

Abstract
HIV protease inhibitors (PI) are fundamental to combination antiretroviral therapy, which has revolutionized HIV clinical care and produced significant reductions in HIV-associated morbidity and mortality. However, PI administration is frequently associated with severe metabolic impairment, including lipodystrophy, dyslipidemia, and insulin resistance; all of which can contribute to cardiovascular and neurologic co-morbidities. Experimental and epidemiological data support a potentially important role for the adipokine adiponectin in both metabolic and neurologic physiology. This study examined if ADP355, a novel, peptide-based adiponectin receptor agonist, could neutralize the detrimental effects of PI treatment in experimental animal models. Adult male C57BL/6 mice were subjected to a clinically relevant, 4- week regimen of lopinavir/ritonavir, with daily injections of ADP355 administered only during the final 2 weeks of PI exposure. Comprehensive metabolic, neurobehavioral, and biochemical analyses revealed that ADP355 administration partially reversed PI-induced loss of subcutaneous adipose tissue, attenuated PI-induced hyperinsulinemia, hypertriglyceridemia, and hypoadiponectinemia, and prevented PI-induced cognitive impairment and brain injury. Collectively, these data reinforce the link between metabolic co-morbidities and cognitive impairment and suggest that pharmacological reactivation of adiponectin pathways could remediate key aspects of PI-induced metabolic syndrome in clinical settings. Furthermore, therapeutic targeting of adiponectin receptors could show utility in reducing the prevalence and/or severity of HIV-associated neurocognitive disorders.
Keywords: HIV-associated neurocognitive disorder, HIV protease inhibitors, Lipodystrophy, Metabolic syndrome, Peptide-based drugs
Introduction
In the US and other developed nations, survival rates associated with HIV infection have improved dramatically since the introduction of combination antiretroviral therapies (ART), which restrict viral replication, raise CD4 cell counts, prevent opportunistic infections, and improve and extend health-related quality of life (Quinn 2008). While ART has revolutionized care of HIV-positive patients, it is well known that these vital drugs have significant metabolic complications. These complications produce clinical syndromes including dyslipidemia, insulin resistance, and lipodystrophy (Anuurad et al. 2009; Barbaro 2007), which not only affect patient health, but also limit ART compliance (Schambelan et al. 2002). Furthermore, clinical studies reveal quite clearly that metabolic dysfunction is associated with perturbed brain function in the context of HIV/ART (Valcour et al. 2006; Bandaru et al. 2007; Foley et al. 2010; Valcour et al. 2011). Thus, there is considerable effort undertaken to prevent or attenuate metabolic co-morbidities in clinical settings. However, current clinical strategies have not produced satisfying outcomes in HIV patients (Gutierrez and Balasubramanyam 2012). For example, the anti-diabetic drug metformin reduces insulin resistance in HIV patients (Mulligan et al. 2007; van Wijk et al. 2011), but does not improve hyperlipidemia (van Wijk et al. 2011) and may actually accelerate lipodystrophy (Kohli et al. 2007). Thiazolidinediones (TZDs) have been shown to improve insulin sensitivity (Mulligan et al. 2007; van Wijk et al. 2011), but also increase hyperlipidemia (Mulligan et al. 2007; van Wijk et al. 2011) and accelerate bone loss in HIV patients (Wei and Wan 2011). Statins are contraindicated for use in HIV because of drug interactions involving the CYP450 system (Jiménez-Nácher et al. 2011). Human recombinant growth hormone (hrGH) has been used off-label to treat lipodystrophy in HIV patients. Indeed, tesamorelin, a synthetic analogue of human growth hormone-releasing hormone and the only FDA-drug approved to treat HIV lipodystrophy, decreases excess accumulation of abdominal fat, reduces triglyceride levels and improves glucose homeostasis (Spooner and Olin 2012). However, studies have shown that hrGH may decrease extremity fat mass, increase insulin resistance, as well as induce arthralgia and edema (Sivakumar et al. 2011). Thus, new therapeutic targets and approaches to significantly and successfully mitigate different co-morbidities in HIV patients are needed.
In this context, therapies that prevent the loss of adipocytes and/or replicate adipocyte function in the face of adiposopathy could provide novel and innovative strategies to preserve physiologic function in HIV patients. Adipocytes regulate many physiologic processes via secretion of adipokines (Rocha and Libby 2008); and in particular, adiponectin may be key to the maintenance of overall health. In terms of physiology, adiponectin is known modulate a number of key processes, including glucose regulation, vascular tone, and fatty acid metabolism (Siasos et al. 2012). Adiponectin also has both vasculoprotective and neuroprotective properties (Chen et al. 2009; Jung et al. 2006; Ouedraogo et al. 2007), and low levels of plasma adiponectin predict both cognitive impairment (Kamogawa et al. 2010) and decreased hippocampal volume (Masaki et al. 2012). Finally, serum adiponectin levels are decreased in HIV patients (Kinlaw and Marsh 2004; Leszczyszyn-Pynka et al. 2005; Veloso et al. 2012), and hypoadiponectinemia correlates with cognitive dysfunction in ART-treated mice (Gupta et al. 2012). These observations suggest that adiponectin replacement therapy could remediate metabolic dysfunction and maintain neurologic performance in the context of chronic ART therapy. However, adiponectin-based therapeutics are presently not available, likely due to difficulties in converting the full size adiponectin protein into a viable drug. Recently, we generated a short adiponectin peptidomimetic, ADP355. The peptide contains adiponectin active site, mimics adiponectin action in vitro acting through the adiponectin receptor 1/2, demonstrates anti-neoplastic efficacy in vivo, and features great stability in biological fluids as well as a favorable toxicity profile (Otvos et al. 2011). As such, ADP355 could be a potential candidate for adiponectin replacement therapy in metabolic conditions characterized by hypoadiponectinemia. As adiponectin-based therapies in lipodystrophy models have never been tested, this study was designed to determine if ADP355 could mitigate PI-induced metabolic dysfunction and preserve CNS physiology in experimental animals. To this end, C57BL/6 mice were subjected to a clinically relevant, 4-week regimen of lopinavir/ ritonavir, with daily injections of ADP355 administered during the final 2 weeks of PI exposure, followed by extensive testing for metabolic and neurologic function.
Materials and Methods
Synthesis and Purification of ADP355 Peptide
The adiponectin-based peptidomimetic ADP355 (H-DAsn-Ile-Pro-Nva-Leu-Tyr-DSer-Phe-Ala-DSer-NH2) was synthesized on the solid-phase by using a CEM Liberty microwave-assisted peptide synthesizer and utilizing Fmoc-chemistry, purified by RP-HPLC, and verified using MALDI-MS as previously reported (Otvos et al. 2011). After purification, ADP355 was lyophilized twice from 2 % aqueous acetic acid solution prior to cellular efficacy studies.
Animal Treatments
The Institutional Animal Care and Use Committee at the Pennington Center approved all experimental protocols, which were compliant with NIH guidelines on the use of experimental animals. 6–8 month–old male C57Bl/6 mice were purchased from Charles River Laboratories (Wilmimgton, MA), and were housed in standard caging with 12:12 light: dark cycle and ad libitum access to food and water. Lopinavir/ritonavir (Kaletra®, Abbott Laboratories), was diluted in a vehicle of 10 % ethanol/15 % propylene glycol, and mice received daily administration of vehicle or lopinavir/ritonavir at 150/37.5 mg/kg via oral gavage. The dose was devised based on dosing guidelines for daily oral lopinavir/ritonavir in adult HIV patients (800/200 total mg or 10/2.5 mg/kg), and on body surface area (BSA) normalization factors (Reagan-Shaw et al. 2007), which translate 10 mg/kg in humans to approximately 125 mg/kg in mice.
