

INTRODUCTION
Functional impairment and depression of behavior are key diagnostic indicators of pain and targets of pain treatment [12,44]. Multiple neural mechanisms likely contribute to the depressive effects of pain, but clinical evidence implicates a hypodopaminergic state as one contributing factor [23,46,52]. Preclinical studies also suggest that reduced dopamine (DA) signaling may contribute to pain-related depression of behavior. For example, intraperitoneal injection of dilute acid (IP acid) is a physiologically relevant chemical noxious stimulus for preclinical studies of pain and analgesia in rodents [7,11,18,35]. In rats, IP acid depresses extracellular DA levels in nucleus accumbens (NAc) and also depresses intracranial self-stimulation (ICSS), an operant behavior that relies on activation of mesolimbic DA neurons that terminate in NAc [25,31,38]. IP acid-induced depression of both NAc DA and ICSS can be blocked by clinically effective analgesics such as the mu opioid receptor agonist morphine and the nonsteroidal anti-inflammatory drug (NSAID) ketoprofen [25,31]. Moreover, IP acid-induced depression of ICSS can also be blocked by DA uptake inhibitors and exacerbated by DA receptor antagonists, further supporting a role for DA in mediating this model of pain-related behavioral depression [34,40].
One implication of these findings is that DA uptake inhibitors, or other drugs that enhance DA signaling, may alleviate pain-related depression of behavior. However, many drugs that block DA transporters (DAT) and inhibit DA uptake also have high abuse liability [32,39]. Development of DA uptake inhibitors as viable therapeutics would benefit from strategies to reduce that abuse liability. One approach to this challenge has been to develop DA uptake inhibitors with reduced selectivity to bind DAT in comparison to serotonin transporters (SERT) and norepinephrine transporters (NET). Selective inhibitors of SERT and NET have little or no abuse liability, and a large body of evidence suggests that serotonin (5HT) in particular may oppose and limit abuse-related effects mediated by DA [22,32,36,51]. Moreover, inhibition of SERT and/or NET might also contribute to analgesic effects by acting on bulbospinal circuits to modulate nociceptive processing in the spinal cord [4,27,43].
Cocaine is illustrative of a nonselective monoamine uptake inhibitor with similar potencies to inhibit DAT, SERT and NET [41], but while cocaine can produce significant analgesia in humans [53], it retains high abuse liability. Consequently, uptake inhibitors of interest as candidate analgesics might optimally have DAT vs. SERT/NET selectivity lower than that of cocaine (to reduce abuse potential) but higher than that of selective SERT or NET inhibitors (to alleviate hypodopaminergic components of pain). Amitifadine is a triple uptake inhibitor that occupies this pharmacological niche. It blocks DAT with approximately 5- to 10-fold weaker potency than it blocks SERT and NET [45], and it increased microdialysis measures of DA<NE<5HT in prefrontal cortex of rats without producing significant locomotor activation [20]. The purpose of the present study was to evaluate effects of amitifadine on NAc DA and ICSS in the absence and presence of the IP acid noxious stimulus. We hypothesized that amitifadine would have sufficient DA activity to block acid-induced depression of NAc DA and ICSS but would not produce abuse-related facilitation of ICSS in the absence of pain.
METHODS
Subjects
Male Sprague-Dawley rats (total N = 27; Harlan, Fredrick, MD, USA) weighing 310–350 g at the time of surgery were used for these studies. Rats were individually housed and maintained on a 12-h light/dark cycle with lights on from 6:00 a.m. to 6:00 p.m. Rats had free access to food and water except during testing. Animal maintenance and research were in compliance with National Institutes of Health guidelines on care and use of animal subjects in research, and all animal use protocols were approved by the Virginia Commonwealth University Institutional Care and Use Committee.
Intracranial Self-Stimulation
Surgery and Training
Rats were implanted with unipolar electrodes (Plastics One, Roanoke, VA) targeting the medial forebrain bundle (2.8 mm posterior to bregma, 1.7 mm lateral to the midsagittal line, and 8.8 mm ventral to skull) and trained to respond for brain stimulation using operant equipment (Med Associates, St. Albans, VT) and procedures similar to those described previously [1,25]. Each lever press resulted in the delivery of a 0.5-s train of square wave cathodal pulses (0.1-ms pulse duration). Responses during the 0.5-s stimulation period did not result in additional stimulation. Following initial training to determine the terminal amplitude of stimulation, frequency manipulations were introduced during sessions that consisted of sequential 10-min components. During each component, a descending series of 10 current frequencies (2.2–1.75 Log Hz in 0.05 log increments) was presented, with a 60-s trial at each frequency. A frequency trial began with a 5-s time out followed by a 5-s “priming” phase during which animals received five non-contingent stimulations with a 0.5-s interval between stimulations. This non-contingent stimulation was followed by a 50-s “response” phase during which responding produced electrical stimulation under a fixed-ratio 1 schedule.
Amitifadine effects on control and acid-depressed ICSS
Amitifadine effects on ICSS in the absence of the noxious stimulus were tested in dose-effect and time-course experiments. Dose-effect test sessions consisted of three “baseline” ICSS components followed first by a 30-min time out period and then by two “test” components. At the beginning of the time out, rats were removed from the ICSS chambers, treated with vehicle or amitifadine (0.32–10 mg/kg, IP; N=5), and placed into their home cages. At the end of the time out, the lactic acid vehicle (bacteriostatic water) was administered (1 ml/kg, IP), and rats were returned to the ICSS chambers for testing. Time-course sessions consisted of three baseline ICSS components followed first by injection of amitifadine (3.2 or 10 mg/kg, IP; N=6) and then by pairs of test components that began 10, 30, 100 and 300 min and 24 hr after injection.
Amitifadine effects on acid-induced depression of ICSS were tested in sessions consisting of three baseline ICSS components followed first by a time out period (30 or 100 min) and then by two test components. At the beginning of the time out, rats were removed from the ICSS chambers, treated with vehicle or amitifadine and placed into their home cages. Following the time out, 1.8% or 5.6% lactic acid was administered, and rats were returned to the ICSS chambers for testing. A 30-fold range of amitifadine doses (0.32–10 mg/kg, IP) was tested as 30-min pretreatment to 1.8% lactic acid (N=5), and the highest amitifadine doses (3.2 and 10 mg/kg, IP) were tested as 100-min pretreatment to 1.8 (N=6) and 5.6% lactic acid (N=5).
Data Analysis
The primary dependent variable was the reinforcement rate in stimulations/trial during each frequency trial. To normalize these data, reinforcement rates from each trial were converted to Percent Maximum Control Rate (%MCR) for that rat on that day. Maximum control rate (MCR) was determined during baseline components of each test session. Data from the first baseline component of each test session were discarded, and MCR was defined as the mean of the maximal rates observed in any frequency trial during the second and third control components. Thus, %MCR for each trial was calculated as: (Reinforcement Rate During a Frequency Trial ÷ Maximum Control Rate) × 100. Normalized data from the frequency trials of each pair of consecutive test components were averaged across rats for statistical analysis using two-way ANOVA, with treatment and brain stimulation frequency as factors. A significant ANOVA was followed by a Holm-Sidak post hoc test, and the criterion for significance was set at p < 0.05.
