

Abstract
Background
The bacterium Pseudomonas fluorescens switches to an alginate-producing phenotype when the pleiotropic anti-sigma factor MucA is inactivated. The inactivation is accompanied by an increased biomass yield on carbon sources when grown under nitrogen-limited chemostat conditions. A previous metabolome study showed significant changes in the intracellular metabolite concentrations, especially of the nucleotides, in mucA deletion mutants compared to the wild-type. In this study, the P. fluorescens SBW25 wild-type and an alginate non-producing mucA- ΔalgC double-knockout mutant are investigated through model-based 13C-metabolic flux analysis (13C-MFA) to explore the physiological consequences of MucA inactivation at the metabolic flux level. Intracellular metabolite extracts from three carbon labelling experiments using fructose as the sole carbon source are analysed for 13C-label incorporation in primary metabolites by gas and liquid chromatography tandem mass spectrometry.
Results
From mass isotopomer distribution datasets, absolute intracellular metabolic reaction rates for the wild type and the mutant are determined, revealing extensive reorganisation of carbon flux through central metabolic pathways in response to MucA inactivation. The carbon flux through the Entner-Doudoroff pathway was reduced in the mucA- ΔalgC mutant, while flux through the pentose phosphate pathway was increased. Our findings also indicated flexibility of the anaplerotic reactions through down-regulation of the pyruvate shunt in the mucA- ΔalgC mutant and up-regulation of the glyoxylate shunt.
Conclusions
Absolute metabolic fluxes and metabolite levels give detailed, integrated insight into the physiology of this industrially, medically and agriculturally important bacterial species and suggest that the most efficient way of using a mucA- mutant as a cell factory for alginate production would be to use non-growing conditions and nitrogen deprivation.
Electronic supplementary material
The online version of this article (doi:10.1186/s12918-015-0148-0) contains supplementary material, which is available to authorized users.
Keywords: Pseudomonas fluorescens, Anti-sigma factor MucA, Fluxome and fluxomics, Carbon labelling experiments, GC-MS/MS, LC-MS/MS, 13C-metabolic flux analysis
Background
Pseudomonas is a bacterial genus containing species of industrial, agricultural and medical relevance due to their metabolic diversity and ability to colonise a wide variety of ecological niches, including not only soil and water but also insects, plants, and animals [1,2]. The biosynthetic diversity of this genus includes production of the polysaccharide alginate, which has many commercial applications, especially in food and medicine [3], but alginate is a complicating factor to the human host during Pseudomonas infections [4].
Alginate is not produced by Pseudomonas fluorescens SBW25 wild type, but its production can be induced by inactivation of the anti-sigma factor MucA [5]. MucA inactivation is a frequent mechanism behind the conversion of P. aeruginosa to a mucoid phenotype infecting the lungs of cystic fibrosis patients [4]. In its active form, MucA prevents alginate biosynthesis by binding and sequestering AlgU, the sigma factor necessary for transcription of the alg operon. The alg operon codes for all of the structural genes needed for alginate synthesis except AlgC [6], the role of which is to convert mannose 6-phosphate (M6P) to mannose 1-phosphate (M1P). M6P is produced from fructose-6-phosphate (F6P), implying that alginate synthesis draws from the hexose-phosphate pool of central metabolism. In both P. aeruginosa and P. fluorescens, MucA inactivation has been shown to affect numerous genes other than those involved in alginate biosynthesis [5,7,8].
In our previous metabolome study [9], P. fluorescens SBW25 wild type and mucA-mutants were cultivated on fructose and glycerol as sole carbon sources. Intracellular concentrations of a wide range of metabolic intermediates, including phosphometabolites, amino acids, organic acids, and nucleotides, were measured. The study revealed significant carbon-source-dependent differences in primary metabolite and nucleotide levels for both the wild type and the mucA mutants. A dramatic decrease in energy charge (EC) was found in MucA mutant strains, which was accompanied by an increased growth yield (YX/S). Interestingly, no counter response action of the energy-deprived mutants’ levels of central intermediates and metabolic genes was observed. This observation led to the hypothesis that mutants do not perceive the low EC as a critical energy limitation. To understand this puzzling robustness, the reorganisation of carbon flows within the biological network of P. fluorescens strains upon MucA inactivation needs to be elucidated. This is the aim of the present study.
In contrast to other “-omics” technologies, metabolic fluxes cannot be directly measured but must be inferred from carbon labelling experiments (CLEs) in combination with mathematical modelling [10]. In CLEs, 13C-enriched carbon substrates are fed to the cells and, driven by the cells’ metabolic and enzymatic activities, isotopically labelled carbon atoms are incorporated into the metabolite pools [11]. The incorporation of labelled material causes all central metabolic intermediates to exist as a set of different isotopomers, which are a collection of isomers that have different labelling patterns of 12C and 13C carbon atoms. Labelling data along with physiological data on observed fluxes (uptake and production fluxes) then enable intracellular fluxes to be quantified through 13C-metabolic flux analysis (13C-MFA) or, analogous to other “omics”-type analyses, fluxomics [12]. During the last few decades, 13C-MFA has become a mature tool to quantify the actual in vivo physiological state of microorganisms, as proven by numerous application studies from the fields of metabolic engineering and systems biology [13-15].
Published fluxome studies for Pseudomonas species are scarce. A flux ratio analysis comparing seven bacterial species (P. fluorescens wild type, P. putida, Agrobacterium tumefaciens, Sinorhizobium meliloti, Rhodobacter sphaeroides, Zymomonas mobilis, and Paracoccus versutus) with the two model organisms E. coli and Bacillus subtilis has been performed with glucose as the carbon source [16]. CLEs were carried out in shake flasks, and samples were taken during exponential growth to derive the relative net flux contribution of converging pathways in selected branch points of the metabolic network from the labelling patterns of protein-bound amino acids. The authors showed that compared to the model organisms, the seven investigated strains, including P. fluorescens, behaved significantly different, as they all mainly relied on the Entner-Doudoroff pathway (EDP) and used the pentose phosphate pathway (PPP) only for biosynthetic functions. In addition, the aerobic bacteria had no by-product formation and to a larger extent used the pyruvate shunt (malate (MAL) to pyruvate (PYR) to oxaloacetate (OAA)) for conversion of MAL to OAA instead of direct conversion by malate dehydrogenase [16]. In another study, intracellular fluxes for P. aeruginosa elucidated alginate production using glucose as the carbon source [17-19]. Significant involvement of the EDP in alginate biosynthesis was shown; glucose was shuttled via triose phosphate glyceraldehyde 3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP) to alginate.