After experimental mice had been treated with lopinavir/ ritonavir or vehicle for 2 weeks, randomly selected mice from each group were treated daily with vehicle (phosphate buffered saline (PBS)) or ADP355 (1 mg/kg in PBS) via intraperitoneal injection for an additional 14 days (28 total days of lopinavir/ritonavir exposure).
Body weight and composition (measured using a Bruker minispec LF90 time domain NMR analyzer, Bruker Optics, Billerica MA) were measured regularly throughout lopinavir/ ritonavir exposure. Blood glucose was measured in tail blood using a glucometer (Ascensia Elite, Bayer, Mishawaka, IN). After cognitive testing (see Section “Fear Conditioning Memory Task”), all mice were humanely euthanatized after a brief (6 h) fast, and blood, adipose tissue, and brain were collected. Data were compiled from 3 separate cohorts of mice, with a total of 9–20 animals in each group.
Fear Conditioning Memory Task
Each mouse was individually evaluated for fear conditioning using an automated, video-based fear conditioning system (Med-Associates, St. Albans, VT). The fear conditioning assay is a type of associative learning task in which subjects are presented with a neutral stimulus (tone) that is paired with an aversive unconditioned stimulus (foot shock), subjects display fear behavior (freezing) in response to the tone. This task does not require the animals to swim, and is not confounded by changes in nociception, food-seeking behavior, hunger, or fatigue. The apparatus consists of a “startle chamber” used on days 1 and 2, which is an 8×15×15-cm acrylic and wire mesh cage located within a custom designed 90×70×70 ventilated sound-attenuating chamber. The floor of each chamber is made of 2.0-mm diameter stainless steel bars spaced 6 mm apart, through which shock is administered, and the unique context is reinforced with an anise-based scent applied to each cage before testing. Animal movement within the apparatus results in displacement of an accelerometer (model U321AO2; PCB Piezotronics, Depew, NY, USA) with the resulting voltage being proportional to the velocity of displacement. For day 3, an entirely separate chamber located in a different room is used to remove all contextual cues. These “trace” chambers contain a flat floor instead of the grid, a”teepee”-shaped insert to modify the chamber dimensions, and an acetic acid odor used to reinforce the novel environment.
Acquisition of fear conditioning on day 1 consisted of 5 min of acclimation to the startle chamber, followed by five consecutive 30 s auditory stimuli (85 dB, 4 KHz) that co-terminated with a mild footshock (0.5 mA×1 s), with 30 s recovery periods between tones. On day 2, the mice were returned to the same chambers, but no stimuli were applied to evaluate freezing responses to context. On day 3, the mice were placed in the novel “trace” chambers, and after habituation of the mice within the chambers for 5 min, a continuous tone (85 dB, 4 KHz) was applied for 5 min. The percent of freezing was recorded as a measure of trace memory of the conditioned response to the tone.
Clinical Chemistry
Whole blood was collected by cardiac puncture of terminally anesthetized mice, and was allowed to clot at 4°C overnight and then centrifuged at 3000xg for 30 min. Serum was collected and either analyzed immediately or aliquoted and stored at −80°C. Levels of total cholesterol, HDL cholesterol, LDL cholesterol, triglycerides, and nonesterified fatty acids (NEFA) in sera were measured colorimetrically using commercially available kits (Wako Chemicals, Richmond, VA). Adiponectin, leptin, and insulin levels in serum were evaluated by ELISA in accordance with the manufacturer's assay protocol (R&D Systems, Minneapolis, MN for adiponectin and leptin; and Crystal Chem Inc., Downers Grove IL for insulin). To measure total pools of adiponectin, serum samples were first denatured (boiled in SDS buffer for 5 min) to break down large complexes.
Evaluation of Brain Injury Markers by Western Blot
Tissue samples were homogenized and processed for Western blot with chemiluminescence as described in previous reports (Pistell et al. 2010b). Blots were processed using the following primary antisera: anti-claudin-5 (1:400, Abcam Inc., Cambridge, MA), anti-ZO-1 (1:100, Abcam Inc.), anti-occludin (1:8000, Abcam Inc.), anti-MMP2 (1:1000, Abcam Inc.), anti-MMP-9 (1:1000, Abcam Inc.), anti-synapsin 1 (1:10000, Thermo Fisher Scientific, Pittsburg, PA), anti-phospho(S553)-synapsin 1 (1:10000, Abcam Inc.), anti-synapse associated protein 97 (1:2500, Abcam Inc.), anti-GFAP (1:5000, Abcam Inc.); anti-Iba-1 (1:500, Wako Chemicals USA Inc., Richmond, VA), and anti-tubulin (1:1000, Wako Chemicals USA Inc.). To ensure accurate quantification across multiple blots, samples from all treatment groups (vehicle, liponavir/ritonavir/PBS, and liponavir/ ritonavir/ADP355) were included in each individual blot. Data were first calculated as a ratio of expression over tubulin expression, which was included as an internal loading control, and then expression in liponavir/ritonavir/PBS and liponavir/ ritonavir/ADP355 was calculated and presented as percent expression in control (vehicle) mice.
Statistical Analyses
All data were analyzed using Prism software (GraphPad Software, Inc., La Jolla, CA), and are displayed as mean ± standard error of measurement. Body weight and composition was analyzed with 2-way repeated measures ANOVA to determine main effects of drug treatment and duration, followed by planned Bonferroni post-hoc comparisons to determine differences between in groups (vehicle, vehicle/ADP355, liponavir/ritonavir/PBS, and liponavir/ritonavir/ADP355). All other data were analyzed by 1-way ANOVA followed by Tukey's Multiple Comparison post-hoc tests to determine specific differences between groups. Statistical significance for all analyses was accepted at p<0.05, and *, **, and *** represent p<0.05, p<0.01, and p<0.001, respectively.
Results
Effects ADP355 on Body Weight and Composition in Lopinavir/Ritonavir-Treated Mice
Previous studies from our laboratory and others have clearly shown that exposure of mice to lopinavir/ritonavir (Kaletra®, Abbott Laboratories, Abbott Park, IL), a protease inhibitor cocktail commonly used in clinical settings to manage HIV produces a severe metabolic derangement also associated with cognitive impairment (Pistell et al. 2010a; Gupta et al. 2012). To determine therapeutic potential ADP355 against PI-induced metabolic and neurologic dysfunction, ADP355 was applied daily to selected 6–8 month old, male C57BL/6 mice for the final 2 weeks of a 4-week regimen of lopinavir/ ritonavir or vehicle as described in Methods. Body weight and body composition measurements taken throughout treatment demonstrated that neither lopinavir/ritonavir nor ADP355 caused significant alterations in body weight (Fig. 1a). However, lopinavir/ritonavir-treated mice did lose significant amounts of total body fat relative to vehicle-treated mice after 2, 3, and 4 weeks of exposure (Fig. 1b). PI-dependent loss of adipose tissue was partially reversed by administration of ADP355 (Fig. 1b). There were no effects, however, of ADP355 on total body fat in vehicle-treated mice (Fig. 1b).
Fig. 1.