To provide a summary measure of performance over the entire range of frequency magnitudes, the total number of stimulations per component was calculated as the average of total stimulations delivered across all 10 frequency trials of each component. Data were expressed as a percentage of the total stimulations earned during the baseline components using the equation: % Baseline Total Stimulations = (Mean Total Stimulations during Test Components ÷ Mean Total Stimulations during Baseline Components) × 100.
Microdialysis
Surgery
Rats were implanted bilaterally with guide cannulae that terminated 1mm above the NAc (1.5mm anterior to Bregma, 1.8mm lateral to midsaggital line, 6.0mm ventral to dura) as described previously [25]. A dummy cannula was inserted into each guide cannula to maintain patency, and guide cannulae were secured to the skull with screws and dental acrylic. Animals were allowed to recover for at least four days before initiation of testing. All cannulae, microdialysis probes, and microdialysis equipment were obtained from Eicom Corp (San Diego, CA).
Procedure
On test days, one dummy cannula was removed, and a microdialysis probe with a 2mm regenerated cellulose membrane (50 KDa molecular weight cutoff) was inserted through the guide cannula and into the NAc. The probe was connected to a two-channel liquid swivel, and the rat was placed in a clear plexiglass chamber (30cm x 30cm x 30 cm). Probes were perfused with a non-buffered artificial cerebrospinal fluid solution (147 mM NaCl, 2.8 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2) at a rate of 1μl/min. Following an equilibration period of at least 60 min, dialysate samples were collected into a 50 μL injector loop at 10-min intervals using an EAS-20s online autoinjector and immediately analyzed for DA and 5HT concentrations by high-pressure liquid chromatography coupled to electrochemical detection. Experiments conducted by probe immersion into a known standard concentration of DA and 5HT indicated a lag time of 24 min for dialysate to traverse the tubing from the probe to the electrochemical detector at the 1μl/min flow rate (data not shown). The mobile phase consisted of 2% methanol (EMD, Gibbstown, NJ), 100 mM phosphate buffer (Sigma Chemicals, St. Louis, MO), 500 mg/L 1-Decane sodium sulfonate (TCI America; Montgomeryville, PA), and 50 mg/L EDTA-2NA (Dojindo Laboratories, Kumamoto, Japan). DA and 5HT were separated using a PP-ODS II reverse phase C18-column and detected using a graphite working electrode and an Ag vs AgCl reference electrode with an applied potential of +450mV. DA and 5HT were identified according to the retention time of the standard, and concentrations were quantified by comparison with peak heights of the standard concentration curve (0.01–100 pg/10uL) determined before each microdialysis experiment to ensure accuracy of standard retention times. Resolution was sufficient to detect DA and 5HT levels as low as 0.1 pg. DA and 5HT levels were considered to have stabilized after collection of six consecutive baseline samples with <10% variability around the mean. Amitifadine or saline was administered IP 100 min before administration of 5.6% lactic acid or vehicle (1 ml/kg, IP), and DA and 5HT levels were detected at 10 min intervals for an additional 200 minutes. Each rat was tested no more than twice at each cannula site, and test days were separated by at least one week. Each test was treated as an independent observation for statistical analysis (N=5–7 per treatment).
Histology
After microdialysis experiments, rats were euthanized by CO2, brains were removed and placed in 10% formalin for at least 1 week. Anatomical placement of guide cannulae and microdialysis probes was verified by gross visual and microscopic examination of photographed coronal sections according to a rat brain atlas (Figure S1) [37]
Data Analysis
The primary dependent variable was the concentration of DA and 5HT in each dialysate fraction. Concentrations in each fraction for each rat were expressed as percent of the average of the six baseline concentrations prior to drug or vehicle administration. Treatment effects on DA and 5HT were analyzed by two-way repeated measures ANOVA with treatment and time as the two factors. Significant ANOVAs were followed by a Holm-Sidak post hoc test, and significance was set at p < 0.05.
Lactic Acid-stimulated Stretching
Five rats were implanted with unipolar electrodes targeting the medial forebrain bundle as described above, but did not receive ICSS training. Test sessions were conducted once per week using procedures described previously [25,40]. Saline or amitifadine (1–10 mg/kg, IP) was administered 30 min prior to treatment with 1.8% lactic acid. Order of administration of doses was determined by Latin square. Immediately after lactic acid treatment, rats were placed into acrylic test chambers (31.0 x 20.1 x 20 cm) for a 30-min observation period. A stretch was operationally defined as a contraction of the abdomen followed by extension of the hind limbs.
Data Analysis
The dependent variable was the number of stretches observed during the 30-min observation period in each rat. Data were averaged across rats, amitifadine effects were analyzed by one-way repeated measures ANOVA, and the significant ANOVA was followed by the Dunnett post-hoc test. Criterion for significance was set at p < .05.
Drugs
Amitifadine was provided by Bruce Blough PhD (Research Triangle Institute, Research Triangle Park, NC), dissolved in saline, and delivered IP in a volume of 1 ml/kg body weight. Lactic acid was purchased from Sigma Chemical Co. (St. Louis, MO), diluted in bacteriostatic water, and delivered IP in a volume of 1 ml/kg body weight.
RESULTS
Effects of amitifadine on control ICSS
Over the course of the ICSS experiments the mean ± SEM baseline maximum control rate of stimulations per trial was 61.51 ± 3.20, and the mean ± SEM baseline total number of stimulations per component delivered across all stimulation frequencies was 266.40 ± 19.98. Figure 1 shows potency and time course of amitifadine effects on control ICSS in the absence of the noxious stimulus. When administered as a 30 min pretreatment, amitifadine had no effect until the highest dose (10 mg/kg), which produced a bi-phasic effect (Figure 1A) expressed as significant depression of ICSS at the two highest frequencies and significant facilitation of ICSS at intermediate frequencies. In time-course studies, 3.2 mg/kg amitifadine had no significant effect on ICSS at any time point (Figure 1B), but 10 mg/kg produced a time-dependent biphasic effect (Figure 1C). After 10 and 30 min, 10 mg/kg amitifadine exclusively depressed ICSS. After 100 min, 10 mg/kg amitifadine depressed ICSS at the two highest frequencies, but facilitated ICSS at intermediate frequencies. Significant effects were no longer apparent at 300 min.
Figure 1.