The objective of the current study is to quantify the effect of MucA inactivation on the primary metabolism of P. fluorescens through determination of the levels of absolute fluxes. For comparability with experiments promoting alginate production [5] on the one hand and the conditions of our previous metabolomics study [9] on the other hand, nitrogen limitation was imposed and fructose was used as the carbon source. To investigate the effect of a mucA mutant, we focus on the flux reorganisation of the primary metabolism. However, alginate production starts upon MucA inactivation [4], which affects the metabolic fluxes due to the removal of the alginate precursor metabolite M6P, concurrently with the effects of MucA inactivation itself. To separate these two effects, we use a mucA- ΔalgC double-knockout strain in which the direct effect of MucA inactivation can be observed without indirect “distortion” by alginate production. It was previously shown that a single deletion of algC does not alter gene expression [5] or metabolite profiles [9] compared to the wild-type strain. The determined absolute flux distributions are compared with data from the previous metabolome study conducted with the same strains. The comparison of both flux maps and metabolite levels generates complementary views of the primary metabolism of P. fluorescens. In particular, flux maps reveal new insights into the mechanisms by which primary metabolism is controlled by MucA, and how MucA may influence alginate production through the re-routing of carbon flow. In summary, this study provides the first integrative perspective on the metabolism of P. fluorescens and lays the groundwork for the future metabolic engineering of this organism.
Results
Chemostat cultivations and carbon labelling experiments (CLE)
To quantify the changes in carbon flows in P. fluorescens upon MucA inactivation, the wild-type strain and an alginate non-producing mucA- ΔalgC double-knockout mutant were grown with fructose as the sole carbon source under nitrogen limitation. For 13C-MFA, it is essential to ensure that cell physiology and metabolism are held in a pseudo-steady state throughout the experiment. Therefore, CLEs are conducted in chemostats with growth rates of 0.04 h-1 under defined environmental conditions. CLEs were performed in two rounds: results derived from one wild-type cultivation (a “pre-CLE”) were used for an experimental design study to determine a statistically maximally informative fructose isotopomer composition, as detailed in the Materials and Methods section. In turn, the calculated mixture was administered in the main CLEs to the two strains.
The specific rates for fructose uptake and biomass production in nitrogen-limited chemostats are shown in Table 1 (carbon source in excess). The wild-type and the mucA- ΔalgC strains show similar specific biomass formation rates. Interestingly, the specific fructose consumption rate is almost 40% lower in the mucA- ΔalgC strain, causing a higher biomass yield on carbon source (YX/S) for the mutant (19% for the mucA-ΔalgC strain compared to 12% for the wild-type).
Table 1.
Measured and calculated cultivation data for P. fluorescens CLEs
| Strain | OD 660 | Specific fructose uptake [mmolC/gDW h] | Specific alginate production [mmolC/ gDW h] | Specific biomass production [mmolC/gDW h] 3) | Specific CO 2 excretion [mmol/gDW h] 4) | Y xs [%] |
|---|---|---|---|---|---|---|
| wild-type1) | 8.0±0.1 | 11.0±0.6 | 0.0 | 1.4±0.13 | 9.6±0.61 | 12 |
| wild-type2) | 7.0±0.1 | 11.8±0.6 | 0.0 | 1.4±0.13 | 10.4±0.61 | 12 |
| Estimated5) | 11.76±0.54 | 1.34±0.13 | 10.41±0.55 | |||
| mucA-ΔalgC 2) | 8.9±0.1 | 7.34±0.4 | 0.0 | 1.4±0.13 | 6.0±0.42 | 19 |
| Estimated5) | 7.39±0.32 | 1.30±0.13 | 6.08±0.34 |
1)Experimental design cultivation on wild type. 2)Main experiment on both strains. 3)Calculated from culture growth rate and biomass composition based on biomass equation. 4)Difference between fructose uptake and biomass production. No additional carbon-containing products (i.e., organic acids) were detected in the culture supernatant. 5) Estimated rates from simulation. Standard deviations are estimated from the process data and used for the 13C modelling.
Mass spectrometry measurements
Metabolite extracts from the CLEs were analysed by LC-MS/MS and GC-MS/MS to generate mass isotopomer datasets. Both chromatographic techniques were used to increase the information content and the coverage of labelling patterns for flux estimation. GC-MS was mainly used to analyse soluble free amino acids, while LC-MS was used for central carbon metabolites (organic acids and sugar phosphates). Based on the MS/MS datasets, intracellular fluxes were estimated for both strains.
Quantification of metabolic fluxes for the wild-type and mucA- ΔalgC strains
A model of P. fluorescens central carbon metabolism was formulated that incorporated reactions for fructose uptake (Carbon Uptake), glycolysis/Embden – Meyerhof – Parnas pathway (EMP), the pentose phosphate pathway (PPP), the Entner-Doudoroff pathway (EDP), the tricarboxylic acid cycle (TCA) and glyoxylate shunt, the anaplerotic reactions (ANA), as well as amino acid biosynthesis pathways (BM/BS), covering the measured metabolite pools (cf. Additional file 1 for specification of the metabolic network model including carbon atom transitions). Details of the 13C-MFA modelling workflow are given in the Materials and Methods section.
Calculated absolute intra- and extracellular fluxes for the wild-type and mucA- ΔalgC strains based on the two main CLEs, are visualised in Figures 1 and 2, respectively, along with the values for net (n) fluxes (difference of forward flux and backward flux rates – see [20] for an in-depth explanation). The flux distributions with bidirectional resolution (i.e., net and exchange fluxes), including the drain from metabolic intermediates to biomass and confidence intervals of the flux estimates, are found in Additional file 2, along with a presentation of all net fluxes for the two strains relative to each other. Net fluxes proceed in the same direction for the two strains (the opposite directions of aldolase (EC 4.1.2.13) and triose phosphate isomerase (EC 5.3.1.1) in glycolysis are not statistically significant). The comparison between the real and the in silico rates (estimated) in Table 1 demonstrate that measurements and model predictions match fairly well.
Figure 1.
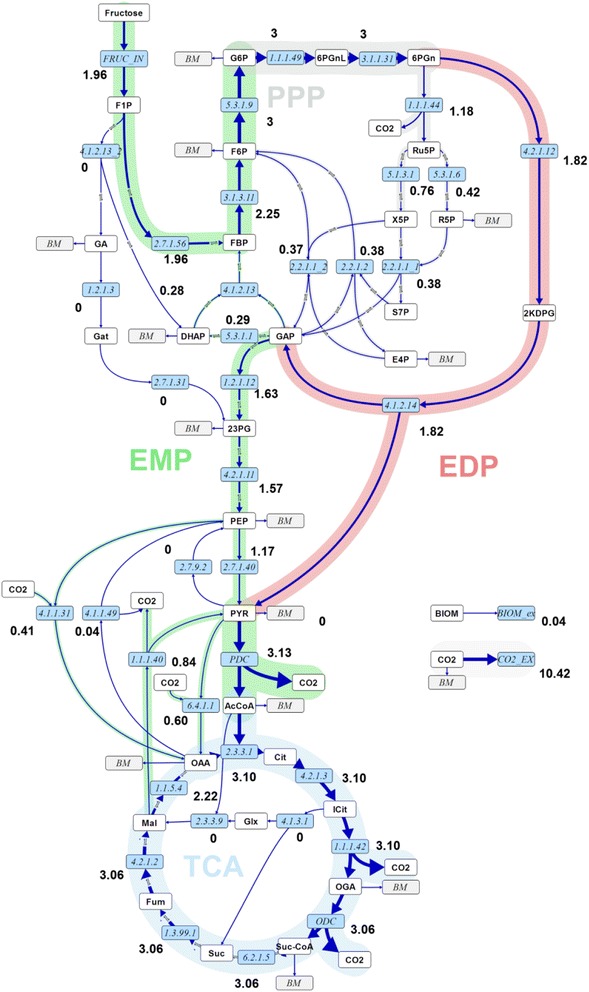
Flux map for P. fluorescens SBW25 wild type. Absolute net fluxes of central carbon metabolism in mmol/(gDW h) and BIOM_ex given as gDW/(gDW h) for P. fluorescens SBW25 wild type. Net fluxes are visualised by the thickness of the reaction arrows (logarithmic scale). Numerical values for the complete flux distribution, consisting of net and exchange fluxes along with their standard deviations, can be found in Additional file 2. For abbreviations of metabolite names, see Additional file 1.