ADP355 preserves total body fat in PI-treated mice. Male C57BL/ 6 mice were given daily oral gavage administration of 10 % ethanol/15 % propylene glycol (vehicle; closed circles) or 150 mg lopinavir/37.5 mg ritonavir/kg (PI; closed circles) for 28 days. Additionally, after 14 days of PI exposure, randomly selected vehicle (vehicle/ADP; closed squares) and PI-treated mice (PI/ADP; open squares) mice were also treated daily with ADP355 (1 mg/kg) or PBS via intraperitoneal injection for the final 14 days of PI exposure. All mice were evaluated regularly for (a) body weight and (b) total body fat mass expressed as percent measured at day 0. All data are mean and S.E.M. of 9–21 mice per group. Data were analyzed by 2-way ANOVA, and *** indicates significant (p<0.001) differences noted in PI and PI/ADP and treated mice as compared to vehicle-treated mice at the same timepoint. ## indicates significant (p<0.01) differences in total body fat between PI and PI/ADP355 mice
To further examine the beneficial effects of ADP355 on lopinavir/ritonavir-induced lipoatrophy, individual subcutaneous (inguinal) and visceral (epididymal and retroperitoneal) adipose depots were collected and were weighed. Lopinavir/ ritonavir treatment resulted in significant reductions in the weight of the subcutaneous inguinal adipose depot (Fig. 2a). However, subcutaneous fat depots were significantly larger in lopinavir/ritonavir-treated mice given ADP355 compared to mice given PBS (Fig. 2a) although inguinal fat pads in ADP355-treated mice remained smaller than those isolated from vehicle-treated mice. Conversely, while lopinavir/ ritonavir significantly reduced the weight of the epididymal (Fig. 2b) and retroperitoneal (Supplemental Fig. 1) adipose depots, ADP355 did not prevent lopinavir/ritonavir-induced loss of visceral fat (Fig. 2b and Supplemental Fig. 1), suggesting a selective beneficial effect of ADP355 on subcutaneous fat. Finally, ADP355 did not affect the weight of any adipose depots in vehicle treated mice (Fig. 2).
Fig. 2.
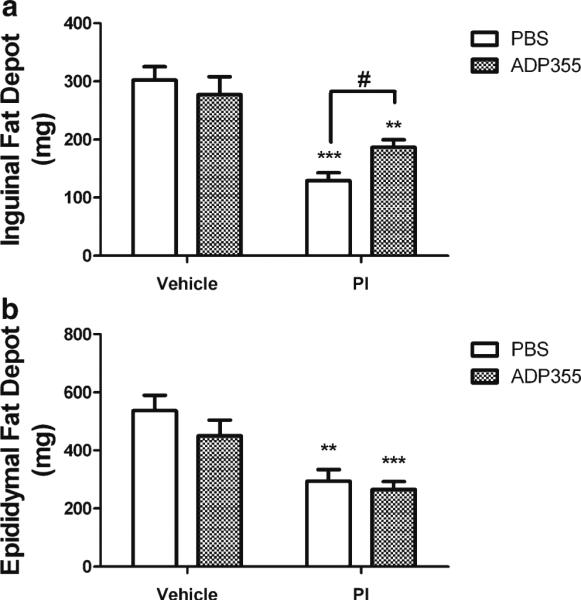
ADP355 preserves subcutaneous, but not visceral fat depots in PI-treated mice. Male C57BL/6 mice were given daily administration of 10 % ethanol/15 % propylene glycol (vehicle; left bars) or 150 mg lopinavir/37.5 mg ritonavir/kg (PI; right bars) for 28 days. Additionally, randomly selected vehicle (open bars) and PI-treated mice (hatched bars) mice were also treated daily with ADP355 (1 mg/kg) or PBS via intraperitoneal injection for the final 14 days of PI exposure, after which all mice were euthanatized and subcutaneous inguinal and visceral epididymal fat pads were collected and weighed. Data are means ± S.E.M. of fat pad mass in milligrams, and were generated from 9–21 mice per group. a Inguinal fat depot weight in vehicle and lopinavir/ritonavir-treated mice following administration of PBS or ADP355. *** and ** indicates significant (p<0.001 and p<0.01) decreases in weight of the inguinal fat depot in lopinavir/ritonavir/PBS and lopinavir/ritonavir/ADP355 mice as compared to vehicle-treated mice, respectively. # indicates significant (p<0.05) increase in weight of the epididymal fat depot in lopinavir/ ritonavir/ADP355 mice compared to lopinavir/ritonavir/PBS mice. b Epididymal fat depots in vehicle and lopinavir/ritonavir-treated mice following administration of PBS or ADP355. *** and ** indicates significant (p<0.001 and p<0.01, respectively) decreases in weight of the inguinal fat depot in lopinavir/ritonavir/ADP355 and lopinavir/ritonavir/PBS mice as compared to vehicle-treated mice, respectively
Effects ADP355 on Serum Markers of Metabolic Syndrome
To determine the effects of ADP355 on PI-induced metabolic syndrome, metabolic data relating specifically to insulin resistance, hyperlipidemia, and adipokine secretion were collected at the end of the 28-day PI exposure period. To document insulin sensitivity, studies focused on measurement of fasting levels of blood glucose and serum insulin, as described in Methods. Lopinavir/ritonavir treatment resulted in moderate but significant increases in fasting glucose levels and dramatic elevations in serum insulin (Fig. 3a and b). While administration of ADP355 did not affect PI-induced hyperglycemia, the peptide completely inhibited PI-dependent hyperinsulinemia (Fig. 3b). ADP355 did not affect glucose or insulin levels in vehicle treated mice (Fig. 3).
Fig. 3.

ADP355 attenuates circulating serum insulin and triglycerides, but not blood glucose, in PI-treated mice. Male C57BL/6 mice were given daily administration of 10 % ethanol/15 % propylene glycol (vehicle; left bars) or 150 mg lopinavir/37.5 mg ritonavir/kg (PI; right bars) for 28 days. Additionally, randomly selected vehicle (open bars) and PI-treated mice (hatched bars) mice were also treated daily with ADP355 (1 mg/kg) or PBS via intraperitoneal injection for the final 14 days of PI exposure, after which all mice were euthanatized and serum was collected from whole blood. Data are means ± S.E.M. and were generated from 9–21 mice per group. a Fasting blood glucose levels measured in vehicle and lopinavir/ ritonavir-treated mice after administration of PBS or ADP355. *** and ** indicates significant (p<0.001 and p<0.01, respectively) increases in fasting blood glucose in lopinavir/ritonavir-treated mice given either PBS or ADP355, respectively, as compared to vehicle-treated mice. b Fasting insulin levels measured in vehicle and lopinavir/ritonavir-treated mice after administration of PBS or ADP355. *** indicates significant (p<0.001) increase in fasting insulin in lopinavir/ritonavir-treated given PBS as compared to vehicle-treated mice, while ## depicts significant (p<0.01) decrease in fasting insulin in lopinavir/ritonavir-treated mice given ADP355 as compared to lopinavir/ritonavir-treated given PBS. c Fasting triglyceride levels measured in vehicle and lopinavir/ritonavir-treated mice after administration of PBS or ADP355. *** indicates significant (p<0.001) increase in triglycerides in both groups of mice given lopinavir/ritonavir, while # depicts significant (p<0.05) decrease in triglycerides measured in lopinavir/ritonavir-treated mice given ADP355 as compared to lopinavir/ritonavir-treated given PBS
Hypertriglyceridemia is a well-established side-effect of protease inhibitor treatment in both mice and humans. Thus our next studies assessed a panel of bioactive serum lipids in PI-treated and control mice. A significant, near 3-fold increase in serum triglycerides was detected in mice given lopinavir/ ritonavir (Fig. 3c). Delayed administration of ADP355 significantly reduced lopinavir/ritonavir-induced hypertriglyceridemia (Fig. 3c), although triglyceride levels in ADP355-treated mice remained elevated relative to vehicle-treated mice. Lopinavir/ritonavir also increased levels of total cholesterol levels, but this effect was not affected by ADP355 (Table 1). Finally, levels of LDL cholesterol, HDL cholesterol, and circulating non-esterified fatty acids (NEFA) were not significantly affected by lopinavir/ritonavir or ADP355 (Table 1).
Table 1.
