ethical committees of the participating centers approved the study.
RESULTS
Three hundred thirty-six subjects fulfilled the inclusion criteria (191 male, 145 female). All were followed by one of the participating clinical centers who were also responsible for referring them for the genetic diagnosis with the exception of 2 boys with Ullrich CMD referred to the genetic labs by a specialist in connective tissue disorders. After diagnosis, both patients were referred and followed by one of the clinical participating centers.
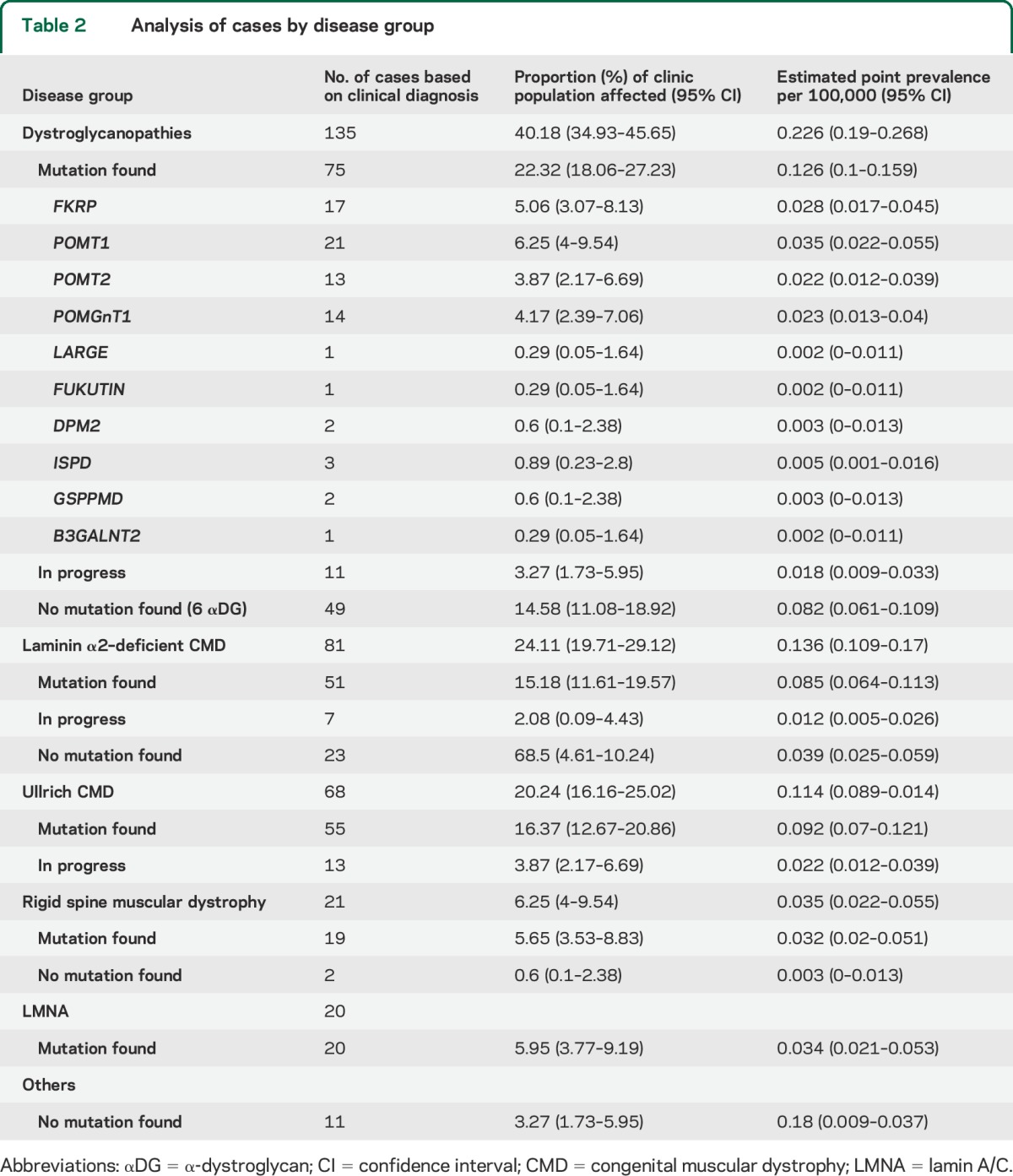
The combined prevalence was 0.563 per 100,000 total population. More than 90% could be classified in one of the major diagnostic groups. Mutations were identified in 220 of the 336. Table 2 shows details for each subgroup.