Figure 2.

Flux map for P. fluorescens SBW25 mucA- Δ algC strain. Absolute net fluxes of central carbon metabolism in mmol/(gDW h) and BIOM_ex given as gDW/(gDW h) for the P. fluorescens mucA- ΔalgC strain. Net fluxes are visualised by the thickness of the reaction arrows (logarithmic scale). Numerical values for the complete flux distribution, consisting of net and exchange fluxes along with their standard deviations, can be found in Additional file 2. For abbreviations of metabolite names, see Additional file 1.
The first branch point in the fructose uptake pathway is fructose 1-phosphate (F1P), which can be converted either to fructose 1,6-bisphosphate (FBP) or to DHAP and glyceraldehyde (GA). GA in turn enters central carbon metabolism either as 3-phosphoglycerate or as 2-phosphoglycerate (symbolised by the pooled entry 23PG) via conversion to glycerate (GAT). The flux maps reveal that fructose uptake via F1P to FBP is the main route for both strains (see EC 2.7.1.56 versus EC 4.1.2.13 in Figures 1 and 2 and the relative comparisons of fructose uptake fluxes in Figure 3A). Furthermore, the main efflux from FBP for both strains is directed to glucose 6-phosphate (G6P) via F6P. The EDP is thus preferred over the glycolytic route for both strains when growing on fructose, in spite of the enzymatic capacity for the direct conversion of FBP to DHAP and GAP (conversion of fructose to these trioses does not rely on the missing phosphofructokinase activity, which is required for conversion of glucose via the glycolytic route).
Figure 3.

Detailed inspection of four important branch points in central carbon metabolism. Relative net fluxes for P. fluorescens SBW25 (blue) and the mucA- ΔalgC strain (red) at (a) Fructose uptake branch point: effluxes as percentage of influx to F1P; (b) EDP versus PPP branch point: effluxes as percentage of influx to 6PGN; (c) TCA versus GLX shunt branch point: effluxes as percentage of influx to ICIT; (d) ANA branch points: net fluxes from MAL to PYR and from PYR to OAA as percentage of influx to MAL (main chart) and absolute values for PEP to OAA fluxes (inset). List of abbreviations given in Additional file 1.
Although the wild-type and the mucA- ΔalgC strains both utilise the same main route for initial fructose catabolism, there are differences between the two strains at important branch points. One of the most striking distinctions occurs at the 6-phosphogluconate (6PGN) node, where the EDP and the PPP split. In the wild-type strain, the major carbon flux from the 6PGN node is directed to the EDP (61 ± 8%, EC 4.2.1.12), whereas the minor proportion goes to the PPP (39 ± 7%, EC 1.1.1.44) (Figures 1 and 3B). However, the situation is reversed for the mucA- ΔalgC strain. As shown in Figures 2 and 3B, the major carbon flux goes to the PPP (71 ± 17%) and the minor efflux for this strain is directed to the EDP (29 ± 5%). Notably, even with an almost 40% decrease in the fructose uptake rate, the mucA- ΔalgC strain still has a higher absolute flux through the PPP (1.6 ± 0.3 mmol/(gDW h) versus 1.18 ± 0.20 mmol/(gDW h) for the wild-type strain). It is thus clear that inactivating the anti-sigma factor MucA leads to a reduced flux through the EDP and an increased flux through the PPP.
The second important branch point that is found to be distinct for the two strains is isocitrate (ICIT) in the TCA. The wild-type strain does not utilise the glyoxylate (GLX) shunt to any significant extent (flux value of 0% from ICIT to GLX with an asymmetric confidence interval [0, 4]% corresponding to an absolute confidence interval of [0.00,0.10] mmol/(gDW h)), while the mucA- ΔalgC strain shuttles 22 ± 7% (0.33 ± 0.16 mmol//(gDW h)) of the ICIT influx to GLX (cf. Figures 1, 2 and 3C). The fructose uptake rate and EDP flux are lower for the mucA- ΔalgC strain than for the wild-type, leading to a significantly lower influx to ICIT compared to the wild type (1.51 ± 0.09 mmol/(gDW h) vs. 3.10 ± 0.17 mmol/(gDW h)). Thus, the utilisation of the GLX shunt in response to MucA inactivation in turn leads to an even further reduction in flux through the remainder of the TCA.
Finally, comparison of the flux maps reveals differences between the two strains, although the current dataset does not allow for the precise estimation of ANA flux values (see broad confidence intervals in Additional file 2). Nevertheless, the results indicate that the mucA- ΔalgC strain might cycle phosphoenolpyruvate (PEP) to OAA and back, with the two net fluxes almost cancelling each other out (to replace the drain of TCA intermediates to biomass, a small net flux to TCA remains). In contrast, for the wild-type strain, the flux from PEP to OAA might be significantly higher than the reverse flux (cf. Figures 1, 2 and inset of Figure 3D portraying the magnitude of the net fluxes). The results also show that although the PYR shunt (MAL → PYR → OAA) is utilised to a significant extent by the wild-type strain, shuttling 28 ± 9% (0.84 ± 0.27 mmol/(gDW h)) of the MAL influx to OAA via PYR, this shunt is significantly less active in the mucA- ΔalgC strain (a flux of 13% or 0.20 mmol/(gDW h) within the confidence range of [0, 0.45] mmol/(gDW h) of the MAL influx goes to PYR) (Figures 1, 2 and main chart of Figure 3D). Thus, down-regulation of the pyruvate shunt must be a consequence of MucA inactivation.
NADH and NADPH production is significantly higher in wild type
Figure 4 displays the net production of ATP, NADH and NADPH in the primary metabolic network based on the estimated absolute fluxes (A) and normalised to fructose uptake for relative (B) comparison between the mutant and the wild type (detailed rates are given in Additional file 2). For both strains, the ATP-consuming reactions almost cancel out ATP production by substrate-level phosphorylation. This leads to a similarly low net ATP production for both strains. Thus, both strains use oxidative phosphorylation for the majority of ATP synthesis, as expected.
Figure 4.

ATP, NADH and NADPH production. Reactions involving ATP and producing reducing power by central carbon metabolism of P. fluorescens SBW25 (blue) and the mucA- ΔalgC strain (red); (a) based on absolute fructose rate, and (b) normalised to the respective strain’s uptake rate. See Additional file 2 for the contributing fluxes.