ADP355 does not affect circulating cholesterol species or NEFA in PI-treated mice
| Vehicle |
PI |
|||
|---|---|---|---|---|
| PBS | ADP355 | PBS | ADP355 | |
| Total cholesterol (mg/dl) | 99.3±3.8 | 104.7±2.6 | 117.4±5.9a | 121.1±6.3a |
| LDL cholesterol(mg/dl) | 14.6±1.0 | 14.9±0.3 | 17.4±1.8 | 18.4±2.0 |
| HDL cholesterol(mg/dl) | 58.9±2.6 | 63.8±2.6 | 62.9±2.6 | 62.6±3.3 |
| NEFA (mEq/L) | 0.928±0.05 | 0.979±0.05 | 0.867±0.04 | 0.967±0.05 |
Male C57BL/6 mice were given daily administration of 10 % ethanol/15 % propylene glycol (vehicle) or 150 mg lopinavir/37.5 mg ritonavir/kg (PI) for 28 days. Additionally, randomly selected vehicle and PI-treated mice were also treated daily with ADP355 (1 mg/kg) or PBS via intraperitoneal injection for the final 14 days of PI exposure, after which all mice were euthanatized and serum lipids were measured as described in Methods
Data are means ± S.E.M. generated from 9-21 mice per group, and were analyzed by 1-way ANOVA followed by planned Tukey posttests to determine the effects of ADP355 in PI- and vehicle-treated mice
Indicates significant (p<0.05) increases in total cholesterol in mice given either lopinavir/ritonavir/PBS or lopinavir/ritonavir/ADP355 as compared to vehicle-treated mice
The loss of adipose tissue lopinavir/ritonavir-treated mice was paralleled with significant and profound decreases in circulating levels of adiponectin and leptin (Fig. 4). PI-induced hypoadiponectinemia was partially reversed by delayed administration of ADP355 (Fig 4a), but ADP355 was not able to prevent lopinavir/ritonavir-induced hypoleptinemia (Fig. 4b) and did not modulate serum resistin levels (Supplemental Fig. 1). Finally, ADP355 did not significantly affect adipokine levels in vehicle-treated mice (Fig. 4).
Fig. 4.
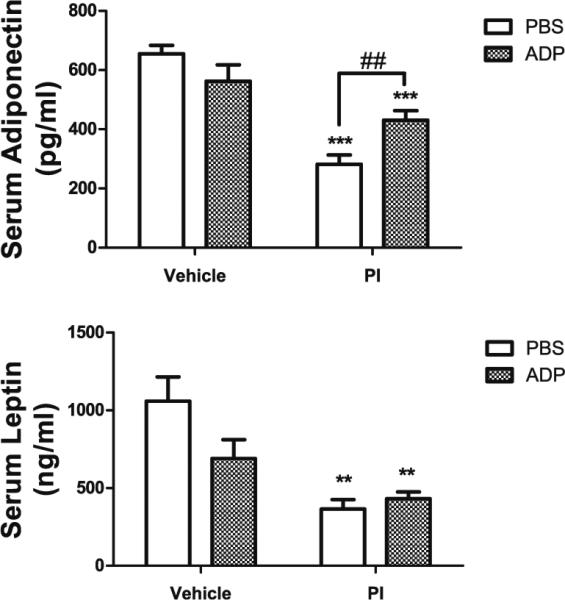
ADP355 preserves circulating serum adiponectin but not leptin in PI-treated mice. Male C57BL/6 mice were given daily administration of 10 % ethanol/15 % propylene glycol (vehicle; left bars) or 150 mg lopinavir/37.5 mg ritonavir/kg (PI; right bars) for 28 days. Additionally, randomly selected vehicle (open bars) and PI-treated mice (hatched bars) mice were also treated daily with ADP355 (1 mg/kg) or PBS via intraperitoneal injection for the final 14 days of PI exposure, after which all mice were euthanatized and serum was collected from whole blood. a Effects of lopinavir/ritonavir with or without ADP355 on serum adiponectin concentration. Data are means ± S.E.M. of adiponectin expressed as pg/μl serum, and were analyzed by 1-way ANOVA. *** indicates significant (p<0.001) decrease in adiponectin in both groups of mice given lopinavir/ritonavir, while ## depicts significant (p<0.01) increase in adiponectin levels in lopinavir/ritonavir-treated mice given ADP355 as compared to lopinavir/ritonavir-treated given PBS. b Effects of lopinavir/ritonavir with or without ADP355 on serum leptin concentration. Data are means ± S.E.M. of leptin expressed as ng/ml serum, and were analyzed by 1-way ANOVA. ** indicates significant (p>0.01) decreases in leptin in lopinavir/ritonavir-treated mice given either PBS or ADP355 as compared to vehicle-treated mice
Effects of ADP355 on Cognitive Performance and Markers of Brain Injury in Lopinavir/Ritonavir-Treated Mice
We have previously shown that chronic administration of combined lopinavir/ritonavir causes significant impairments in memory performance, but does not affect motor function (Pistell et al. 2010a; Gupta et al. 2012). To determine the effects of ADP355 on PI-induced memory impairment, the fear conditioning assay was implemented as described in Methods. Significant differences in freezing behavior were observed on the third day of the fear conditioning test, when the “tone test” that provides a measure of associative learning was deployed (Fig. 5). Specifically, freezing behavior in response to the tone was dramatically decreased in lopinavir/ ritonavir-treated mice as compared to vehicle-treated mice (Fig. 5). However, this PI-induced memory impairment was completely prevented by ADP355 administration (Fig. 5).
Fig. 5.
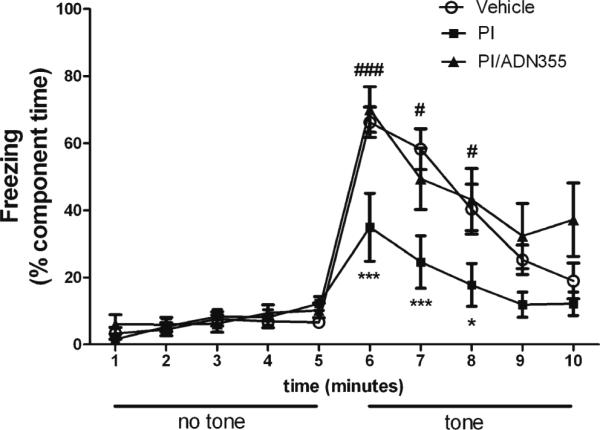
ADP355 preserves cognitive performance in PI-treated mice. Male C57BL/6 mice were treated daily with vehicle or lopinavir/ritonavir (150/37.5 mg/kg body weight) for 28 days, after which mice were evaluated behaviorally for memory performance using the fear conditioning assay as described in Methods. Experiments were conducted in 12–20 animals per group over 2 separate cohorts. Data are means ± S.E.M. of composite freezing behavior, and were analyzed by 2-way ANOVA. *and *** indicate significant (p<0.05, p<0.001, respectively) decreases in freezing behavior in lopinavir/ritonavir/PBS-treated mice as compared to vehicle-treated mice, while # and ### depict significant (p<0.05, p<0.001, respectively) increases in freezing in lopinavir/ritonavir-treated mice given ADP355 as compared to lopinavir/ritonavir-treated given PBS
PI-induced brain injury was quantified by evaluating the expression of specific brain proteins, as described previously (Gupta et al. 2012). Analyses were thematically split into evaluations of cerebrovascular integrity, synaptic density, and reactive gliosis conducted in the cerebral cortex as cortical injury has been shown to be caused by PI exposure in mice (Gupta et al. 2012), and also to perturb performance in the fear conditioning task in rodents (Han et al. 2012). Cerebrovascular and blood–brain barrier integrity were evaluated by measuring the expression of tight junction proteins ZO-1 and occludin, as well as the matrix metalloproteinases MMP2 and MMP9, as described in Methods. Data show that lopinavir/ ritonavir-treated mice given PBS showed significantly decreased expression of occludin and increased expression of MMP2 as compared to vehicle-treated mice (Fig. 6a). Conversely, lopinavir/ritonavir-treated mice given ADP355 showed significant reversals in the expression of occludin and MMP2, and indeed, no markers of cerebrovascular injury in lopinavir/ritonavir/ADP355-treated were statistically different from vehicle-treated mice (Fig. 6a).