Table 2.
Analysis of cases by disease group
Major diagnostic categories.
Dystroglycanopathies.
This group comprised 135 patients, 40.18% of the CMD population, with a prevalence of 0.226 per 100,000. In these patients, the diagnosis of dystroglycanopathy followed the evidence of predominant weakness and wasting of the upper compared with the lower limbs, and markedly elevated serum CK levels. In a minority of cases, the diagnosis followed clinical and radiologic evidence of CNS involvement in the first months of life, associated with highly increased values of CK serum levels (more than 5-fold the normal values). In all, muscle biopsy showed absent or reduced αDG.
The form with mental retardation and microcephaly with or without cerebellar involvement and MEB were the most frequent phenotypes.
Mutations were identified in 75 of the 135 patients with αDG reduction included in the present study. All 135 were screened for the 6 genes identified before 2012 (namely, FKRP, POMT1, POMT2, POMGnT1, Fukutin, LARGE). Sixty-seven patients (49.62%) had 2 allelic mutations identified in one of these 6 genes. By subtype of mutations, POMT1 was the most common, representing 15.5% of the total dystroglycanopathies, followed by FKRP, POMGnT1, and POMT2 (12.59%, 10.37%, and 9.62%, respectively). Details of the occurrence of each form are shown in table 2. The other categories were individually much smaller with only isolated cases with mutations in LARGE or Fukutin.
Of the 68 subjects who did not have mutations in the first 6 genes identified, only a subset of 18 was screened for the more recently discovered genes. Mutations were found in 8 of these 18, raising the number of patients with identified mutation from 67 to 75 of 135. Three subjects had mutations in ISPD, 2 in DPM2, 2 in GSPPMD, and 1 in B3GALNT2. Because genetic analysis for the remaining 50 is in progress, we cannot therefore yet fully establish the prevalence of mutations in the new genes.
Nine of the 135 patients were familial cases (5 sets of siblings; one of them had a sibling not included in the present study). All the patients in this group were of Italian origin with the exception of 16 who were resident in Italy but with parents originally from other countries (8 from Northern Africa, 4 from Eastern Europe, and 4 from Asia).
Merosin-deficient CMD.
Eighty-one patients were classified as laminin α2 (merosin)-deficient CMD (24.11% of the whole CMD cohort) with a prevalence of 0.136 per 100,000 for Italy.
All had a phenotype consistent with the classic course of typical phenotype reported for merosin-deficient CMD including the involvement of the white matter on brain MRI,8,11 and increased CK (more than 5-fold the normal values).
LAMA2 chain gene mutations were found in 51 of 81. In 7 patients, genetic analysis for LAMA2 is in progress or consent was not obtained by the parents.
In the remaining 23 patients, mutations were not found; of these, 21 patients had absent or markedly reduced laminin α2 and 2 partial deficiency on muscle biopsy. In the 2 cases with partial deficiency, the reduction in laminin α2 expression was greater than the reduction observed in αDG protein expression. Both were also screened for the first 6 genes identified for dystroglycanopathies and no mutation was found.
Sixteen cases were familial (6 sets of siblings and 2 couples of first cousins). Sixty patients in this group were of Italian origin while the other 21 were resident in Italy but had parents originally from other countries (14 from Northern Africa, 3 from Eastern Europe, 3 from Asia, and 1 from South America).
Ullrich CMD.
Sixty-eight patients (20.24%) had a phenotype compatible with Ullrich CMD and reduced collagen on skin and or muscle biopsy. CK levels were normal or only mildly elevated. Muscle MRI was available in 32 of the 64 and showed the typical pattern previously reported in association with Ullrich CMD.12 The point prevalence was 0.114 per 100,000.