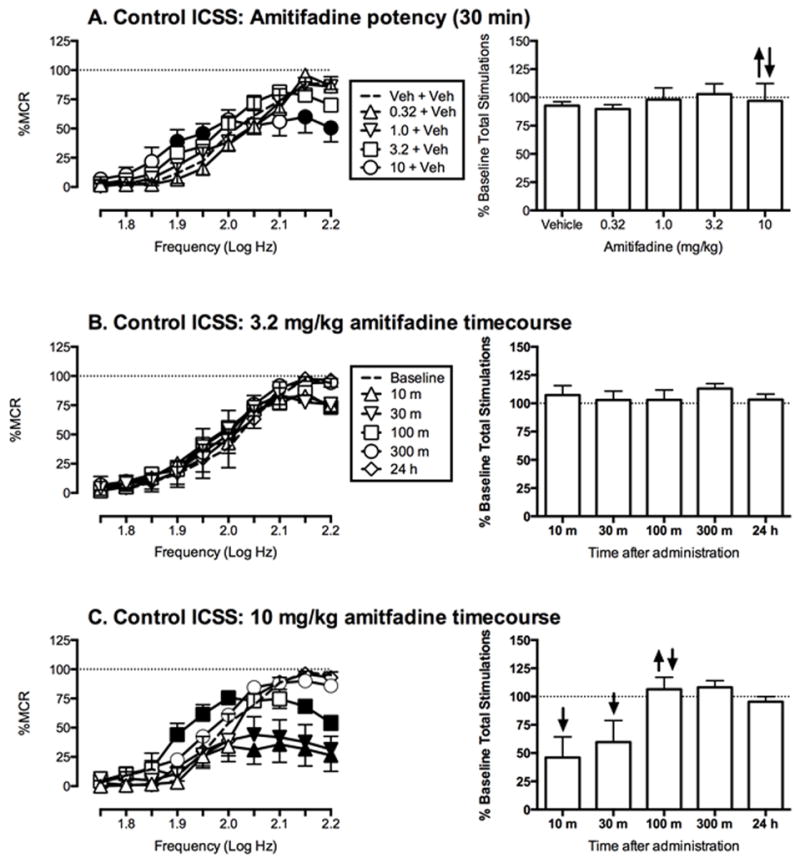
Effects of amitifadine on control ICSS in the absence of the noxious stimulus. Left column shows drug effects on full ICSS frequency–rate curves. Abscissae: Frequency of electrical brain stimulation in Log Hz. Ordinates: Percent maximum control reinforcement rate (%MCR). Filled points represent frequencies at which reinforcement rates were statistically different from vehicle rates as determined by a two-way ANOVA followed by a Holm–Sidak post hoc test, P < 0.05. Right column shows summary ICSS data expressed as percent pre-drug baseline number of stimulations per component delivered across all frequencies of brain stimulation. Abscissae: drug dose in mg/kg. Ordinates: Percent baseline total number of stimulations per component. Upward/downward arrows indicate significant drug-induced increase/decrease in ICSS relative to vehicle for at least one brain stimulation frequency as determined by analysis of full frequency–rate curves. All data show mean ± SEM for five to six rats. Statistical results for data in left panels are as follows: (A) Amitifadine potency at a 30 min pretreatment time (N=5): significant main effect of frequency [F(9,36) = 27.9, P < 0.001], no significant main effect of dose [F(4,16) = 0.7, P = 0.59], and a significant frequency X dose interaction [F(36,144) = 2.7, P = < 0.0001]. (B) 3.2 mg/kg amitifadine timecourse (n = 6): significant main effect of frequency [F(9,45) = 32.1, P < 0.0001] but not of time [F(5,25) = 0.7, P = 0.61], and no significant interaction [F(45,225) = 1.4, P = 0.05] (C) 10 mg/kg amitifadine timecourse (n = 6): significant main effect of frequency [F(9,45) = 36.6, P < 0.0001], time [F(5,25) = 4.8, P < 0.003] and significant frequency X time interaction [F(45,225) = 5.8, P < 0.0001].
Effects of amitifadine on acid-depressed ICSS
Figure 2 shows effects of amitifadine on IP acid-induced depression of ICSS. IP injection of 1.8% lactic acid immediately prior to ICSS sessions produced a rightward shift in the ICSS frequency-rate curve with significant depression of ICSS at intermediate frequencies (Figure 2A). Doses of amitifadine that had no effect on control ICSS (1.0 and 3.2 mg/kg) significantly attenuated acid-induced depression of ICSS when administered as a 30 min pretreatment (Figure 2B). The highest dose, 10 mg/kg, produced a biphasic effect expressed as attenuation of acid-induced depression of ICSS at low and intermediate frequencies, and exacerbation of acid-induced depression of ICSS at the highest frequencies. When administered as a 100 min pretreatment (Figure 2C), 3.2 and 10 mg/kg dose-dependently attenuated acid-induced depression of ICSS, but exacerbation of acid-induced depression of ICSS by 10 mg/kg was no longer apparent.
Figure 2.
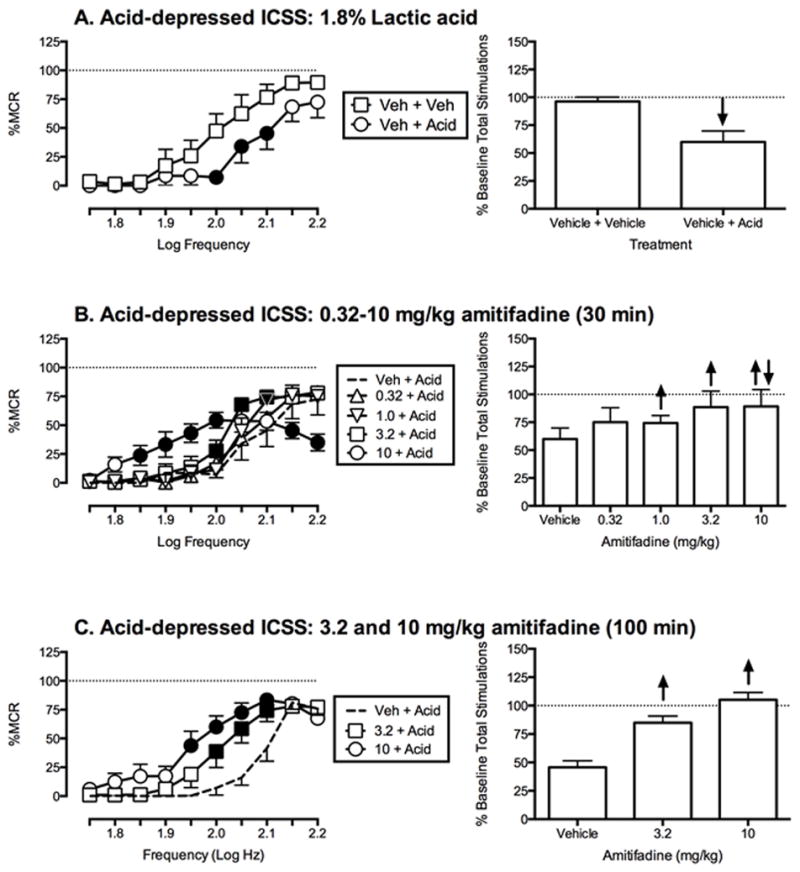
Effects of amitifadine on 1.8% acid-induced depression of ICSS. Left column shows effects on full ICSS frequency–rate curves. Abscissae: Frequency of electrical brain stimulation in Log Hz. Ordinates: Percent maximum control reinforcement rate (%MCR). Right columns show summary ICSS data expressed as percent pre-treatment baseline total number of stimulations per component delivered across all frequencies of brain stimulation. Abscissae: drug dose in mg/kg. Ordinates: Percent baseline number of stimulations per component. All other details as described for Figure 1. All data show mean ± SEM for five to six rats. Statistical results for data in left panels are as follows: (A) 1.8% Acid effect (n = 5): significant main effect of frequency [F(9,36) = 19.4, P < 0.001], treatment [F(1,4) = 25.1, P < 0.01], and significant frequency X treatment interaction [F(9,36) = 3.2, P < 0.01]. (B) Amitifadine effects determined at 30 min post-treatment (n = 5): significant main effect of frequency [F(9,36) = 53.8, P < 0.0001], no significant main effect of dose [F(4,16) = 1.8, P = 0.17], and significant frequency X treatment interaction [F(36,144) = 4.9, P = < 0.0001]. (C) Amitifadine effects determined at 100 min post-treatment (n = 6): significant main effect of frequency [F(9,45) = 54.8, P < 0.0001], dose [F(2,10) = 25.83, P < 0.0001] and significant frequency X treatment interaction [F(18,90) = 4.7, P = < 0.0001].