In contrast, the net production of NADH and NADPH differs for the two strains, both being higher for the wild type (Figure 4A). However, the relative proportion of NADH to NADPH production is approximately the same, even though there are large differences in major intracellular carbon flow between the two strains (Figure 4B, with data normalised to the fructose uptake rate). In the wild-type strain, the production of NADH is significantly higher because of higher uptake of fructose, more of which is shuttled through the EDP and to the TCA cycle than in the mucA- ΔalgC strain. Likewise, NADPH production is higher for the wildtype, again predominantly because of the wildtype’s higher TCA flux (isocitrate dehydrogenase is NADPH-dependent, EC. 1.1.1.42). On a relative scale, compared to the influx of the network (Table 1), NADPH production is similar in both strains. Because biomass production compared to substrate uptake is higher for the mucA- ΔalgC strain, the wild-type strain generates a surplus of NADPH that is not needed for anabolic purposes.
Discussion
Pseudomonas fluorescens is an attractive organism in industrial, medical and agricultural biotechnology. The first step towards optimising alginate production with this microorganism is to accurately characterise the influencing factors and underlying metabolic regulation mechanisms that reorganise the carbon flow of alginate-producing mutant strains in comparison to the wildtype. This study investigated how the effect of MucA modulates the carbon flow under conditions favouring alginate production. To this end, information-optimised CLEs were designed and, in turn, conducted in well-controlled chemostats under steady-state conditions. Mass isotopomer data were collected from LC- as well as GC-coupled MS devices, generating comprehensive labelling datasets with high coverage. Labelling data, in combination with measured uptake and biomass formation rates, enable the precise determination of absolute intracellular flux rates by 13C-MFA. Finally, the fluxome of P. fluorescens is related to previously determined transcriptome and metabolome data.
Our fluxome study revealed significant reorganisation of major intracellular metabolic fluxes due to inactivation of the pleiotropic anti-sigma factor MucA. The most significant findings from the mucA- ΔalgC double-knockout mutant are as follows: 1. a reduction in EDP flux, 2. an increase in PPP flux, and 3. an activation of the GLX shunt. The data for the wildtype pre-experiment and the two cultivations of the main experiment (Table 1) coincide well with previously published data for P. fluorescens cultivations under similar conditions [5,9], thus confirming that the results from these down-scaled CLE cultivations (120 mL cultivations) can be correlated with transcriptome and metabolome results generated at standard lab-scale operating conditions (750 mL). The experimental set-up with nitrogen-limited chemostats provides the sole carbon source, fructose, in excess. The cells can therefore adjust their carbon source consumption to their metabolic requirements.
Reduced fructose uptake rate and EP pathway flux in the mucA- ΔalgC mutant with lowest energy charge
Interestingly, the wild-type strain has a 60% higher fructose uptake rate than the mucA- ΔalgC strain (Figures 1, 2 and Table 1). Because the growth rate of both strains is the same, the biomass yield on substrate (YXS) is 60% higher in the mucA- ΔalgC strain. Thus, the wildtype either uses more of the fructose for maintenance processes, has a less energy-efficient metabolism (i.e., lower P/O ratio), and/or disposes of surplus energy through futile cycling, which was frequently described for other organisms such as Escherichia coli, Bacillus subtilis, and Corynebacterium glutamicum [16,21-24].
The reduction in fructose uptake in the mucA- ΔalgC strain cannot be a direct effect of MucA inactivation alone, as an alginate-producing mucA- single-deletion mutant had a two-fold increased fructose uptake rate in the metabolome study (18.3 mmol C/(gDW h) in the mucA- mutant vs. 9.4 mmol C/(gDW h) in the wild type) [9]. MucA inactivation thus seems to exert its effect on the EDP, the PPP and the TCA rather than on the uptake system and the initial reactions of F6P and G6P alone.
The previous metabolome study [9] showed metabolite pool changes by MucA inactivation in the presence and absence of alginate production. The most striking result was a significantly decreased EC ((ATP + 0.5ADP)/(ATP + ADP + AMP)) in both the alginate-non-producing mucA- ΔalgC mutant used in this fluxome study and an alginate-producing mucA- single-knockout mutant [9]. The reduction in EC was caused both by a decrease in ATP concentration and an increase in ADP and AMP concentrations, the latter almost 10 times higher. The higher EC of the wild type correlates with its larger NADH production (Figure 4). However, the mucA- ΔalgC strain does not exploit the full capacity to increase its EC in the way the wild type does by, for example, increasing fructose uptake. The likely reason for this is that mucA- mutants do not perceive their relatively low EC as a stress situation, and therefore, no cellular adaptation response is stimulated.
In theory, P. fluorescens could utilise glycolysis alone for fructose assimilation, converting FBP to PYR and generating two net ATP. By converting FBP to PYR via the EDP, no net ATP is generated, as no ATP is formed via fructose-1,6-bisphosphatase (EC 3.1.3.11). From this perspective, both P. fluorescens strains preferably use the less energy-efficient EDP route. Flamholz and co-workers analysed glycolysis and the EDP in terms of thermodynamic and kinetic constraints and concluded that glycolysis incurs a higher protein cost (i.e., more enzymatic proteins are needed to sustain the same flux) than the EDP [25]. The lower protein cost of the EDP might especially favour this pathway under the studied conditions because the chemostats were nitrogen limited, raising the pressure on protein synthesis. Thus, nitrogen limitation seems advantageous for alginate production on fructose.
Concerning the preferred route for fructose uptake, it is also interesting to note that FBPase in P. fluorescens is negatively regulated by AMP, as it is in other bacteria, e.g., E. coli [26]. Because alginate synthesis starts with the conversion of F6P to M6P, the induction of alginate synthesis in the mucA- mutant must alleviate the possible rate-limitation of the FBPase. The eight-fold increase in the mucA- mutants’ AMP concentration could have led to the direct conversion of FBP to DHAP and GAP by FBP aldolase (EC 4.1.2.13) in the mucA- ΔalgC strain, but that was not observed in the current fluxome study.
Disposal of surplus energy in the wild-type strain
According to our fluxome study, a large proportion of the excess fructose uptake of the wild type is shuttled via EDP to TCA and complete oxidation to CO2, with energy conservation taking place through oxidative phosphorylation. This occurs with no obvious use of this extra energy compared to the mucA- ΔalgC strain. Based on current knowledge of co-factor dependencies [5], the NADPH as well as NADH production are higher in the wild type (Figure 4). Strikingly, the NADPH production is higher than needed for anabolic processes in the wild type because the biomass production rate is the same for both strains. The cyclic operation of anaplerotic reactions in the wild type points to futile cycling dissipating energy by hydrolysing energy equivalents, as is also hypothesised for Corynebacterium species [27-29]. The wild type uses the PYR shunt, which generates one NADPH and consumes one ATP, which is a less energy-conserving solution than the direct oxidation of MAL to OAA with the NADH-producing MAL dehydrogenase reaction. It has been previously demonstrated that in batch cultivations growing on glucose, P. fluorescens primarily relies on the PYR shunt for conversion of MAL to OAA in contrast to other Gram-negative (E. coli, P. putida, A. tumefaciens, R. sphaeroides and Z. mobilis) and Gram-positive (B. subtilis and P. versutus) bacteria [16]. Thus, the pyruvate shunt might be active in situations where NADH production is not required.