Mutations were found in 55 of the 68. Regarding the phenotypes, 11 cases were familial (5 sets of siblings) and none had other familial cases not included in the present study. All the patients in this group were of Italian origin with the exception of 9 who were resident in Italy but with parents originally from other countries (2 from South America, 2 from Northern Africa, 2 from Eastern Europe, 1 from Asia, and 2 from Turkey).
Fifty-five cases with COL6 mutations in whom ambulation was achieved by 24 months and the onset of weakness or contractures occurred after that, were not classified as CMD and therefore not included in the present study.
Rigid spine muscular dystrophy type 1.
Twenty-one patients (6.25%) had delayed ambulation, developed rigid spine and early respiratory impairment, with a clinical and MRI phenotype compatible with rigid spine muscular dystrophy type 1 (RSMD1). Serum CK levels were normal or only mildly elevated. Muscle MRI was available in 18 and showed the typical pattern previously reported in association with SEPN1 mutations.12 Mutations in SEPN1 were found in 19 of the 21. The point prevalence was 0.035 per 100,000.
They were all sporadic cases, with the exception of 2 siblings. All the patients in this group were of Italian origin with the exception of 2 whose parents were originally from South Asia.
Laminopathies.
Twenty patients (5.95%) had congenital presentation with dropped head or other clinical signs suggestive of the form of CMD associated with laminopathy. All but 4 achieved independent ambulation. Mutations in LAMA A/C were found in all 20. The point prevalence was 0.034 per 100,000.
They were all sporadic cases with the exception of a girl whose father was also affected and had died of cardiac arrhythmia. All were of Italian origin with the exception of one child whose parents were originally from South Asia.
Other cases.
Eleven cases fulfilled the inclusion criteria for CMD but did not fit into any of the previous major categories. All had increased CK, ranging from 2- to 8-fold the normal values. Three had signs of CNS involvement but normal laminin α2 and αDG on muscle biopsy.
DISCUSSION
The population prevalence figure for CMD in our study was 0.563 per 100,000. Because all the Italian tertiary care centers for neuromuscular disorders participated in this study, this figure is likely to include all cases with a clinical presentation evocative of a CMD phenotype or at least all those for whom a possible diagnosis of CMD was considered. We cannot exclude that in some cases with milder phenotypes or later onset, the diagnosis of CMD may not have been considered and these patients may have never been referred to a tertiary care center or had any diagnostic test for CMD performed.
In this study, we subdivided our cohort according to the most recent classification for CMD. Because of this, the results are not easily comparable with previous population studies as most of them only included small cohorts and/or were performed before the identification of new genes.
In 1996, the incidence of CMD was estimated at between 4.7 × 105 live births in the north of Italy,5 but only 20 patients with CMD were included. Another study reports a point prevalence of 6.3 × 105 in 20 patients identified in Western Sweden as part of a larger epidemiologic study in neuromuscular disorders in childhood.3 In all these cases, no subtyping of the different forms of CMD was available. A more recent study4 exploring the prevalence of all the genetic muscle disease in Northern England reports a prevalence of CMD of 0.76 × 105 with 26 CMD cases subdivided into 4 major subtypes.
Other recent studies have investigated larger cohorts of CMDs reporting the relative frequencies of CMD subtypes,13,14 but these mainly reflected the activity of diagnostic centers and were not population studies.
More than 85% of our patients are of Italian origin, and when we analyzed their postcodes, we were unable to identify any obvious correlation between individual forms and local regional areas.
Dystroglycanopathies were the most common group of CMD in our database (40.18%), followed by merosin-deficient CMD (24.11%) and Ullrich CMD (20.24%). No data on the prevalence of the individual forms are available for comparison in the population studies3,5 published before the last decade. Our data appear to be different from the large diagnostic UK study reporting that dystroglycanopathies were present in 12% of the whole CMD cohort, although that study only considered patients in whom a final genetic diagnosis was reached between 2001 and 2008.14 It is possible that regional differences may also have a role in the variability observed in different countries.