Figure 3 shows effects of amitifadine on acid-induced depression of ICSS using a higher intensity noxious stimulus, 5.6% lactic acid. IP injection of 5.6% lactic acid immediately prior to ICSS sessions produced a rightward and downward shift in the ICSS frequency-rate curve (Figure 3A). When administered as a 100 min pretreatment to 5.6% lactic acid, amitifadine produced a biphasic effect (Figure 3B). Specifically, 3.2 and 10 mg/kg attenuated acid-induced depression of ICSS at low and intermediate frequencies, but exacerbated acid-induced depression of ICSS at the highest frequency.
Figure 3.
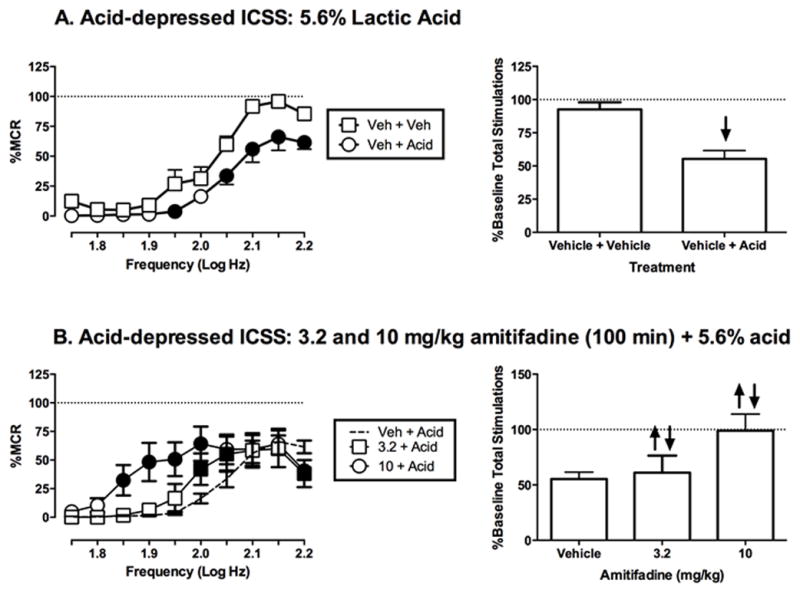
Effects of amitifadine on 5.6% acid-induced depression of ICSS. Left column shows effects on full ICSS frequency–rate curves. Abscissae: Frequency of electrical brain stimulation in Log Hz. Ordinates: Percent maximum control reinforcement rate (%MCR). Right column shows summary ICSS data expressed as percent baseline number of stimulations per component delivered across all frequencies of brain stimulation. Abscissae: drug dose in mg/kg. Ordinates: Percent baseline total number of stimulations per component. All other details as described for Figure 1. All data show mean ± SEM for five to six rats. Statistical results for data in left panels are as follows: (A) 5.6% Acid effect (n = 5): significant main effect of frequency [F(9,36) = 83.3, P < 0.0001], treatment [F(1,4) = 17.0, P < 0.05], and significant frequency X treatment interaction [F(9,36) = 2.4, P < 0.05]. (B) Amitifadine effects determined at 100 min post-treatment (n = 6): significant main effect of frequency [F(9,36) = 24.2, P < 0.0001], no significant main effect of dose [F(2,8) = 2.4, P = 0.15], and a significant frequency X treatment interaction [F(18,72) = 3.7, P = < 0.0001].
Effects of amitifadine on control and acid-depressed NAc extracellular DA
Over the course of the microdialysis experiments the mean ± SEM baseline DA and 5HT levels within NAc were 1.82 ± 0.06 and 1.22 ± 0.03 pg/9μl, respectively. Amitifadine produced dose- and time-dependent increases in DA levels within NAc (Figure 4A). Vehicle and 1.0 mg/kg had no significant effect, but 3.2 and 10 mg/kg significantly increased DA levels beginning 40 min after injection, and DA remained elevated for 260 min (3.2 mg/kg) and 300 min (10 mg/kg) after injection. Amitifadine also significantly increased NAc 5HT levels (Figure 4B). Vehicle and 1.0 mg/kg amitifadine had no significant effect, but 3.2 and 10 mg/kg significantly increased 5HT beginning 30 to 40 min after injection. NAc 5HT remained elevated for 100–110 min following administration.
Figure 4.
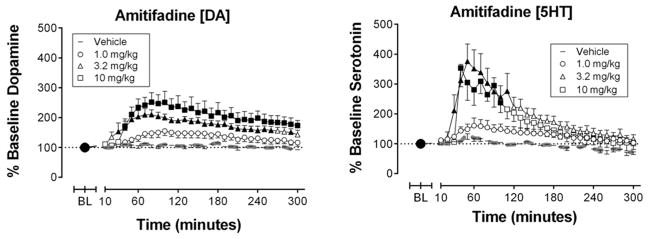
Effects of amitifadine on control levels of nucleus accumbens extracellular dopamine (left panel) and serotonin (right panel) in the absence of the noxious stimulus. Abscissae: Time in min after injection of amitifadine or vehicle. Ordinates: Percent baseline neurotransmitter levels. Filled points represent time points at which neurotransmitter levels were statistically different from vehicle levels as determined by a two-way ANOVA followed by a Holm–Sidak post hoc test, P < 0.05. All data show mean ± SEM for five to six rats. Statistical results for data in left panels are as follows: (A) Amitifadine effects on dopamine (n = 6): significant main effect of time [F(30,600) = 15.1, P < 0.01] and treatment [F(3,20) = 10.2, P < 0.001], and significant time X treatment interaction [F(90,300) = 2.5, P = < 0.01]. (B) Amitifadine effects on serotonin (n = 5–7): significant main effect of time [F(30,600) = 22.0, P < 0.001] and treatment [F(3,20) = 3.8, P < 0.05], and significant time X treatment interaction [F(90,300) = 3.6, P = < 0.001].
IP acid (5.6%) produced a time-dependent decrease in NAc DA with significant decreases first observed 70 min after injection of IP acid (Figure 5A). DA was significantly depressed for the remainder of the experiment except for the 260 and 300 min post-injection time points. Amitifadine (3.2 mg/kg) blocked IP acid-induced depression of NAc DA (Figure 5B). Specifically, in rats treated with 3.2 mg/kg amitifadine, NAc DA levels did not differ between animals that received IP acid vehicle and IP acid, and did not fall below baseline levels. IP acid had no significant effect on NAc 5HT levels in the absence or presence of amitifadine pretreatment (Figure S2).
Figure 5.