Fig. 6.
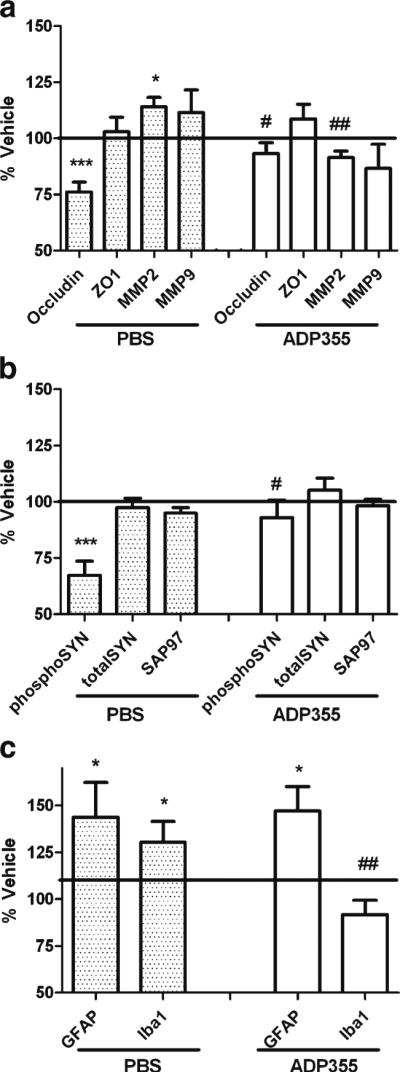
ADP355 preserves markers of cerebrovascular integrity, synaptic density, and modulates reactive gliosis in PI-treated mice. Male C57BL/6 mice were treated daily with vehicle or lopinavir/ritonavir (150/ 37.5 mg/kg body weight) for 28 days, after which markers of cerebrovascular integrity, synaptic density, and reactive gliosis were evaluated in tissue homogenates prepared from the frontal cortex as described in Methods. Data depict mean ± SEM expression in lopinavir/ritonavir-treated mice presented as % vehicle (100 % line) on graph. Data were obtained from 12–20 mice/group, and were analyzed by 1-way ANOVA. a Expression of the tight junction proteins claudin-5, ZO-1, and occludin; and the matrix metalloproteinases MMP2 and MMP9. * and ** indicate significant (p<0.05 and 0.01, respectively) changes in expression in lopinavir/ritonavir/PBS-treated mice as compared to vehicle, while # and ## depict significant (p<0.05, p<0.01, respectively) changes in expression in lopinavir/ritonavir-treated mice given ADP355 as compared to lopinavir/ritonavir-treated given PBS. b Expression of the post-synaptic marker synapse associated protein 97 (SAP97), the pre-synaptic protein synapsin 1, and phosphorylated synapsin 1. *** indicates significant (p<0.001) the significant decrease in phosphorylated synapsin 1 expression in lopinavir/ritonavir/PBS-treated mice relative to vehicle mice, while # depicts the significant (p<0.05) reversal in phosphorylated synapsin 1 expression in lopinavir/ritonavir-treated mice given ADP355. c Expression of the glial markers GFAP and Iba-1. * indicates significant (p<0.05) increases in GFAP and Iba-1 expression in lopinavir/ritonavir-treated mice relative to vehicle-treated mice, while # depicts the significant (p<0.05) reversal in Iba-1 expression in lopinavir/ritonavir-treated mice given ADP355
Evaluations of synaptic density were based on altered expression of the post-synaptic protein synapse associated protein 97 (SAP97) and total and phosphorylated forms of the pre-synaptic protein synapsin 1 (SYN1). Total levels of SAP97 and total SYN1 were similar in all groups (Fig. 6b), but phosphorylated SYN1 expression was significantly reduced in liponavir/ritonavir mice relative to controls (Fig. 6b). However, SYN1 phosphorylation was completely preserved in liponavir/ritonavir-treated mice given ADP355
To determine the effects of ADP355 on liponavir/ritonavir-induced reactive gliosis, the expression of astrocyte and microglial markers were evaluated using Western blot. PI treatment significantly increased the expression of glial fibrillary acidic protein (GFAP), an intermediate filament protein and a marker of astrocyte hypertrophy. Administration of ADP355, however, did not modulate astrocyte reactivity (Fig. 6c). Microgliosis was evaluated by measuring expression of Iba-1, a 17-kDa calcium binding protein specifically expressed in macrophages/microglia that can be detected in denatured samples. Liponavir/ritonavir caused significant increases in Iba-1 expression relative to vehicle controls (Fig. 6c), while lopinavir/ritonavir mice given ADP355 showed complete reversal of PI-induced microgliosis (Fig. 6c).
Discussion
It is well-established that ART frequently causes a pathologic sequela of metabolic derangement in HIV patients that likely contributes to co-morbidities in this patient population. Data in this manuscript demonstrate the protective effects of a novel, peptide-based adiponectin receptor agonist against metabolic and neurologic dysfunction caused by the lopinavir/ritonavir cocktail in mice. ADP355 administration moderated PI-induced loss of subcutaneous adipose, attenuated PI-induced hyperinsulinemia, hypertriglyceridemia, and hypoadiponectinemia, and completely prevented PI-induced cognitive impairment and brain injury. Collectively, these data indicate that ART-induced lipodystrophy and the resulting hypoadiponectinemia may be key players driving metabolic dysfunction in HIV patients, which could sensitize patients to HIV-associated neurocognitive disturbances. This scenario is in general agreement with clinical studies showing that HIV patients with metabolic compromise have a greater risk of developing neurologic complications (Valcour et al. 2006; Bandaru et al. 2007; Valcour et al. 2011; Foley et al. 2010). Furthermore, cognitive impairment in adult HIV patients has been shown to correlate with cardiovascular disease, hyper-tension, and cholesterolemia; but not with conventional risk factors for dementia, including CD4 cell counts, viral load, CNS penetration of ART, hepatitis C infection, or alcohol abuse (Wright et al. 2010). As epidemiological studies indicate, HIV remains the most common preventable and treatable cause of neurologic impairment in patients under the age of 50 (Ances and Ellis 2007). Additionally, recent clinical trials using neuroprotective or anti-inflammatory drugs for treatment of HIV-associated neurocognitive disorders have generally proven unsuccessful (Tan and McArthur 2012). Thus, there is a critical need to develop novel and innovative therapies to preserve neurologic function in HIV patients. Data in this manuscript raise the exciting possibility that pharmacological activation of adiponectin receptors in HIV patients could not only mitigate key aspects of ART-induced metabolic syndrome but also decrease the incidence and/or severity of HIV-associated neurocognitive disorders.