Yet there remain open questions: how does the cell regenerate or allocate NADP if needed? Activation of a transhydrogenase (EC 1.6.1.1) may provide an explanation to this question [30]. Due to the lower fructose uptake rate of the mucA- ΔalgC strain, it is unlikely that this mutant is energy limited. It is puzzling that the mutant activates the GLX shunt, which is mostly associated with growth on two-carbon substrates, and generates one net OAA from acetyl-CoA without any energy conservation. Additionally, and similarly to the wild type, the ANA cycling of the mucA- ΔalgC strain through PEP carboxylase (EC 4.1.1.31) and PEP carboxykinase (EC 4.1.1.49) could be regarded as a futile cycle because one net ATP is consumed. However, the extent of futile cycling could not be quantified because, as mentioned, ANA reactions were not resolved in the forward and backward directions with statistical confidence. To this end, further CLEs with specially designed 13C-label mixtures are needed [28,31].
Comparison with other -omics data
The comparative investigation of changes in the transcriptome [5] and the fluxome of the investigated strains reveals that the strong reorganisation of central metabolic carbon flows is not tightly coupled to expression levels of the corresponding metabolic genes, which remained remarkably constant.
Additionally, and not surprisingly, direct comparison of changes in the metabolome and the fluxome do not show any clear correlation: while the net flux through TCA was reduced by 50% in the mucA- ΔalgC strain and the glyoxylate shunt was activated, the pool sizes of the TCA metabolites were not altered, except for the pool size of succinate, which increased by a factor of 2.4 (cf. Figure 5). Arguably, there was a weak positive correlation between the minor increase in both PPP metabolite pools and PPP flux in the mucA- ΔalgC strain compared to the wild type. The concentration of the F1P pool increased eight-fold in the mucA- ΔalgC strain, with a 40% reduced fructose uptake rate compared to wild type, but the same F1P pool increase was also observed in an alginate-producing mucA- mutant with a doubled fructose uptake rate compared to wild type. This change in the F1P pool could have global metabolic effects through modulating the actions of the catabolite repressor/activator protein Cra (also known as FruR). Cra is a global sensor and regulator of primary metabolic fluxes in Gram-negative bacteria, with a dual role as both a transcriptional repressor and an activator [32,33]. For example, both PPP and EDP genes were up-regulated in an E. coli cra- mutant, indicating that Cra represses these pathways [34]. F1P is the preferred effector of Cra in P. putida, and FBP is probably a minor effector as well [35]. No strong effect on the gene expression levels of metabolic genes, either through F1P mediated by Cra, or directly through MucA inactivation, was observed in P. fluorescens [5], but this work indicates a link, at the metabolic level, between F1P pool changes and metabolic flux reorganisation. Hence, metabolome and fluxome data provide a “true” orthogonal view of the metabolism of P. fluorescens, in the sense that understanding how cells modulate metabolic fluxes in response to interventions requires integrative multi-omics studies.
Figure 5.

Changes in flux values and metabolite concentrations. Red fluxes/metabolite pools indicate a more than 150% increase in the mutant mucA- ΔalgC strain compared to the wild type. Blue fluxes edges/metabolite pools indicate a decrease to less than 50% in the mutant mucA- ΔalgC strain compared to the wild type.
Conclusion
In this work, a mucA- ΔalgC double-knockout P. fluorescens mutant was compared to the wild-type strain at the metabolic flux level to illuminate the global metabolic response to MucA inactivation on central carbon metabolism without a biasing carbon re-routing by alginate production. Corroborating transcriptome and metabolome results from former studies, a strong influence of the mucA- ΔalgC double-knockout mutant was shown, involving fine-tuned and significant reorganisation of carbon utilisation. Salient alterations lead to a decrease of the overall fructose uptake and an activation of the PPP and the GLX shunt that is accompanied by a deactivation of the EDP. Although both strains produce a similar amount of energy (NADH and ATP) per mole of substrate, the mutant seems to exhibit more energy-efficient behaviour than the wildtype.
Clearly, to mechanistically elucidate the metabolic control mechanisms of alginate production on fluxes, mucA- mutants need to be further investigated. These experiments bear the potential to elucidate the complex control mechanisms of MucA on alginate production and central carbon metabolism. These control points may become important targets for drug development to prevent the formation of the mucoid phenotype, which leads to pulmonary infection in cystic fibrosis patients, or to increase industrial alginate production. This requires extending the multi-omics characterisation to further mutants, including catabolite repressor/activator cra mutants, and experimental conditions, i.e., growth rates and carbon sources, that are clinically and industrially relevant. From the modelling point of view, the application of non-stationary 13C-MFA in combination with advanced experimental design strategies would be advantageous to advance the integrative analysis of metabolite levels and fluxes and increase the statistical significance of the results, particularly with regard to anaplerosis.
Methods
Strains
The strains used in this study were P. fluorescens SBW25 and P. fluorescens SBW25 mucA-ΔalgC. Construction of the mutant strain was previously presented in detail [5]. Flux distribution results for these strains can be compared to metabolome results [9] for the same strains grown under identical conditions. A confounding factor when using the mucA-ΔalgC strain to look for effects of MucA inactivation other than alginate production is that it produces all biosynthetic enzymes required for alginate production other than AlgC.
Cultivations and sample preparation
Chemostat cultivations were performed as described earlier [5], except that the working volume was scaled down from 750 mL to 120 mL in a custom-made 0.5-L reactor vessel and the carbon source concentration was scaled down from 40 g/L to be less in excess, using the equivalent of 25 g/L and 20 g/L of naturally labelled fructose for the wild type and the mucA- ΔalgC strain, respectively. These carbon source concentrations lead to a fructose concentration of 2.5 g/L or above in the fermenters at steady state. In brief, the chemostat cultivations were nitrogen limited using fructose as a carbon source with a dilution rate of 0.04 h-1. Temperature was maintained at 25°C, pH was maintained at 7.8 by automated sodium hydroxide addition, air flow was 1.5 VVM, and oxygen saturation was maintained above 20% by adjustment of the stirring speed. Cultivations were operated as batches for 24 h before initiating continuous operation with a feed containing naturally labelled fructose. After 1.5-2.0 volume exchanges, the feed stock was changed. Afterwards, the feedstock contained 13C-labelled fructose (two different lots of 1-13C-fructose with chemical purities of 99.1 and 98.0% (w/w) and isotope enrichments of 99% (mole/mole); and one lot of U-13C-fructose with chemical purity of 100% (w/w) and isotopic enrichment of 99.5% (mole/mole), Cambridge Isotope Laboratories, Inc, Andover). Sampling was performed after an additional 3 volume exchanges. Process data of double knock out mutant is given in Additional file 3.
Three cultivations were performed; the only difference between the first and the two last was the fructose isotopomer composition used in the 13C-labelled feed. In the first cultivation, the wild-type strain was grown using a 60.04% 1-13C-, 20.31% U-13C- and 19.65% 12C-fructose isotopomer composition (values based on experimental weight of dry substances adjusted for chemical impurities). In the two main cultivations, the wild type and the mucA-ΔalgC strain were grown with the designed fructose isotopomer mixture of 60.08% 1-13C-, 39.92% U-13C-fructose.