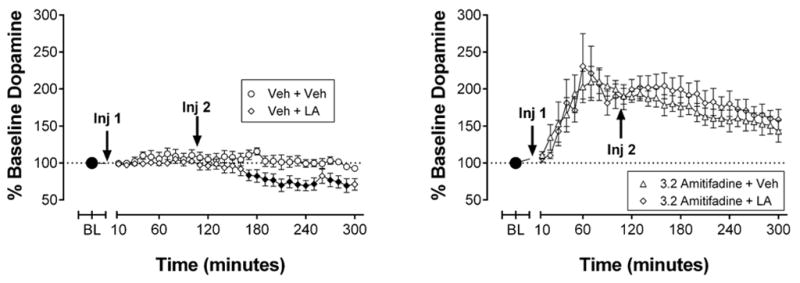
Effects of IP 5.6% acid on extracellular dopamine in the absence or presence of 3.2 mg/kg amitifadine. Abscissae: Time after injection of amitifadine or vehicle. Ordinates: Percent baseline neurotransmitter levels. “Inj 1” represents time of vehicle or amitifadine injection. “Inj 2” represents time of vehicle or acid injection. Filled points represent time points at which neurotransmitter levels were statistically different from vehicle levels as determined by a two-way ANOVA followed by a Holm–Sidak post hoc test, P < 0.05. All data show mean ± SEM for six rats. Statistical results for data in left panels are as follows: (A) 5.6% acid effects on dopamine levels after vehicle pretreatment (n = 6): significant main effect of time [F(20,200) = 8.8, P < 0.001], treatment [F(1,10) = 5.2, P < 0.05], and significant time X treatment interaction [F(20,200) = 3.1, P = < 0.001]. (B) 5.6% acid effects on dopamine levels after amitifadine pretreatment (n = 6): significant main effect of time [F(20,200) = 18.5, P < 0.001].
Effects of amitifadine on lactic acid-stimulated stretching
Figure 6 shows effects of amitifadine in the assay of lactic acid-stimulated stretching. Following pretreatment with saline, 1.8% lactic acid stimulated a stretching response. Pretreatment with amitifadine produced a dose-dependent decrease in acid-stimulated stretching, and this effect achieved statistical significance at a dose of 10 mg/kg amitifadine.
Figure 6.
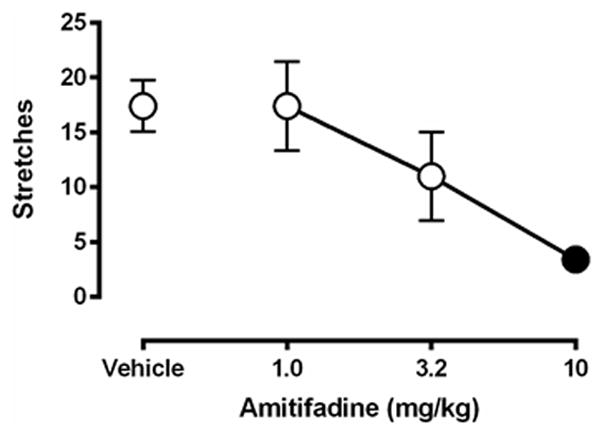
Effects of amitifadine on 1.8% acid-induced stretching. Abscissa: dose of amitifadine administered 30 min before lactic acid. Ordinate: number of stretches during 30-min observation period. Filled point represents dose at which stretching was significantly decreased compared to amitifadine vehicle pretreatment as determined by one-way ANOVA followed by Dunnetts test, p < 0.05. All data show mean ± SEM for five rats. One-way ANOVA indicated a significant effect of amitifadine [F(3,12) = 7.8, P < 0.005]
DISCUSSION
This study examined effects of amitifadine on pain-related depression of ICSS and NAc DA in rats. This work addressed the hypotheses that, based on its selectivity for blocking monoamine uptake (5HT≥NE≥DA), amitifadine would block pain-related depression of ICSS and NAc DA, and do so with minimal abuse-related effects. Amitifadine blocked pain-related depression of ICSS and NAc extracellular DA. Amitifadine also facilitated control ICSS, but this effect was modest compared to that of more DAT-selective monoamine uptake inhibitors [34], and was accompanied by effects which may be viewed as abuse-limiting [32]. Moreover, amitifadine was more potent to block pain-related depression of ICSS than to facilitate control ICSS or increase NAc DA.
Pain-related depression of ICSS and NAc DA
In the present study, IP acid served as a visceral chemical noxious stimulus that depressed ICSS. This agrees with previous studies of pain-related depression of behavior [5,6,16,24,29,30,42]. In particular, IP acid administration and other noxious stimuli depress ICSS [19,26,31,32], and studies examining drug effects on pain-depressed ICSS have yielded results that are largely consistent with the clinical analgesic efficacy of these drugs [24,25,31,33,34,38,40]. ICSS is used for research on the neurobiology of motivation and mood, and depression of ICSS is often interpreted as evidence of amotivational and anhedonic dimensions of depressed mood [13,48]. This suggests that acid-induced depression of ICSS may serve as one preclinical model of pain-related depression of behavior and mood [31].
The reinforcing effects of brain stimulation in ICSS are mediated in part by activation of the mesolimbic DA system [32], and depression of ICSS is often associated with depression of DA signaling. The present results confirmed our previous finding that IP acid can depress NAc DA release [25]. The results are also consistent with findings on the temporal relationship between acid-induced depression of ICSS and NAc DA release. IP acid produces an initial phase of declining DA followed by sustained significant reduction. Acid-induced depression of ICSS appears better correlated with the phase of declining DA levels than the sustained reduction. Similar temporal relationships have been described for effects of kappa agonists, which also depress NAc DA levels and ICSS [14,25]. One possible explanation for these findings is that the period of decline reflects reduced synaptic DA release, and the sustained reduction reflects slow recovery of extrasynaptic DA levels after recovery of synaptic DA release. These preclinical findings are consistent with data from humans suggesting that impaired mesolimbic DA signaling may contribute to some motivational and emotional aspects of pain [10,23,46,52], and support the hypothesis that promotion of DA signaling may alleviate motivational and emotional aspects of pain.
In addition to depressing ICSS and NAc DA, treatment with IP acid stimulated a stretching response in rats. This agrees with studies showing IP administration of acid or other irritants can elicit a stretching response in rodents, and this response is often used as a dependent measure for studies of antinociception [35]. One goal of the present study was to compare amitifadine effects on endpoints of pain-depressed vs. pain-stimulated behavior.
Amitifadine-induced antinociception
Amitifadine produced antinociception in the present study insofar as it blocked acid-induced depression of ICSS and NAc DA, and acid-induced stimulation of stretching. These results agree with findings that DA uptake inhibitors blocked both acid-induced depression of ICSS and stimulation of stretching [34,40]. For example, RTI-113 is a highly selective inhibitor of DAT, and a relatively low dose of RTI-113 was sufficient to block acid-induced depression of ICSS without altering control ICSS in the absence of the noxious stimulus [40]. In contrast, selective inhibitors of SERT or NET failed to block acid-induced depression of ICSS [39]. Together, these findings provide evidence to suggest that inhibition of DAT, and not SERT or NET, mediates amitifadine antinociception in the assay of acid-induced depression of ICSS.
In contrast to the effects of amitifadine in the present study, higher doses of the selective DAT inhibitor RTI-113 blocked acid-induced depression of ICSS, and also facilitated ICSS to levels that exceeded control rates. Moreover, higher RTI-113 doses also significantly facilitated ICSS in the absence of IP acid, and this type of effect is associated with high abuse potential [32]. Similar effects are produced by other drugs with prominent effects as DAT inhibitors, such as cocaine [34], methylphenidate (K. Freitas and S. Negus, unpublished observations) and bupropion [40]. This profile of effects is consistent with clinical findings that DAT inhibitors and DA releasers can produce analgesic effects in humans under some conditions, but therapeutic potential is limited by high abuse potential [50,53].