Among our cohort of dystroglycanopathies, the most frequent phenotypes observed were microcephaly and mental retardation, with or without cerebellar abnormalities, previously labeled as Italian MEB, and the typical MEB phenotype.15 These phenotypes were also found to be among the most frequent in the large UK study.16–18
All the cases identified as dystroglycanopathies were further subdivided according to the gene mutations. POMT1 mutations were the most frequent, followed by FKRP, POMGnT1, and POMT2. In 14.58%, a mutation could not be identified, but most of them have not yet been systematically screened for all the recently discovered genes. The detection rate of mutations in the subset of cases that had already been screened for all genes suggests that the full screening is likely to markedly reduce the number of cases in whom mutations cannot be identified.
The next major group was laminin α2–deficient CMD (24.11% of the whole CMD cohort) with an estimated point prevalence of 0.136 per 100,000. This form has often been reported to be the most frequent form of CMD, with values up to 40% of the CMD cases.11,19 More recent studies13,14,20 report values similar to those found in our cohort.
Twenty-three of the 81 cases in our cohort (28%) had a typical clinical phenotype consistent with merosin-deficient CMD, associated with absent or marked reduction of laminin α2 on muscle biopsy and classic white matter changes on brain MRI, but no mutation could be identified. With the exception of 7 previous cases in whom DHPLC (denaturing high-performance liquid chromatography) was used, Sanger sequencing of the entire coding region was performed in all. It is possible that these patients have deep intronic variants, which affected splicing or exon deletions or duplications, not easily detectable by the standard mutation detection methods used.
Forms with congenital presentation and collagen VI involvement were found in 20.24% of our cohort with similar values to that observed in the large UK series14 (21%). Mutations were found in all but one of those in whom the analysis has been completed. The high rate of detection may be explained by the fact that all these cases had the typical phenotype characterized by distal laxity, skin changes, rigid spine, and early respiratory impairment. The combination of these clinical findings with reduction of collagen 6 on skin cultures and muscle biopsy and, when performed, of the typical muscle MRI changes, appear to increase the possibility of finding mutations in the COL6 genes. Because of our strict inclusion criteria, we may have missed some of the milder Ullrich CMD or of the recently described intermediate phenotype21 that had normal early milestones, including age of ambulation and onset of clinical signs after the age of 2 years. All the patients with Bethlem myopathy had onset of clinical signs after ambulation was achieved and were therefore not included.
Mutations in SEPN1 were found in 19 of the 21 patients in whom the clinical phenotype was consistent with that reported in RSMD22,23 even though it was not always easy to establish retrospectively whether these should be classified as minicore myopathies or RSMD1 because of the marked clinical and pathologic overlap between these forms.24
Because of its retrospective nature, our study has some limitations as the patients included had biopsies performed in different laboratories and from the reports it was not always easy to compare the severity of changes on muscle biopsies. Similarly, data on the antibodies used for assessing laminin α2 were not always consistent because the samples were processed at different times. This should be systematically reassessed, revaluating the biopsies using similar classification criteria.
Mutations were identified in 220 of the 336 (65%), but in a proportion of cases, analysis of newly identified genes is still in progress. It is of interest that, even though genetic confirmation was not achieved in all, approximately 90% of our cases could be ascribed to one of the major clinical categories. This is probably related to the fact that the diagnosis was based on an integrated approach including clinical, pathologic, genetic, and often MRI features rather than on the analysis of samples sent by other centers with reported information. This approach has also allowed identification of a number of patients, formerly labeled as CMD in the past, with mutations in other genes responsible for other disorders such as congenital myopathies or congenital myasthenia, that have a clinical and pathologic overlapping with CMD.
Another strength is that the genetic testing for the major categories such as dystroglycanopathies or collagenopathies is centralized and this guarantees that all the cases in whom the diagnosis of these categories was suspected have been included. Work is in progress to improve the detection rate of merosin-deficient CMD, using RNA studies or other genomic techniques such as MLPA (multiplex ligation-dependent probe amplification) or DNA arrays. Should this more comprehensive approach not detect any mutation, these patients would allow exploring possible genetic heterogeneity in this subgroup.