Immediately prior to sampling for metabolite extracts, an aliquot of 5 mL was taken from the culture for OD660 and residual fructose measurement. The CO2 concentration in the off gas was measured with a Rosemount Binos 100 CO2 analyser. These data were used to calculate the carbon balance as described previously [5]. For preparation of metabolite extracts, the cultivations were sampled two (wild-type experimental design cultivation samples for LC-MS/MS analysis) or three (for all other conditions) times for each of the two MS-methods (biological replicates) to be employed, using fast vacuum filtration and quenching of metabolism in 37.5% cold methanol (-20°C). Details of the sampling method are as described in our previous metabolome study [9], except that only one filter with pore size 0.8 μm (Millipore cat#AAWP04700) was used instead of a stack of three filters and the culture volume filtered was 6 mL instead of 2 mL, leading to metabolites from 3 mL of culture being present in the final freeze-dried metabolite extracts.
Sample and evaluation of mass isotopomer datasets
Metabolite extracts were analysed by an LC-MS/MS method and a GC-MS/MS method to generate mass isotopomer distribution datasets for the P. fluorescens SBW25 wild-type and the mucA-ΔalgC strains. Prior to LC-MS/MS analysis, samples were reconstituted in 500 μL 60% (v/v) methanol. Analysis was performed on an Agilent 1200 series LC connected via an electrospray ion source to an Agilent 6410 triple-quadrupole MS using an adaptation of the reverse phase tributylamine ion exchange method introduced by Luo et al. [36]. The same LC-MS/MS method was used in the metabolome study of P. fluorescens [9]. A total of eight central carbon metabolism metabolites were analysed for mass isotopomer distributions by LC-MS/MS and used in the study. For each compound, the number of precursor-to-fragment transitions (multiple reaction monitoring (MRM)-transitions) was expanded to account for all mass isotopomers of the compound, i.e., (n-m + 1)(m + 1) MRM-transitions for each compound, where n is the number of carbons in the precursor ion and m is the number of carbons in the product ion, respectively [37]. A list of all MRM-transitions and accompanying MS-settings for the LC-MS/MS method can be found in Additional file 4.
Samples for GC-MS/MS analysis were derivatised prior to analysis as described previously [9] using an adaptation of the methyl chloroformate derivatisation protocol [38,39]. Analysis was performed on an Agilent 7890A GC – 7000 triple-quadrupole MS interfaced with a chemical ionisation (CI) ion source based on a previously described MS/MS method [40]. The GC method was as described previously [9], with the exception that the injected volume was increased from 1 μL to 3 μL. The MS method used positive CI with methane as the reagent gas and nitrogen as the collision gas. MRM-transitions to account for all mass isotopomers of the 14 detected compounds (central carbon metabolism metabolites and amino acids) were included in the fluxomics models. A list of all MRM-transitions and accompanying GC-MS/MS-settings can be found in Additional file 4.
The resulting mass isotopomer fractions are listed in Additional file 5. Average relative standard deviations (STD) for the mass isotopomers are calculated from the biological replicates for the estimation of intracellular fluxes. The obtained LC-MS/MS STD values were 13% for the wild-type experimental design cultivation, and 10% and 8% for the main experiment cultivations on wild type and mucA-ΔalgC, respectively. For mass isotopomers detected by the GC-MS/MS method, these STD values were 10%, 12% and 15%, respectively.
Computational 13C-MFA: modelling, flux estimation and design of informative CLEs
A biochemical reaction network of central carbon metabolism of P. fluorescens SBW25 was derived from the genome-scale metabolic model iSB1139 reconstructed by Borgos et al. [5] as well as from the KEGG database (http://www.genome.jp/kegg). Construction of our model was guided by 1) representing degradation pathways by effluxes from the system and excluding low-abundance biomass components (see Additional file 1: Tables S.1.2 and S.1.4); 2) pooling metabolites due to limited measurement information, e.g., 1,3-bisphosphoglycerate, 2-phosphoglycerate and 3-phosphoglycerate, which are represented by one lumped pool 23PG while preserving the carbon atom transitions; and 3) aggregating parallel fluxes, such as isoenzymes. Like other Pseudomonas strains, P. fluorescens does not possess a phosphofructokinase enzyme for conversion of F6P to FBP; thus, only the gluconeogenetic conversion of FBP to F6P is included in the model (EC 3.1.3.11).
The model of P. fluorescens central carbon metabolism consists of routes for fructose uptake (Carbon Uptake), EMP, EDP, PPP, TCA cycle including the GLX shunt, the ANA section, as well as the biomass equation, which defines the net fluxes of metabolites into amino acid and fatty acid synthesis. To formulate the latter effluxes, the biomass composition for P. putida devised by Nogales et al. [41] was adopted and integrated as a set of constraints for the fluxes into building block synthesis (Additional file 1). In total, the model contains 66 metabolites and 116 reactions (18 thereof reversible), each supplemented with carbon atom transitions (Additional file 1). The model has 29 degrees of freedom (13 net and 15 exchange fluxes) to be estimated from the measured datasets generated for the P. fluorescens wild type and the mucA-ΔalgC mutant strain.
The generation of isotopomer balances, the simulation of measurements, the experimental mixture design, the flux estimation (minimisation of a weighted least squares objective repeated 100 times with randomly sampled starting values combined with a globalised optimisation strategy to detect multiple equally good but essentially different flux solutions), and the statistical assessment of resulting flux confidence intervals were performed using the software tool 13CFLUX2 [12]. For further details of the computational 13C-MFA protocols, we refer to classical review papers, e.g., [42]. Finally, the resulting flux estimations are represented in the context of the metabolic network model using the visualisation software Omix [43].
An optimal experimental design (OED) study to determine an information-optimised fructose label mixture was performed based on the fluxes estimated from the pre-CLE with P. fluorescens SBW25 wild type. The main CLEs for the wild type and the mucA-ΔalgC strain were performed using the calculated OED mixture consisting of 60.08% 1-13C-, 39.92% U-13C-fructose isotopomers. When inspecting the data thoroughly with the isotopomer network at hand, some peak measurements have to be excluded due to low peak quality. Specifically, we used the following procedure to achieve an acceptable fit: 1) we assumed a minimum STD of 1% to account for the noise of measurements close to the detection limit of the MS instruments, 2) we obtained the best fit by minimising the difference between simulated and measured data, and 3) we excluded those measurements with low abundance that have extraordinary large discrepancies (values for experimental and simulated measurements are found in Additional file 6). Optimisation runs provided a unique optimum for each dataset and flux estimation. For the best-fitting flux maps, the statistical analyses were performed with constraining several main exchange fluxes (as indicated by the absence of uncertainties for fluxes in Additional file 2.
Acknowledgments
This study was financed by an internal grant at NTNU (SKL) and the Norwegian Research Council via the Era-Net SysMO program. The authors thank R. Aune for technical assistance during the 13C-fructose cultivations.
Additional files
Metabolic network model of central carbon conversion routes of Pseudomonas fluorescens SBW25 (Figure S.1.1). Metabolic network model of the central carbon metabolism of P. fluorescens SBW25 used for 13C metabolic flux analysis (Table S.1.2). Biomass equation for P. putida from Nogales, Palsson et al. [41] used in the model of P. fluorescens SBW25 central carbon metabolism (Table S1.3). Constraint from biomass equation for P. putida from Nogales, Palsson et al. [41] used in the model of P. fluorescens SBW25 central carbon metabolism based on Additional file 1: Table S1.3 (Table S.1.4). List of main central carbon metabolism metabolites of the P. fluorescens SBW25 (Table S.1.5).