In comparison to effects of these DAT inhibitors, the profile of antinociceptive effects produced by amitifadine more closely resembled effects of the clinically effective analgesics morphine and ketoprofen. Morphine is a mu opioid receptor agonist that produces strong analgesia and also has abuse liability [21]. However, the abuse potential of morphine depends in part on a history of prior opioid exposure [2], and in opioid-naïve humans, morphine can produce analgesia without prominent abuse-related effects [17,55]. Similarly, in opioid-naïve rats, morphine can block acid-induced depression of both ICSS and NAc DA without producing an abuse-related increase in either ICSS or NAc DA in the absence of the noxious stimulus [25,38]. Ketoprofen is a clinically effective NSAID, and ketoprofen also blocks acid-induced depression of ICSS and NAc DA without producing abuse-related effects [25,34]. This similarity in effects of amitifadine to effects of two clinically effective analgesics supports the possibility that amitifadine may also have utility for treatment of pain.
The present study showed depression of ICSS by a noxious stimulus, but ICSS can also be depressed by other manipulations associated with negative affective states and behavioral depression, such as withdrawal from abused drugs [48]. The present finding that amitifadine blocked acid-induced depression of ICSS is consistent with a previous study showing that amitifadine reversed depression of ICSS produced by alcohol withdrawal in rats [49]. Amitifadine also produced antidepressant-like effects in preclinical screens of other depression-related behaviors [45], and in a clinical trial in patients with major depressive disorder, amitifadine was associated with improvement on multiple measures [47]. Together, these results suggest that amitifadine may have utility to alleviate depression of behavior and mood resulting not only from pain, but also from other causes.
Of the three pain assays used here, amitifadine was least potent in the assay of acid-stimulated stretching. Moreover, 10 mg/kg recruited ICSS rate-decreasing effects, which may indicate that antinociception in the stretching assay is in part a product of non-specific behavioral disruption rather than analgesia. The higher potency of amitifadine to block acid-induced depression of ICSS vs. acid-stimulated stretching is also consistent with effects produced in these procedures by other DA-selective uptake inhibitors and non-selective uptake inhibitors [34,40], and suggests that stimulation of DA transmission may be better for alleviating pain-depressed than pain-stimulated behaviors.
Amitifadine effects on control ICSS and NAc DA
Effects of amitifadine in the absence of the noxious stimulus are consistent with the characterization of amitifadine as a mixed monoamine uptake inhibitor with low abuse liability. In the microdialysis procedure, amitifadine significantly increased NAc DA levels, an effect produced by many abused drugs [15]. However, doses of amitifadine that increased NAc DA produced even larger increases in NAc 5HT levels. Similar results were obtained for effects of amitifadine on DA and 5HT levels in prefrontal cortex [20], and evidence suggests that 5HT can oppose and limit abuse-related effects of DA [22,32,36,51]. In the ICSS procedure, amitifadine doses up to 3.2 mg/kg had no effect on control ICSS despite the efficacy of 3.2 mg/kg amitifadine to alleviate acid-induced depression of ICSS. A higher dose of 10 mg/kg amitifadine did produce significant facilitation of low ICSS rates maintained by low frequencies of brain stimulation, and facilitation of ICSS has been linked to abuse potential [32]. However, the magnitude of this facilitation was small relative to effects of abused DAT inhibitors such as cocaine or methylenedioxypyrovalerone (MDPV) [9,34], and it was accompanied by decreases in high ICSS rates maintained by high brain stimulation frequencies. We have reported previously that this profile of mixed rate-increasing and rate-decreasing effects is associated with lower abuse liability than profiles of exclusive ICSS facilitation [8,32]. Moreover, a previous ICSS study with amitifadine reported no abuse-related effects [49]. Reinforcing effects of amitifadine have not been examined in drug self-administration procedures; however, the triple uptake inhibitor bicifadine, which has similar selectivity for blocking uptake of DA vs 5HT/NE to that of amitifadine, functioned as a weak reinforcer in non-human primates [36].
Conclusion
This study adds to a literature examining the expression, neurobiology, and treatment of pain-related depression of behavior that may aid in the development of new therapeutics. The findings replicate previous studies demonstrating the sensitivity of ICSS in detecting pain-related disruption of behavior, provide evidence of the role of mesolimbic dopamine systems in mediating these effects, and support the use of these types of assays in bridging knowledge gaps in the pain field. Moreover, these data suggest that triple uptake inhibitors may be efficacious in the treatment of pain-related depression of behavior, with minimal recruitment of abuse-related effects [40].
Supplementary Material
Coronal section depictions of rat brain showing the positions of microdialysis probes. Numbers to the left of the image indicate anterior-posterior positions relative to bregma.
Effects of IP 5.6% acid on extracellular serotonin in the absence or presence of 3.2 mg/kg amitifadine. Abscissae: Time after injection of amitifadine or vehicle. Ordinates: Percent baseline neurotransmitter levels. “Inj 1” represents time of vehicle or amitifadine injection. “Inj 2” represents time of vehicle or acid injection. Filled points represent time points at which neurotransmitter levels were statistically different from vehicle levels as determined by a two-way ANOVA followed by a Holm–Sidak post hoc test, P < 0.05. All data show mean ± SEM for six to seven rats. Statistical results for data in left panels are as follows: (A) 5.6% acid effects on serotonin levels after vehicle pretreatment (n = 6): significant main effect of time [F(20,200) = 8.8, P < 0.001], treatment [F(1,10) = 5.2, P < 0.05], and significant time X treatment interaction [F(20,200) = 3.1, P = < 0.001]. (B) 5.6% acid effects on dopamine levels after amitifadine pretreatment (n= 6–7): significant main effect of time [F(20,200) = 18.5, P < 0.001].
Acknowledgments
The authors acknowledge Ms. Christina Johnson and Dr. Matthew Lazenka for outstanding technical assistance.
Dr. Banks declares that his research has been funded by NIH. During the past 3 years, he has received compensation as a collaborator with the pharmaceutical companies Abbott and Purdue for projects related to opioid pharmacology and analgesic drug development. Dr. Banks declares that the present study was not related to this professional relationship and should not be perceived as constituting a potential conflict of interest.
Dr. Negus declares that his research has been funded by NIH. During the past 3 years, he has received compensation as a consultant for or collaborator with Alkermes Pharmaceutical Company for projects related to abuse liability assessment.
Research reported in this publication was supported by the National Institute on Neurological Disorders and Stroke and the National Institute on Drug Abuse of the National Institutes of Health under award numbers R01-NS070715 and F32-DA033920. In addition, Mr. Leitl was supported in part by T32-DA007027. Dr. Bough was supported by R01-DA12970.
Footnotes
Dr. Miller and Mr. Leitl declare no conflict of interest.
Dr. Blough declares no conflict of interest other than funding from the NIH.