Lipodystrophy is highly common in HIV patients and is tightly associated with other metabolic co-morbidities, including insulin resistance and hyperlipidemia (Paruthi et al. 2013). Indeed, there is evidence that lipodystrophy, which has a prevalence of up to 83 % and was widely reported only after the introduction of ART, may actually precede and precipitate the development of broader metabolic complications (dyslipidemia, insulin resistance) in HIV- patients (Barbaro 2007; Arai et al. 2011). Thus, therapies that prevent the loss of adipocytes and/or replicate adipocyte function in the face of lipodystrophy could preserve metabolic function in HIV patients. Evidence of decreased adiponectin levels in HIV-positive patients with lipodystrophy (Kinlaw and Marsh 2004; Barbaro 2007; Chen et al. 2009), and the negative correlation between adiponectin and insulin resistance (Arama et al. 2013) support investigation of the protective effects of adiponectin in models of HIV. Data in this manuscript show that delayed treatment with ADP355 peptide was able to partially reverse adipose tissue depletion in HIV pro-tease inhibitor-treated mice, and that this partial preservation of adipose mass was associated with preservation of insulin and triglyceride levels. Furthermore, the protective effects of ADP355 against lipoatrophy appeared preferentially directed towards subcutaneous rather than visceral fat. Such findings could have important and exciting clinical ramifications as subcutaneous fat has intrinsic beneficial metabolic properties (Gil et al. 2011), but is particularly vulnerable to ART-induced lipoatrophy (de Waal et al. 2013). Subcutaneous and visceral adipocytes derive from different progenitor cells that exhibit a different gene expression pattern, and thus may respond quite differently to the effects of adiponectin receptor stimulation. Indeed, gene expression of AdipoR1 has been reported to be 2- fold higher, and AdipoR2 expression as much as ~10-fold higher, in subcutaneous as compared to visceral fat (Nannipieri et al. 2007), which could explain in part the preferential preservation of subcutaneous adipose by ADP355.
Delayed treatment with ADP355 peptide completely prevented neurologic decline in PI-treated mice, supporting existing data showing that adiponectin receptor signaling can support neurologic health (Oomura et al. 2006; Harvey 2007; Ouchi and Walsh 2007; Chen et al. 2009; Jung et al. 2006). The protective effect of ADP355 on neurologic function is likely mediated through multiple mechanisms but could include direct actions on neurons. While the exact role of adiponectin in the brain is still subject to debate, there is substantial evidence of adiponectin receptor expression in brain cells (Yamauchi et al. 2003; Fry et al. 2006; Rodriguez-Pacheco et al. 2007). Furthermore, studies showing that peripheral adiponectin activates hypothalamic AMPK (Kubota et al. 2007), and that systemic and ventricular administrations of adiponectin produce similar reductions in hyperglycemia and hyperlipidemia (Qi et al. 2004) all support a direct effect of adiponectin in the CNS. However, it is also possible that CNS health is bolstered by ADP355-induced reductions in metabolic dysfunction, including effects on hypertryglyceridemia (Fruebis et al. 2001) and/or insulin sensitivity and dyslipidemia (Duntas et al. 2004). Perhaps more importantly, our data show that ADP355 preserved the expression of markers of cerebrovascular homeostasis, which is in keeping with multiple lines of evidence linking adiponectin to microvascular homeostasis (Ouedraogo et al. 2007; (Vachharajan et al. 2012). Vascular pathology has received considerable attention as a participant in HIV-associated neurocognitive disorders (Foley et al. 2010), and it is quite reasonable to propose that metabolic co-morbidities exacerbate HIV-related brain injury via disruption of cerebrovascular and blood brain barrier integrity. For example, published studies have shown that neurocognitive impairment in adult HIV patients is correlated with cardiovascular disease, hyper-tension, and hypercholesterolemia; but not with more conventional risk factors for dementia, including hepatitis C infection, alcohol abuse, CD4 cell counts, viral load, or CNS penetration of ART regimens (Wright et al. 2010). Other studies show that HIV-positive subjects with untreated cardiovascular disease have significantly reduced processing speed, recall, and executive functioning relative to those on medication (Foley et al. 2010). Data also suggest that cerebrovascular disease plays a greater role in the cognitive compromise of aging HIV-infected individuals as compared to the normal population (McMurtray et al. 2007). Indeed, a recent imaging study of coronary artery calcium accumulation in HIV-infected patients suggested that vascular “age” was increased in over 40 % of patients, with a mean increase of 15 years over chronological age (Guaraldi et al. 2009). Evaluated collectively, these data strongly suggest that the improved neurocognitive performance noted in ADP355-treated mice may be mediated at least in part via preservation of cerebrovascular homeostasis. This physiologic scenario is especially significant because it suggests that attenuation of HIV-associated neurocognitive disorders could be achieved with drugs that need not penetrate into brain.
This manuscript complements a growing body of literature providing proof of concept data on the benefit of adiponectin replacement therapy in multiple disease states. For example, injection of recombinant forms of adiponectin have been reported to improve insulin sensitivity and alleviate hyperlipidaemia (Xu et al. 2003), as well as preserve metabolic function in experiential models of high-fat/sucrose diets (Fruebis et al. 2001), leptin deficiency (Takahashi et al. 2005), and ritonavir-induced hyperlipidemia in mice (Xu et al. 2004). A critical caveat to these studies, however, is whether or not recombinant adiponectin is able to recapitulate the physiologic actions of endogenous adiponectin, which is released into the circulation as full-length trimers, hexamers, and high-molecular weight multimers. As the metabolic stabilizing effects of adiponectin are generally attributed to the high molecular weight species (Waki et al. 2003; Pajvani et al. 2003), it is likely that the lack of multimerization and/or post-translational modifications of bacterially produced adiponectin compromises its in vivo efficacy. Thus, to circumvent these difficulties, we utilized an adiponectin-based peptidomimetic to directly activate adiponectin receptors. Verification of ADP355 specificity and pharmacology has been previously reported, and data show that ADP355 restricts proliferation of adiponectin receptor-positive cancer cells in a dose-dependent manner, and modulates key adiponectin-based signaling pathways (AMPK, Akt, STAT3, ERK1/2) through both adiponectin receptors, with a greater contribution of AdipoR1 (Otvos et al. 2011). Furthermore, no adverse side-effects or off-target toxicity of ADP355 could be found in treated mice, even at doses of up to 50-fold higher than the clinically effective dose range (Otvos et al. 2011). Finally, while many peptides pass the blood–brain barrier and accumulate in the brain in amounts sufficient to produce physiological effects, it is unknown if ADP355 permeates the blood brain barrier. However, as described in the preceding paragraph, data suggest that ADP355 need not penetrate fully into the brain to confer significant neuroprotection. The ability of ADP355 to preserve subcutaneous adipose tissue, decrease hyperinsulinemia, and prevent cerebrovascular dysfunction could undoubtedly translate into improved CNS physiology in clinical settings of HIV. The potential for a safe and effective adjunctive therapy to reduce the prevalence/severity of HIV-associated neurocognitive decline would be tremendously beneficial in clinical settings, as essentially all anti-inflammatory and/or neuroprotective agents that have been investigated as potential adjuvant therapies have proven ineffective in clinical trials (Tan and McArthur 2012). Thus, these data provide strong support for the development of adiponectin-based therapies such as ADP355 as adjunctive regimens to bolster both metabolic and neurologic function in clinical settings of HIV infection.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Barry Robert for expert veterinary assistance related to lopinavir/ritonavir administration, and also thank Prof. John Wade (Florey Neuroscience Institutes, Melbourne, Australia) for peptide synthesis. This work was supported by a grant from the NIH (MH099944), and also used PBRC Core facilities (Animal Phenotyping) that are funded by the NIH (P20-RR021945 and P30-DK072476).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11481-014-9529-1) contains supplementary material, which is available to authorized users.
Conflict of Interest The authors declare that they have no conflict of interest.
Contributor Information
Jennifer K. Pepping, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, LA 70808, USA Department of Pathobiological Sciences, School of Veterinary Medicine, Louisiana State University, Baton Rouge, LA 70803, USA.
Laszlo Otvos, Jr., Department of Biology, Temple University, Philadelphia, PA 19122, USA
Eva Surmacz, Sbarro Institute for Cancer Research and Molecular Medicine, Temple University, Philadelphia, PA 19122, USA.
Sunita Gupta, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, LA 70808, USA.
Jeffrey N. Keller, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, LA 70808, USA
Annadora J. Bruce-Keller, Pennington Biomedical Research Center, Louisiana State University System, Baton Rouge, LA 70808, USA Inflammation and Neurodegeneration Laboratory, Pennington Biomedical Research Center/LSU, 6400 Perkins Road, Baton Rouge, LA 70808, USA.