Further work has also been planned to perform further sequencing including next-generation sequencing to detect the more rare forms of CMD such as nesprin that have not been systematically investigated in our unresolved cases.
Our results clearly show how the detection rate of mutations has increased following the increased number of new genes identified. We foresee that even screening for all the known genes, a proportion will still remain without a diagnosis and that further CMD subtypes and genes are yet to be discovered.
GLOSSARY
- αDG
α-dystroglycan
- CK
creatine kinase
- CMD
congenital muscular dystrophy
- MEB
muscle-eye-brain disease
- RSMD1
rigid spine muscular dystrophy type 1
AUTHOR CONTRIBUTIONS
Alessandra Graziano: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Flaviana Bianco: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, study supervision. Adele D'Amico: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Isabella Moroni: study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Sonia Messina: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Claudio Bruno: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Elena Pegoraro: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Marina Mora: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Guja Astrea: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Francesca Magri: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Giacomo Pietro Comi: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Angela Lucia Berardinelli: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Maurizio Moggio: study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Lucia Morandi: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Antonella Pini: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval. Roberta Petillo: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Giorgio Tasca: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Mauro Monforte: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Carlo Minetti: drafting/revising the manuscript, study concept or design, accepts responsibility for conduct of research and will give final approval, acquisition of data. Tiziana Mongini: study concept or design, accepts responsibility for conduct of research and will give final approval, acquisition of data. Enzo Ricci: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Ksenija Gorni: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Roberta Battini: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Marcello Villanova: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data. Luisa Politano: study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, study supervision, obtaining funding. Francesca Gualandi: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data. Alessandra Ferlini: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients. Francesco Muntoni: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, study supervision, obtaining funding. Filippo Santorelli: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients, acquisition of data. Enrico Silvio Bertini: study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, obtaining funding. Marika Pane: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients, acquisition of data. Eugenio Mercuri: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, acquisition of data, obtaining funding.
STUDY FUNDING
The study is supported by a Telethon UILDM grant (GUP11001). The authors are grateful to TREAT NMD and Care CMD for their support in developing CMD registries in Italy. F. Muntoni is supported by the Great Ormond Street Children's charity and the Great Ormond Street Hospital Biomedical Research Center.
DISCLOSURE
A. Graziano, F. Bianco, A. D'Amico, I. Moroni, S. Messina, and C. Bruno report no disclosures relevant to the manuscript. E. Pegoraro has served on a scientific advisory board for BioMarin Pharmaceutical Inc.; has received funding for travel from Genzyme Corporation; has received speaker honoraria from MedaPharma; and receives research support from Wellstone and Telethon Italy. M. Mora, G. Astrea, and F. Magri report no disclosures relevant to the manuscript. G. Comi is site principal investigator for the PTC extension study of Ataluren in DMD, for the GSK study on exon skipping; receives research support from Telethon Italy and SMA Europe. A. Berardinelli, M. Moggio, L. Morandi, A. Pini, R. Petillo, G. Tasca, M. Monforte, and C. Minetti report no disclosures relevant to the manuscript. T. Mongini has served on a scientific advisory board for Telethon Italy; has received funding for travel from Genzyme Corporation; and has received research support from AIFA (Italian Government Drug Agency) and Telethon