References
- 1.Altarifi AA, Miller LL, Negus SS. Role of μ-opioid receptor reserve and μ-agonist efficacy as determinants of the effects of μ-agonists on intracranial self-stimulation in rats. Behav Pharmacol. 2012;23:678–692. doi: 10.1097/FBP.0b013e328358593c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altarifi AA, Negus SS. Some determinants of morphine effects on intracranial self-stimulation in rats: dose, pretreatment time, repeated treatment, and rate dependence. Behav Pharmacol. 2011;22:663–673. doi: 10.1097/FBP.0b013e32834aff54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amato D, Natesan S, Yavich L, Kapur S, Muller CP. Dynamic regulation of dopamine and serotonin responses to salient stimuli during chronic haloperidol treatment. Int J Neuropsychopharmacol. 2011;14:1327–1339. doi: 10.1017/S1461145711000010. [DOI] [PubMed] [Google Scholar]
- 4.Andrews JS, Wu N, Chen S-Y, Yu X, Peng X, Novick D. Real-world treatment patterns and opioid use in chronic low back pain patients initiating duloxetine versus standard of care. J Pain Res. 2013;6:825–835. doi: 10.2147/JPR.S50323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews N, Harper S, Issop Y, Rice ASC. Novel, nonreflex tests detect analgesic action in rodents at clinically relevant concentrations. Annals of the New York Academy of Sciences. 2011;1245:11–13. doi: 10.1111/j.1749-6632.2011.06342.x. [DOI] [PubMed] [Google Scholar]
- 6.Andrews N, Legg E, Lisak D, Issop Y, Richardson D, Harper S, Pheby T, Huang W, Burgess G, Machin I, Rice ASC. Spontaneous burrowing behaviour in the rat is reduced by peripheral nerve injury or inflammation associated pain. Eur J Pain. 2012;16:485–495. doi: 10.1016/j.ejpain.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacological Reviews. 2001;53:597. [PubMed] [Google Scholar]
- 8.Bauer C, Banks M, Blough B, Negus S. Use of intracranial self-stimulation to evaluate abuse-related and abuse-limiting effects of monoamine releasers in rats. British Journal of Pharmacology. 2013;168:850–862. doi: 10.1111/j.1476-5381.2012.02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonano JS, Glennon RA, Felice LJD, Banks ML, Negus SS. Abuse-related and abuse-limiting effects of methcathinone and the synthetic “bath salts” cathinone analogs methylenedioxypyrovalerone (MDPV), methylone and mephedrone on intracranial self-stimulation in rats. Psychopharmacology. 2014;231:199–207. doi: 10.1007/s00213-013-3223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borsook D, Becerra L, Carlezon WA, Jr, Shaw M, Renshaw P, Elman I, Levine J. Reward-aversion circuitry in analgesia and pain: implications for psychiatric disorders. Eur J Pain. 2007;11:7–20. doi: 10.1016/j.ejpain.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Bray GE, Ying Z, Baillie LD, Zhai R, Mulligan SJ, Verge VMK. Extracellular pH and neuronal depolarization serve as dynamic switches to rapidly mobilize trkA to the membrane of adult sensory neurons. J Neurosci. 2013;33:8202–8215. doi: 10.1523/JNEUROSCI.4408-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brennan F, Carr DB, Cousins M. Pain Management: A Fundamental Human Right. Anesth Analg. 2007;105:205–221. doi: 10.1213/01.ane.0000268145.52345.55. [DOI] [PubMed] [Google Scholar]
- 13.Carlezon WA, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protocols. 2007;2:2987–2995. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- 14.Carlezon WA, Jr, Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY-W, Cohen BM. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;316:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- 15.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cobos EJ, Ghasemlou N, Araldi D, Segal D, Duong K, Woolf CJ. Inflammation-induced decrease in voluntary wheel running in mice: A nonreflexive test for evaluating inflammatory pain and analgesia. PAIN. 2012;153:876–884. doi: 10.1016/j.pain.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conley KM, Toledano AY, Apfelbaum JL, Zacny JP. Modulating effects of a cold water stimulus on opioid effects in volunteers. Psychopharmacology (Berl) 1997;131:313–320. doi: 10.1007/s002130050298. [DOI] [PubMed] [Google Scholar]
- 18.Deval E, Gasull X, Noel J, Salinas M, Baron A, Diochot S, Lingueglia E. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther. 2010;128:549–558. doi: 10.1016/j.pharmthera.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Ewan EE, Martin TJ. Differential suppression of intracranial self-stimulation, food-maintained operant responding, and open field activity by paw incision and spinal nerve ligation in rats. Anesth Analg. 2014;118:854–862. doi: 10.1213/ANE.0000000000000119. [DOI] [PubMed] [Google Scholar]
- 20.Golembiowska K, Kowalska M, Bymaster FP. Effects of the triple reuptake inhibitor amitifadine on extracellular levels of monoamines in rat brain regions and on locomotor activity. Synapse. 2012;66:435–444. doi: 10.1002/syn.21531. [DOI] [PubMed] [Google Scholar]
- 21.Gutstein HB, Akil H. Opioid Analgesics. In: Parker KL, Brunton LL, Lazo JS, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill Professional; 2005. [Google Scholar]
- 22.Howell LL. Nonhuman primate neuroimaging and cocaine medication development. Exp Clin Psychopharmacol. 2008;16:446–457. doi: 10.1037/a0014196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarcho JM, Mayer EA, Jiang ZK, Feier NA, London ED. Pain, affective symptoms, and cognitive deficits in patients with cerebral dopamine dysfunction. PAIN. 2012;153:744–754. doi: 10.1016/j.pain.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwilasz AJ, Negus SS. Dissociable effects of the cannabinoid receptor agonists Δ9-tetrahydrocannabinol and CP55940 on pain-stimulated versus pain-depressed behavior in rats. J Pharmacol Exp Ther. 2012;343:389–400. doi: 10.1124/jpet.112.197780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leitl MD, Onvani S, Bowers MS, Cheng K, Rice KC, Carlezon WA, Banks ML, Negus SS. Pain-Related Depression of the Mesolimbic Dopamine System in Rats: Expression, Blockade by Analgesics, and Role of Endogenous Kappa Opioids. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2013.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leitl MD, Potter DN, Cheng K, Rice KC, Carlezon WA, Jr, Negus SS. Chronic pain-related depression of behavior: effects of intraplantar formalin and complete Freund’s adjuvant on intracranial self-stimulation (ICSS) and endogenous kappa opioid biomarkers in rats. Molecular Pain. doi: 10.1186/1744-8069-10-62. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev. 2014;1:CD007115. doi: 10.1002/14651858.CD007115.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marinelli S, Pascucci T, Bernardi G, Puglisi-Allegra S, Mercuri NB. Activation of TRPV1 in the VTA excites dopaminergic neurons and increases chemical- and noxious-induced dopamine release in the nucleus accumbens. Neuropsychopharmacology. 2005;30:864–870. doi: 10.1038/sj.npp.1300615. [DOI] [PubMed] [Google Scholar]
- 29.Martin TJ, Buechler NL, Kahn W, Crews JC, Eisenach JC. Effects of laparotomy on spontaneous exploratory activity and conditioned operant responding in the rat: a model for postoperative pain. Anesthesiology. 2004;101:191–203. doi: 10.1097/00000542-200407000-00030. [DOI] [PubMed] [Google Scholar]