References
- Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27:86–92. doi: 10.1055/s-2006-956759. [DOI] [PubMed] [Google Scholar]
- Anuurad E, Semrad A, Berglund L. Human immunodeficiency virus and highly active antiretroviral therapy-associated metabolic disorders and risk factors for cardiovascular disease. Metab Syndr Relat Disord. 2009;7:401–410. doi: 10.1089/met.2008.0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y, Takayama M, Abe Y, Hirose N. Adipokines and aging. J Atheroscler Thromb. 2011;18:545–545. doi: 10.5551/jat.7039. [DOI] [PubMed] [Google Scholar]
- Arama V, Tiliscan C, Streinu-Cercel A, Ion D, Mihailescu R, Munteanu D, Hristea A, Arama SS, group. S-As Insulin resistance and adipokines serum levels in a caucasian cohort of hiv-positive patients undergoing antiretroviral therapy: a cross sectional study. BMC Endocr Disord. 2013;13:4. doi: 10.1186/1472-6823-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru VV, McArthur JC, Sacktor N, Cutler RG, Knapp EL, Mattson MP, Haughey NJ. Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology. 2007;68:1481–1487. doi: 10.1212/01.wnl.0000260610.79853.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaro G. Visceral fat as target of highly active antiretroviral therapy-associated metabolic syndrome. Curr Pharm Des. 2007;13:2208–2213. doi: 10.2174/138161207781039661. [DOI] [PubMed] [Google Scholar]
- Chen B, Liao WQ, Xu N, Xu H, Wen JY, Yu CA, Liu XY, Li CL, Zhao SM, Campbell W. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res. 2009;1273:129–137. doi: 10.1016/j.brainres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- de Waal R, Cohen K, Maartens G. Systematic review of antiretroviral-associated lipodystrophy: lipoatrophy, but not central fat gain, is an antiretroviral adverse drug reaction. PLoS One. 2013;8:e63623. doi: 10.1371/journal.pone.0063623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duntas LH, Popovic V, Panotopoulos G. Adiponectin: novelties in metabolism and hormonal regulation. Nutr Neurosci. 2004;7:195–200. doi: 10.1080/10284150400009998. [DOI] [PubMed] [Google Scholar]
- Foley J, Ettenhofer M, Wright MJ, Siddiqi I, Choi M, Thames AD, Mason K, Castellon S, Hinkin CH. Neurocognitive functioning in HIV-1 infection: effects of cerebrovascular risk factors and age. Clin Neuropsychol. 2010;24:265–285. doi: 10.1080/13854040903482830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M, Smith PM, Hoyda TD, Duncan M, Ahima RS, Sharkey KA, Ferguson AV. Area postrema neurons are modulated by the adipocyte hormone adiponectin. J Neurosci. 2006;26:9695–9702. doi: 10.1523/JNEUROSCI.2014-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil A, Olza J, Gil-Campos M, Gomez-Llorente C, Aguilera CM. Is adipose tissue metabolically different at different sites? Int J Pediatr Obes. 2011;6(Suppl 1):13–20. doi: 10.3109/17477166.2011.604326. [DOI] [PubMed] [Google Scholar]
- Guaraldi G, Zona S, Alexopoulos N, Orlando G, Carli F, Ligabue G, Fiocchi F, Lattanzi A, Rossi R, Modena M, Esposito R, Palella F, Raggi P. Coronary aging in HIV-infected patients. 2009;49:1756–1762. doi: 10.1086/648080. [DOI] [PubMed] [Google Scholar]
- Gupta S, Knight AG, Losso BY, Ingram DK, Keller JN, Bruce-Keller AJ. Brain injury caused by HIV protease inhibitors: role of lipodystrophy and insulin resistance. Antiviral Res. 2012;95:19–29. doi: 10.1016/j.antiviral.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez AD, Balasubramanyam A. Dysregulation of glucose metabolism in HIV patients: epidemiology, mechanisms, and management. Endocrine. 2012;41:1–10. doi: 10.1007/s12020-011-9565-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Hong S, Lee D, Lee MH, Choi JS, Koh MJ, Sun W, Kim H, Lee HW. Altered expression of synaptotagmin 13 mRNA in adult mouse brain after contextual fear conditioning. Biochem Biophys Res Commun. 2012;425:880–885. doi: 10.1016/j.bbrc.2012.07.166. [DOI] [PubMed] [Google Scholar]
- Harvey J. Leptin regulation of neuronal excitability and cognitive function. Curr Opin Pharmacol. 2007;7:643–647. doi: 10.1016/j.coph.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Nácher I, Alvarez E, Morello J, Rodriguez-Nóvoa S, de Andrés S, Soriano V. Approaches for understanding and predicting drug interactions in human immunodeficiency virus-infected patients. Expert Opin Drug Metab Toxicol. 2011;7:457–477. doi: 10.1517/17425255.2011.558839. [DOI] [PubMed] [Google Scholar]
- Jung TW, Lee JY, Shim WS, Kang ES, Kim JS, Ahn CW, Lee HC, Cha BS. Adiponectin protects human neuroblastoma SH-SY5Y cells against MPP + -induced cytotoxicity. Biochem Biophys Res Commun. 2006;343:564–570. doi: 10.1016/j.bbrc.2006.02.186. [DOI] [PubMed] [Google Scholar]
- Kamogawa K, Kohara K, Tabara Y, Uetani E, Nagai T, Yamamoto M, Igase M, Miki T. Abdominal fat, adipose-derived hormones and mild cognitive impairment: the J-SHIPP study. Dement Geriatr Cogn Disord. 2010;30:432–439. doi: 10.1159/000321985. [DOI] [PubMed] [Google Scholar]
- Kinlaw WB, Marsh B. Adiponectin and HIV-lipodystrophy: taking HAART. Endocrinology. 2004;145:484–486. doi: 10.1210/en.2003-1513. [DOI] [PubMed] [Google Scholar]
- Kohli R, Shevitz A, Gorbach S, Wanke C. A randomized placebo-controlled trial of metformin for the treatment of HIV lipodystrophy. HIV Med. 2007;8:420–426. doi: 10.1111/j.1468-1293.2007.00488.x. [DOI] [PubMed] [Google Scholar]
- Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, Ezaki O, Nagai R, Tobe K, Terauchi Y, Ueki K, Minokoshi Y, Kadowaki T. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007;6:55–68. doi: 10.1016/j.cmet.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Leszczyszyn-Pynka M, Pynka S, Boron-Kaczmarska A, Pilarska K. Serum leptin and adiponectin concentrations in patients infected with human immunodeficiency virus type 1 (HIV-1) on antiretroviral therapy. Endokrynol Pol. 2005;56:19–24. [PubMed] [Google Scholar]
- Masaki T, Anan F, Shimomura T, Fujiki M, Saikawa T, Yoshimatsu H. Association between hippocampal volume and serum adiponectin in patients with type 2 diabetes mellitus. Metabolism. 2012 doi: 10.1016/j.metabol.2012.01.016. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- McMurtray A, Nakamoto B, Shikuma C, Valcour V. Small-vessel vascular disease in human immunodeficiency virus infection: the Hawaii aging with HIV cohort study. Cerebrovasc Dis. 2007;24:236–241. doi: 10.1159/000104484. [DOI] [PubMed] [Google Scholar]
- Mulligan K, Yang Y, Wininger DA, Koletar SL, Parker RA, Alston-Smith BL, Schouten JT, Fielding RA, Basar MT, Grinspoon S. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS. 2007;21:47–57. doi: 10.1097/QAD.0b013e328011220e. [DOI] [PubMed] [Google Scholar]
- Nannipieri M, Bonotti A, Anselmino M, Cecchetti F, Madec S, Mancini E, Baldi S, Santini F, Pinchera A, Rossi M, Ferrannini E. Pattern of expression of adiponectin receptors in human adipose tissue depots and its relation to the metabolic state. Int J Obes (Lond) 2007;31:1843–1848. doi: 10.1038/sj.ijo.0803676. [DOI] [PubMed] [Google Scholar]