The complete flux distributions (net and exchange fluxes) and confidence intervals for P. fluorescens SBW25 wild type strain (Table S.2.1) and mucA- ΔalgC strain (Table S.2.2). Relative net fluxes for the mucA- ΔalgC strain to the wild-type strains on a logarithmic scale (Figure S.2.3 (a)) and scaled by fructose uptake (Figure S.2.3 (b)) on a logarithmic scale. Reactions involving ATP and producing reducing power (NADH/NADPH) by central carbon metabolism of P. fluorescens SBW25 wild type (blue) and the mucA-ΔalgC strain (red) are presented in Figure S.2.4.
Figure S.3 shows the time profile of the 13 C-labelled fructose P. fluorescens algC -ΔmucA double knock out cultivation.
Mass spectrometry settings for the LC-MS/MS (Table S.4.1) and GC-MS/MS (Table S.4.2) MRM acquisition methods and the model format of the measurements (last column).
Mass isotopomer distributions detected by LC-MS/MS ( Table S.5.1 ) and GS-MS/MS ( Table S.5.2 ) for three P. fluorescens cultivations with labeled fructose isotopomer mixtures.
Table S.6.1 presents values of simulated measurements for the main CLEs.
Footnotes
Stina K Lien and Sebastian Niedenführ contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SKL performed the 13C-fructose cultivations and the off-line analyses, (including GC-MS/MS and LC-MS/MS analyses for mass isotopomer determination,) participated in the construction of the metabolic network model, the data analysis, the interpretation of data and the drafting of the manuscript. SN performed the 13CFLUX2 simulations and data visualisation using OMIX software and participated in the interpretation of the data and the drafting of the manuscript. HS was involved in the 13C-cultivations and interpretation of the data and commented on the manuscript. KN supervised model building and simulations, interpreted data and was involved in the drafting, editing and critical revision of the manuscript in preparation for submission. PB conceived of the study and was involved in the design, data interpretation and writing of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Stina K Lien, Email: stina.lien@ntnu.no.
Sebastian Niedenführ, Email: s.niedenfuehr@fz-juelich.de.
Håvard Sletta, Email: havard.sletta@sintef.no.
Katharina Nöh, Email: K.Noeh@fz-juelich.de.
Per Bruheim, Email: Per.Bruheim@ntnu.no.
References
- 1.Mahajan-Miklos S, Rahme LG, Ausubel FM. Elucidating the molecular mechanisms of bacterial virulence using non-mammalian hosts. Mol Microbiol. 2000;37(5):981–8. doi: 10.1046/j.1365-2958.2000.02056.x. [DOI] [PubMed] [Google Scholar]
- 2.Poblete-Castro I, Becker J, Dohnt K, dos Santos VM, Wittmann C. Industrial biotechnology of Pseudomonas putida and related species. Appl Microbiol Biotechnol. 2012;93(6):2279–90. doi: 10.1007/s00253-012-3928-0. [DOI] [PubMed] [Google Scholar]
- 3.Skjåk-Bræk G, Draget KI. Alginates: properties and applications. In: Matyjaszewski K, Möller M, editors. Polymer science: a comprehensive reference. Amsterdam: Elsevier BV; 2012. pp. 213–20. [Google Scholar]
- 4.Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JIA, Jensen P, et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology-Sgm. 1999;145:1349–57. [DOI] [PubMed]
- 5.Borgos SE, Bordel S, Sletta H, Ertesvag H, Jakobsen O, Bruheim P, et al. Mapping global effects of the anti-sigma factor MucA in Pseudomonas fluorescens SBW25 through genome-scale metabolic modeling. BMC Syst Biol. 2013;7:19. doi: 10.1186/1752-0509-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morea A, Mathee K, Franklin MJ, Giacomini A, O’Regan M, Ohman DE. Characterization of algG encoding C5-epimerase in the alginate biosynthetic gene cluster of Pseudomonas fluorescens. Gene. 2001;278(1–2):107–14. doi: 10.1016/S0378-1119(01)00685-0. [DOI] [PubMed] [Google Scholar]
- 7.Firoved AM, Deretic V. Microarray analysis of global gene expression in mucoid Pseudomonas aeruginosa. J Bacteriol. 2003;185(3):1071–81. doi: 10.1128/JB.185.3.1071-1081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones AK, Fulcher NB, Balzer GJ, Urbanowski ML, Pritchett CL, Schurr MJ, et al. Activation of the Pseudomonas aeruginosa AlgU Regulon through mucA Mutation Inhibits Cyclic AMP/Vfr Signaling. J Bacteriol. 2010;192(21):5709–17. doi: 10.1128/JB.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lien SK, Sletta H, Ellingsen TE, Valla S, Correa E, Goodacre R, et al. Investigating alginate production and carbon utilization in Pseudomonas fluorescens SBW25 using mass spectrometry-based metabolic profiling. Metabolomics. 2013;9(2):403–17. doi: 10.1007/s11306-012-0454-0. [DOI] [Google Scholar]
- 10.Sauer U. Metabolic networks in motion: 13C-based flux analysis. Mol Syst Biol. 2006;2:62. doi: 10.1038/msb4100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiechert W. 13C metabolic flux analysis. Metab Eng. 2001;3(3):195–206. doi: 10.1006/mben.2001.0187. [DOI] [PubMed] [Google Scholar]
- 12.Weitzel M, Nöh K, Dalman T, Niedenführ S, Stute B, Wiechert W. 13CFLUX2 - high-performance software suite for 13C-metabolic flux analysis. Bioinformatics (Oxford, England) 2013;29(1):143–5. doi: 10.1093/bioinformatics/bts646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woolston BM, Edgar S, Stephanopoulos G. Metabolic engineering: past and future. In: Prausnitz JM, editor. Annual review of chemical and biomolecular engineering, Vol 4. vol. 4. Palo Alto: Annual Reviews; 2013. pp. 259–88. [DOI] [PubMed] [Google Scholar]
- 14.Nöh K, Wiechert W. The benefits of being transient: isotope-based metabolic flux analysis at the short time scale. Appl Microbiol Biotechnol. 2011;91(5):1247–65. doi: 10.1007/s00253-011-3390-4. [DOI] [PubMed] [Google Scholar]
- 15.Zamboni N, Sauer U. Novel biological insights through metabolomics and 13C-flux analysis. Curr Opin Microbiol. 2009;12(5):553–8. doi: 10.1016/j.mib.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Fuhrer T, Fischer E, Sauer U. Experimental identification and quantification of glucose metabolism in seven bacterial species. J Bacteriol. 2005;187(5):1581–90. doi: 10.1128/JB.187.5.1581-1590.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynn AR, Sokatch JR. Incorporation of isotope from specifically labeled glucose into alginates of Pseudomonas aeruginosa and Azotobacter vinelandii. J Bacteriol. 1984;158(3):1161–2. doi: 10.1128/jb.158.3.1161-1162.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narbad A, Hewlins MJE, Gacesa P, Russell NJ. The use of 13C-NMR spectroscopy to monitor alginate biosynthesis in mucoid Pseudomonas aeruginosa. Biochem J. 1990;267(3):579–84. doi: 10.1042/bj2670579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narbad A, Russell NJ, Gacesa P. Radiolabelling patterns in alginate of Pseudomonas aeruginosa synthesized from specifically-labelled 14C-monosaccharide precursors. Microbios. 1988;54(220–21):171–9. [PubMed] [Google Scholar]