- 30.Miller LL, Picker MJ, Umberger MD, Schmidt KT, Dykstra LA. Effects of alterations in cannabinoid signaling, alone and in combination with morphine, on pain-elicited and pain-suppressed behavior in mice. J Pharmacol Exp Ther. 2012;342:177–187. doi: 10.1124/jpet.112.191478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Negus SS. Expression and treatment of pain-related behavioral depression. Lab Anim (NY) 2013;42:292–300. doi: 10.1038/laban.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Negus SS, Miller LL. Intracranial Self Stimulation to Evaluate Abuse Potential of Drugs. Pharmacological Reviews. 2014 doi: 10.1124/pr.112.007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Negus SS, Morrissey EM, Rosenberg M, Cheng K, Rice KC. Effects of kappa opioids in an assay of pain-depressed intracranial self-stimulation in rats. Psychopharmacology (Berl) 2010;210:149–159. doi: 10.1007/s00213-009-1770-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Negus SS, O’Connell R, Morrissey E, Cheng K, Rice KC. Effects of peripherally restricted κ opioid receptor agonists on pain-related stimulation and depression of behavior in rats. J Pharmacol Exp Ther. 2012;340:501–509. doi: 10.1124/jpet.111.186783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical Assessment of Candidate Analgesic Drugs: Recent Advances and Future Challenges. J Pharmacol Exp Ther. 2006;319:507–514. doi: 10.1124/jpet.106.106377. [DOI] [PubMed] [Google Scholar]
- 36.Nicholson KL, Balster RL, Golembiowska K, Kowalska M, Tizzano JP, Skolnick P, Basile AS. Preclinical Evaluation of the Abuse Potential of the Analgesic Bicifadine. J Pharmacol Exp Ther. 2009;330:236–248. doi: 10.1124/jpet.109.150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Amsterdam; Boston: Elsevier; 2007. [Google Scholar]
- 38.Pereira Do Carmo G, Stevenson GW, Carlezon WA, Negus SS. Effects of pain- and analgesia-related manipulations on intracranial self-stimulation in rats: further studies on pain-depressed behavior. Pain. 2009;144:170–177. doi: 10.1016/j.pain.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ritz MC, Lamb RJ, Goldberg, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg MB, Carroll FI, Negus SS. Effects of Monoamine Reuptake Inhibitors in Assays of Acute Pain-Stimulated and Pain-Depressed Behavior in Rats. The Journal of Pain. 2013;14:246–259. doi: 10.1016/j.jpain.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 42.Rutten K, Robens A, Read Sj, Christoph T. Pharmacological validation of a refined burrowing paradigm for prediction of analgesic efficacy in a rat model of sub-chronic knee joint inflammation. European Journal of Pain. 2014;18:213–222. doi: 10.1002/j.1532-2149.2013.00359.x. [DOI] [PubMed] [Google Scholar]
- 43.Sawynok J, Esser MJ, Reid AR. Antidepressants as analgesics: an overview of central and peripheral mechanisms of action. J Psychiatry Neurosci. 2001;26:21–29. [PMC free article] [PubMed] [Google Scholar]
- 44.Sinatra R. Causes and Consequences of Inadequate Management of Acute Pain. Pain Medicine. 2010;11:1859–1871. doi: 10.1111/j.1526-4637.2010.00983.x. [DOI] [PubMed] [Google Scholar]
- 45.Skolnick P, Popik P, Janowsky A, Beer B, Lippa AS. Antidepressant-like actions of DOV 21,947: a “triple” reuptake inhibitor. Eur J Pharmacol. 2003;461:99–104. doi: 10.1016/s0014-2999(03)01310-4. [DOI] [PubMed] [Google Scholar]
- 46.Tiemann L, Heitmann H, Schulz E, Baumkotter J, Ploner M. Dopamine precursor depletion influences pain affect rather than pain sensation. PLoS ONE. 2014;9:e96167. doi: 10.1371/journal.pone.0096167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran P, Skolnick P, Czobor P, Huang NY, Bradshaw M, McKinney A, Fava M. Efficacy and tolerability of the novel triple reuptake inhibitor amitifadine in the treatment of patients with major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Psychiatr Res. 2012;46:64–71. doi: 10.1016/j.jpsychires.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 48.Vlachou S, Markou A. Intracranial Self-Stimulation. In: Olmstead MC, editor. Animal Models of Drug Addiction. Neuromethods. Humana Press; 2011. [Accessed 15 Feb 2013]. pp. 3–56. Available: http://link.springer.com/protocol/10.1007/978-1-60761-934-5_1. [Google Scholar]
- 49.Warnock KT, Yang ARST, Yi HS, June HL, Jr, Kelly T, Basile AS, Skolnick P, June HL. Amitifadine, a triple monoamine uptake inhibitor, reduces binge drinking and negative affect in an animal model of co-occurring alcoholism and depression symptomatology. Pharmacology Biochemistry and Behavior. 2012;103:111–118. doi: 10.1016/j.pbb.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Webb SS, Smith GM, Evans WO, Webb NC. Toward the development of a potent, nonsedating, oral analgesic. Psychopharmacology (Berl) 1978;60:25–28. doi: 10.1007/BF00429174. [DOI] [PubMed] [Google Scholar]
- 51.Wee S, Anderson KG, Baumann MH, Rothman RB, Blough BE, Woolverton WL. Relationship between the serotonergic activity and reinforcing effects of a series of amphetamine analogs. J Pharmacol Exp Ther. 2005;313:848–854. doi: 10.1124/jpet.104.080101. [DOI] [PubMed] [Google Scholar]
- 52.Wood PB. Role of central dopamine in pain and analgesia. Expert Rev Neurother. 2008;8:781–797. doi: 10.1586/14737175.8.5.781. [DOI] [PubMed] [Google Scholar]
- 53.Yang JC, Clark WC, Dooley JC, Mignogna FV. Effect of intranasal cocaine on experimental pain in man. Anesth Analg. 1982;61:358–361. [PubMed] [Google Scholar]
- 54.Young AMJ. Increased extracellular dopamine in nucleus accumbens in response to unconditioned and conditioned aversive stimuli: studies using 1 min microdialysis in rats. Journal of Neuroscience Methods. 2004;138:57–63. doi: 10.1016/j.jneumeth.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 55.Zacny JP, Lichtor SA. Within-subject comparison of the psychopharmacological profiles of oral oxycodone and oral morphine in non-drug-abusing volunteers. Psychopharmacology (Berl) 2008;196:105–116. doi: 10.1007/s00213-007-0937-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coronal section depictions of rat brain showing the positions of microdialysis probes. Numbers to the left of the image indicate anterior-posterior positions relative to bregma.
Effects of IP 5.6% acid on extracellular serotonin in the absence or presence of 3.2 mg/kg amitifadine. Abscissae: Time after injection of amitifadine or vehicle. Ordinates: Percent baseline neurotransmitter levels. “Inj 1” represents time of vehicle or amitifadine injection. “Inj 2” represents time of vehicle or acid injection. Filled points represent time points at which neurotransmitter levels were statistically different from vehicle levels as determined by a two-way ANOVA followed by a Holm–Sidak post hoc test, P < 0.05. All data show mean ± SEM for six to seven rats. Statistical results for data in left panels are as follows: (A) 5.6% acid effects on serotonin levels after vehicle pretreatment (n = 6): significant main effect of time [F(20,200) = 8.8, P < 0.001], treatment [F(1,10) = 5.2, P < 0.05], and significant time X treatment interaction [F(20,200) = 3.1, P = < 0.001]. (B) 5.6% acid effects on dopamine levels after amitifadine pretreatment (n= 6–7): significant main effect of time [F(20,200) = 18.5, P < 0.001].