Italy. E. Ricci, K. Gorni, R. Battina, M. Villanova, L. Politano, F. Gualandi, A. Ferlini, F. Muntoni, and F. Santorelli report no disclosures relevant to the manuscript. E. Bertini is site principal investigator for the PTC extension study of Ataluren in DMD and for the GSK study on exon skipping. He also receives funds from the Italian Telethon, the Italian Ministry of Health, and SMA Europe for observational studies on outcome measures. M. Pane reports no disclosures relevant to the manuscript. E. Mercuri is site principal investigator for the PTC extension study of Ataluren in DMD and for the GSK study on exon skipping. He also receives funds from the Italian Telethon and SMA Europe. He has acted on the advisory board for Acceleron Pharma, Prosensa, Ely Lilly, and PTC Therapeutics, Inc. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Dubowitz V. 22nd ENMC sponsored workshop on congenital muscular dystrophy held in Baarn, The Netherlands, 14–16 May 1993. Neuromuscul Disord 1994;4:75–81. [DOI] [PubMed] [Google Scholar]
- 2.Mercuri E, Muntoni F. The ever-expanding spectrum of congenital muscular dystrophies. Ann Neurol 2012;72:9–17. [DOI] [PubMed] [Google Scholar]
- 3.Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscul Disord 2000;10:1–9. [DOI] [PubMed] [Google Scholar]
- 4.Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009;132:3175–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mostacciuolo ML, Miorin M, Martinello F, Angelini C, Perini P, Trevisan CP. Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet 1996;97:277–279. [DOI] [PubMed] [Google Scholar]
- 6.Bonnemann CG, Wang CH, Quijano-Roy S, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord 2014;24:289–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nadeau A, Muntoni F. Skin changes in Ullrich congenital muscular dystrophy. Neuromuscul Disord 2008;18:982. [DOI] [PubMed] [Google Scholar]
- 8.Geranmayeh F, Clement E, Feng LH, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010;20:241–250. [DOI] [PubMed] [Google Scholar]
- 9.Messina S, Bruno C, Moroni I, et al. Congenital muscular dystrophies with cognitive impairment: a population study. Neurology 2011;75:898–903. [DOI] [PubMed] [Google Scholar]
- 10.Mercuri E, Messina S, Bruno C, et al. Congenital muscular dystrophies with defective glycosylation of dystroglycan: a population study. Neurology 2009;72:1802–1809. [DOI] [PubMed] [Google Scholar]
- 11.Tome FM, Evangelista T, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III 1994;317:351–357. [PubMed] [Google Scholar]
- 12.Mercuri E, Clements E, Offiah A, et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol 2010;67:201–208. [DOI] [PubMed] [Google Scholar]
- 13.Peat RA, Smith JM, Compton AG, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology 2008;71:312–321. [DOI] [PubMed] [Google Scholar]
- 14.Clement EM, Feng L, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord 2012;22:522–527. [DOI] [PubMed] [Google Scholar]
- 15.Villanova M, Mercuri E, Bertini E, et al. Congenital muscular dystrophy associated with calf hypertrophy, microcephaly and severe mental retardation in three Italian families: evidence for a novel CMD syndrome. Neuromuscul Disord 2000;10:541–547. [DOI] [PubMed] [Google Scholar]
- 16.Clement E, Mercuri E, Godfrey C, et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol 2008;64:573–582. [DOI] [PubMed] [Google Scholar]
- 17.Godfrey C, Foley AR, Clement E, Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev 2011;21:278–285. [DOI] [PubMed] [Google Scholar]
- 18.Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130:2725–2735. [DOI] [PubMed] [Google Scholar]
- 19.Philpot J, Sewry C, Pennock J, Dubowitz V. Clinical phenotype in congenital muscular dystrophy: correlation with expression of merosin in skeletal muscle. Neuromuscul Disord 1995;5:301–305. [DOI] [PubMed] [Google Scholar]
- 20.Bönnemann CG. Congenital muscular dystrophy. In: Squire LR, editor. Encyclopedia of Neuroscience. Oxford, UK: Academic Press; 2009:67–74. [Google Scholar]
- 21.Foley AR, Quijano-Roy S, Collins J, et al. Natural history of pulmonary function in collagen VI-related myopathies. Brain 2013;136:3625–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flanigan KM, Kerr L, Bromberg MB, et al. Congenital muscular dystrophy with rigid spine syndrome: a clinical, pathological, radiological, and genetic study. Ann Neurol 2000;47:152–161. [PubMed] [Google Scholar]
- 23.Scoto M, Cirak S, Mein R, et al. SEPN1-related myopathies: clinical course in a large cohort of patients. Neurology 2011;76:2073–2078. [DOI] [PubMed] [Google Scholar]
- 24.Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 2002;71:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]