- 20.Wiechert W. The thermodynamic meaning of metabolic exchange fluxes. Biophys J. 2007;93(6):2255–64. doi: 10.1529/biophysj.106.099895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emmerling M, Dauner M, Ponti A, Fiaux J, Hochuli M, Szyperski T, et al. Metabolic flux responses to pyruvate kinase knockout in Escherichia coli. J Bacteriol. 2002;184(1):152–64. [DOI] [PMC free article] [PubMed]
- 22.Petersen S, de Graaf AA, Eggeling L, Möllney M, Wiechert W, Sahm H. In vivo quantification of parallel and bidirectional fluxes in the anaplerosis of Corynebacterium glutamicum. J Biol Chem. 2000;275(46):35932–41. [DOI] [PubMed]
- 23.Sauer U, Hatzimanikatis V, Bailey JE, Hochuli M, Szyperski T, Wüthrich K. Metabolic fluxes in riboflavin-producing Bacillus subtilis. Nat Biotechnol. 1997;15(5):448–52. [DOI] [PubMed]
- 24.Yang C, Hua Q, Baba T, Mori H, Shimizu K. Analysis of Escherichia coli anaplerotic metabolism and its regulation mechanisms from the metabolic responses to altered dilution rates and phosphoenolpyruvate carboxykinase knockout. Biotechnol Bioeng. 2003;84(2):129–44. [DOI] [PubMed]
- 25.Flamholz A, Noor E, Bar-Even A, Liebermeister W, Milo R. Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc Natl Acad Sci U S A. 2013;110(24):10039–44. doi: 10.1073/pnas.1215283110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hines JK, Kruesel CE, Fromm HJ, Honzatko RB. Structure of inhibited fructose-1,6-bisphosphatase from Escherichia coli - Distinct allosteric inhibition sites for AMP and glucose 6-phosphate and the characterization of a gluconeogenic switch. J Biol Chem. 2007;282(34):24697–706. [DOI] [PubMed]
- 27.Blombach B, Riester T, Wieschalka S, Ziert C, Youn JW, Wendisch VF, et al. Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol. 2011;77(10):3300–10. [DOI] [PMC free article] [PubMed]
- 28.Petersen S, Mack C, de Graaf AA, Riedel C, Eikmanns BJ, Sahm H. Metabolic consequences of altered phosphoenolpyruvate carboxykinase activity in Corynebacterium glutamicum reveal anaplerotic regulation mechanisms in vivo. Metab Eng. 2001;3(4):344–61. [DOI] [PubMed]
- 29.Sauer U, Eikmanns BJ. The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. Fems Microbiol Rev. 2005;29(4):765–94. doi: 10.1016/j.femsre.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 30.Bruheim P, Butler M, Ellingsen TE. A theoretical analysis of the biosynthesis of actinorhodin in a hyper-producing Streptomyces lividans strain cultivated on various carbon sources. Appl Microbiol Biotechnol. 2002;58(6):735–42. [DOI] [PubMed]
- 31.Möllney M, Wiechert W, Kownatzki D, de Graaf AA. Bidirectional reaction steps in metabolic networks: IV, Optimal design of isotopomer labeling experiments. Biotechnol Bioeng. 1999;66(2):86–103. doi: 10.1002/(SICI)1097-0290(1999)66:2<86::AID-BIT2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 32.Saier MH, Ramseier TM. The catabolite repressor/activator (Cra) protein of enteric bacteria. J Bacteriol. 1996;178(12):3411–7. doi: 10.1128/jb.178.12.3411-3417.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kochanowski K, Volkmer B, Gerosa L, van Rijsewijk BRH, Schmidt A, Heinemann M. Functioning of a metabolic flux sensor in Escherichia coli. Proc Natl Acad Sci U S A. 2013;110(3):1130–5. [DOI] [PMC free article] [PubMed]
- 34.Sarkar D, Siddiquee KAZ, Arauzo-Bravo MJ, Oba T, Shimizu K. Effect of cra gene knockout together with edd and iclR genes knockout on the metabolism in Escherichia coli. Arch Microbiol. 2008;190(5):559–71. [DOI] [PubMed]
- 35.Chavarria M, Santiago C, Platero R, Krell T, Casasnovas JM, de Lorenzo V. Fructose 1-phosphate is the preferred effector of the metabolic regulator Cra of Pseudomonas putida. J Biol Chem. 2011;286(11):9351–9. [DOI] [PMC free article] [PubMed]
- 36.Luo B, Groenke K, Takors R, Wandrey C, Oldiges M. Simultaneous determination of multiple intracellular metabolites in glycolysis, pentose phosphate pathway and tricarboxylic acid cycle by liquid chromatography-mass spectrometry. J Chromatogr A. 2007;1147:153–64. doi: 10.1016/j.chroma.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 37.Choi J, Antoniewicz MR. Tandem mass spectrometry: a novel approach for metabolic flux analysis. Metab Eng. 2011;13(2):225–33. doi: 10.1016/j.ymben.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 38.Smart KF, Aggio RBM, Van Houtte JR, Villas-Boas SG. Analytical platform for metabolome analysis of microbial cells using methyl chloroformate derivatization followed by gas chromatography-mass spectrometry. Nat Protoc. 2010;5(10):1709–29. doi: 10.1038/nprot.2010.108. [DOI] [PubMed] [Google Scholar]
- 39.Villas-Bôas SG, Delicado DG, Åkesson M, Nielsen J. Simultaneous analysis of amino and nonamino organic acids as methyl chloroformate derivatives using gas chromatography-mass spectrometry. Anal Biochem. 2003;322:134–8. doi: 10.1016/j.ab.2003.07.018. [DOI] [PubMed] [Google Scholar]
- 40.Kvitvang HFN, Andreassen T, Adam T, Villas-Boas SG, Bruheim P. Highly sensitive GC/MS/MS method for quantitation of amino and nonamino organic acids. Anal Chem. 2011;83(7):2705–11. doi: 10.1021/ac103245b. [DOI] [PubMed] [Google Scholar]
- 41.Nogales J, Palsson BO, Thiele I. A genome-scale metabolic reconstruction of Pseudomonas putida KT2440: iJN746 as a cell factory. BMC Syst Biol. 2008;2:79. [DOI] [PMC free article] [PubMed]
- 42.Zamboni N, Fendt SM, Ruhl M, Sauer U. 13C-based metabolic flux analysis. Nat Protoc. 2009;4(6):878–92. doi: 10.1038/nprot.2009.58. [DOI] [PubMed] [Google Scholar]
- 43.Droste P, Nöh K, Wiechert W. Omix - a visualization tool for metabolic networks with highest usability and customizability in focus. Chemie Ingenieur Technik. 2013;85(6):849–62. doi: 10.1002/cite.201200234. [DOI] [Google Scholar]