- Oomura Y, Hori N, Shiraishi T, Fukunaga K, Takeda H, Tsuji M, Matsumiya T, Ishibashi M, Aou S, Li X, Kohno D, Uramura K, Sougawa H, Yada T, Wayner M, Sasaki K. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;11:2738–2749. doi: 10.1016/j.peptides.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Otvos LJ, Haspinger E, La Russa F, Maspero F, Graziano P, Kovalszky I, Lovas S, Nama K, Hoffmann R, Knappe D, Cassone M, Wade J, Surmacz E. Design and development of a peptide-based adiponectin receptor agonist for cancer treatment. BMC Biotechnol. 2011;11:90. doi: 10.1186/1472-6750-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380:24–30. doi: 10.1016/j.cca.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, Chan L, Goldstein BJ, Scalia R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest. 2007;117:1718–1726. doi: 10.1172/JCI29623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications fpr metabolic regulation and bioactivity. J Biol Chem. 2003;278:9073–9085. doi: 10.1074/jbc.M207198200. [DOI] [PubMed] [Google Scholar]
- Paruthi J, Gill N, Mantzoros CS. Adipokines in the HIV/HAART-associated lipodystrophy syndrome. Metabolism. 2013;24:S0026–S0495. doi: 10.1016/j.metabol.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Pistell PJ, Gupta S, Knight AG, Domingue M, Uranga RM, Ingram DK, Kheterpal I, Ruiz C, Keller JN, Bruce-Keller AJ. Metabolic and neurologic consequences of chronic lopinavir/ritonavir administration to C57BL/6 mice. Antiviral Res. 2010a;88:334–342. doi: 10.1016/j.antiviral.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol. 2010b;219:25–32. doi: 10.1016/j.jneuroim.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, Scherer PE, Ahima RS. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004;10:524–529. doi: 10.1038/nm1029. [DOI] [PubMed] [Google Scholar]
- Quinn TC. HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS Patient Care STDS. 2008;22(Suppl 3):S7–S12. doi: 10.1097/01.aids.0000327510.68503.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2007;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Rocha VZ, Libby P. The multiple facets of the fat tissue. Thyroid. 2008;18:175–183. doi: 10.1089/thy.2007.0296. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pacheco F, Martinez-Fuentes AJ, Tovar S, Pinilla L, Tena-Sempere M, Dieguez C, Castaño JP, Malagon MM. Regulation of pituitary cell function by adiponectin. Endocrinology. 2007;148:401–410. doi: 10.1210/en.2006-1019. [DOI] [PubMed] [Google Scholar]
- Schambelan M, Benson CA, Carr A, Currier JS, Dubé MP, Gerber JG, Grinspoon SK, Grunfeld C, Kotler DP, Mulligan K, Powderly WG, Saag MS, Society-USA. IA Management of metabolic complications associated with antiretroviral therapy for HIV-1 infection: recommendations of an International AIDS Society-USA panel. J Acquir Immune Defic Syndr. 2002;31:257–275. doi: 10.1097/00126334-200211010-00001. [DOI] [PubMed] [Google Scholar]
- Siasos G, Tousoulis D, Kollia C, Oikonomou E, Siasou Z, Stefanadis C, Papavassiliou AG. Adiponectin and cardiovascular disease: mechanisms and new therapeutic approaches. Curr Med Chem. 2012;19:1193–1209. doi: 10.2174/092986712799320583. [DOI] [PubMed] [Google Scholar]
- Sivakumar T, Mechanic O, Fehmie DA, Paul B. Growth hormone axis treatments for HIV-associated lipodystrophy: a systematic review of placebo-controlled trials. HIV Med. 2011;12:453–462. doi: 10.1111/j.1468-1293.2010.00906.x. [DOI] [PubMed] [Google Scholar]
- Spooner LM, Olin JL. Tesamorelin: a growth hormone-releasing factor analogue for HIV-associated lipodystrophy. Ann Pharmacother. 2012;46:240–247. doi: 10.1345/aph.1Q629. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Saegusa S, Sumino H, Nakahashi T, Iwai K, Morimoto S, Kanda T. Adiponectin replacement therapy attenuates myocardial damage in leptin-deficient mice with viral myocarditis. J Int Med Res. 2005;33:207–214. doi: 10.1177/147323000503300208. [DOI] [PubMed] [Google Scholar]
- Tan IL, McArthur JC. HIV-associated neurological disorders: a guide to pharmacotherapy. CNS Drugs. 2012;26:123–134. doi: 10.2165/11597770-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Vachharajan IV, Cunningham C, Yoza B, Carson JJ, Vachharajani TJ, McCall C. Adiponectin-deficiency exaggerates sepsis- induced microvascular dysfunction in the mouse brain. Obesity (Silver Spring) 2012;20:498–504. doi: 10.1038/oby.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcour VG, Sacktor NC, Paul RH, Watters MR, Selnes OA, Shiramizu BT, Williams AE, Shikuma CM. Insulin resistance is associated with cognition among HIV-1-infected patients: the Hawaii aging with HIV cohort. J Acquir Immune Defic Syndr. 2006;43:405–410. doi: 10.1097/01.qai.0000243119.67529.f5. [DOI] [PubMed] [Google Scholar]
- Valcour V, Maki P, Bacchetti P, Anastos K, Crystal H, Young M, Mack W, Cohen M, Golub E, Tien P. Insulin resistance and cognition among HIV-infected and HIV-uninfected adult women—the women’s interagency HIV study. AIDS Res Hum Retroviruses. 2011 doi: 10.1089/aid.2011.0159. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk JP, Hoepelman AI, de Koning EJ, Dallinga-Thie G, Rabelink TJ, Cabezas MC. Differential effects of rosiglitazone and metformin on postprandial lipemia in patients with HIV-lipodystrophy. Arterioscler Thromb Vasc Biol. 2011;31:228–233. doi: 10.1161/ATVBAHA.110.216192. [DOI] [PubMed] [Google Scholar]
- Veloso S, Escoté X, Ceperuelo-Mallafré V, López-Dupla M, Peraire J, Viladés C, Domingo P, Castro A, Olona M, Sirvent JJ, Leal M, Vendrell J, Richart C, Vidal F. Leptin and adiponectin, but not IL18, are related with insulin resistance in treated HIV-1-infected patients with lipodystrophy. Cytokine. 2012;58:253–260. doi: 10.1016/j.cyto.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, Kimura S, Nagai R, Kadowaki T. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem. 2003;278:40352–40363. doi: 10.1074/jbc.M300365200. [DOI] [PubMed] [Google Scholar]
- Wei W, Wan Y. Thiazolidinediones on PPARγ: the roles in bone remodeling. PPAR Res. 2011;2011:867180. doi: 10.1155/2011/867180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright EJ, Grund B, Robertson K, Brew BJ, Roediger M, Bain MP, Drummond F, Vjecha MJ, Hoy J, Miller C, de Oliveira AC P, Pumpradit W, Shlay JC, El-Sadr W, Price RW, Group. ISS Cardiovascular risk factors associated with lower baseline cognitive performance in HIV-positive persons. Neurology. 2010;75:864–873. doi: 10.1212/WNL.0b013e3181f11bd8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Yin S, Wong L, Chan KW, Lam KS. Adiponectin ameliorates dyslipidemia induced by the human immunodeficiency virus protease inhibitor ritonavir in mice. Endocrinology. 2004;145:487–494. doi: 10.1210/en.2003-1140. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno N, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.