

Abstract
The environment can influence human health and disease in many harmful ways. Many epidemiological studies have been conducted with the aim of elucidating the association between environmental exposure and human disease at the molecular and pathological levels, and such associations can often be through induced epigenetic changes. One such mechanism for this is through environmental factors increasing oxidative stress in the cell, and this stress can subsequently lead to alterations in DNA molecules. The two cellular organelles that contain DNA are the nucleus and mitochondria, and the latter are particularly sensitive to oxidative stress, with mitochondrial functions often disrupted by increased stress. There has been a substantial increase over the past decade in the number of epigenetic studies investigating the impact of environmental exposures upon genomic DNA, but to date there has been insufficient attention paid to the impact upon mitochondrial epigenetics in studying human disease with exposure to environment.
Here, in this review, we will discuss mitochondrial epigenetics with regards to epidemiological studies, with particular consideration given to study design and analytical challenges. Furthermore, we suggest future directions and perspectives in the field of mitochondrial epigenetic epidemiological studies.
Keywords: Mitochondria, epigenetics, epidemiology, DNA methylation, disease
Why mitochondrial epigenetics?
The environment plays a critical role in human health and disease. Environments hazardous to human health can range from natural conditions to pollutants generated by human activities, e.g. those associated with the air, water, temperature, and food. Humans are exposed to a multitude of environmental pollutants at differing intensities during their lifetimes, and the list of diseases linked to different types of pollutants is continuously being updated. Individuals have different susceptibilities to these environmental exposures; a part of the general population may be protected from their effects, or only develop mild symptoms, while a variable proportion may go on to present severe disease phenotypes.
Environmental pollutants may cause damage at the molecular level in the cell, which can disrupt cellular function. The most widely studied impact of environmental exposures is that of DNA damage. There are two cellular organelles that contain DNA: the nucleus, which contains genomic DNA; and the mitochondria, each of which contains multiple copies of its own genome of approximately 16kb in length. Mitochondria play an important role in the cellular response to environmental stressors (Manoli et al. 2007). Damaged mitochondrial DNA (mtDNA) co-exists with normal mtDNA copies in cells, and the influence of mtDNA mutations can range between normal, mild and severe according to the proportion of abnormal mtDNA copies. The proportion of these diseased mitochondria that are passed on from mother to child is random, and this is referred to as the mitochondrial ‘bottle neck’ (Figure 1). If the child inherits fewer diseased than normal mitochondria from the mother, disease symptoms may not be appear in the child. Non-Mendelian mitochondrial diseases can be maternally inherited, but can also be acquired in later life.
Figure 1. Lifetime environmental exposures and mitochondria-related disease.

Human mitochondria are maternally inherited, leading to the ‘bottleneck’ theory. As humans are exposed to the environment throughout their lifetime, the onset of diseases related to mitochondrial function can be at any stage, and with different phenotypes. M: maternal, P: paternal, C: Child, COPD: Chronic Obstructive Pulmonary Disease
There is growing evidence to support the hypothesis that mitochondrial dysfunction can be caused by environmental pollutants that are known to contribute to common human diseases, and the symptoms of such diseases may become more severe with chronic exposure. Mitochondrial oxidative damage, DNA copy number and DNA mutations have been widely studied in relation to environmental exposure and disease outcomes, but many mitochondrial diseases cannot be fully understood solely by genetic studies due to their non-Mendelian inheritance. Epigenetic elements can regulate gene expression without changing the DNA sequence. Unlike the ‘fixed’ genetic code, epigenetics can be reversibly modified by internal and external cellular stimuli including but not limited to pollutants, oxidative stress, temperature, nutrients, UV, and aging. It is therefore clear that more focus is required upon mitochondrial epigenetics in order to understand the development of non-Mendelian mitochondrial dysfunction in response to the environmental exposures that are implicated in disease.
Existence of mitochondrial epigenetics: End of controversy and beginning of functional studies
The mitochondrial genome differs from the nuclear in several key ways. Of particular relevance to epigenetic studies is that it contains fewer CpG dinucleotides than genomic DNA, with only 435 present in its 16kb length (Figure 2). These are almost evenly dispersed, lacking the conventional ‘CpG islands’ that are found in genomic DNA. Furthermore, the mitochondrial genome lacks retrotransposons, such as LINE-1 and Alus, that compromise 40% of the human nuclear genome.
Figure 2.
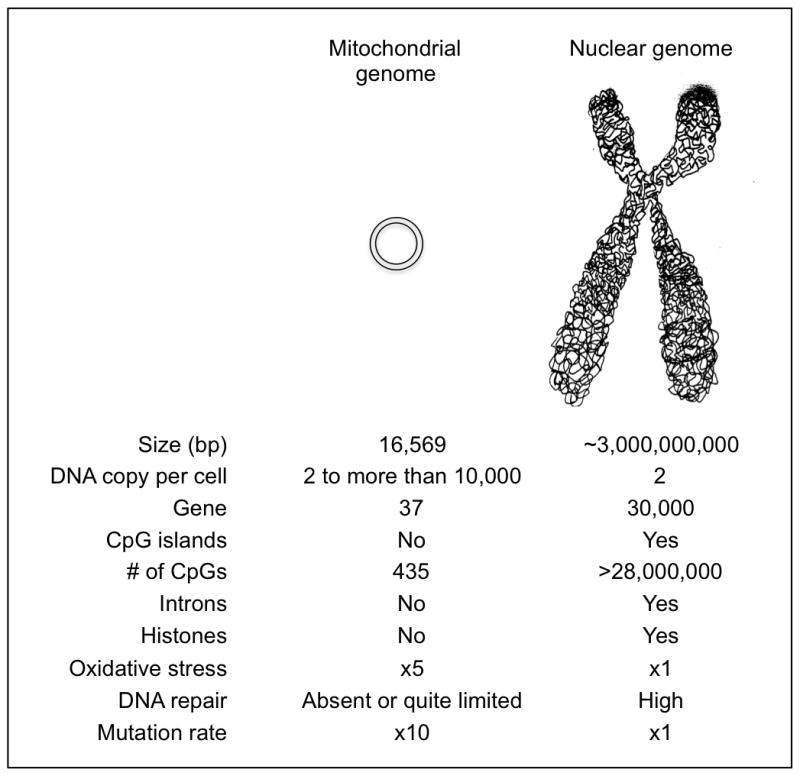
Comparison between mitochondrial genome and nuclear genome
In 1971, Belozersky et al. published the first report on mtDNA methylation, as well of associated DNA methyltransferase (DNMT) activity, using loach embryos. The authors reported that mtDNA was less methylated than nuclear DNA, and they also showed evidence for the presence of DNMT activity that could be distinguished from other cellular DNMT activity (Nass 1973). Vaniushin et al. and Nass et al. similarly identified evidence to suggest the presence of a mitochondrial DNMT that differs from the nuclear ones, including the nature of recognized DNA sequences (Kudriashova et al. 1976). While other groups have also reported mtDNA methylation in humans (Shmookler Reis and Goldstein 1983) and in other mammals (Mushkambarov et al. 1976), the existence of mtDNA methylation has been far more controversial than that of genomic DNA methylation, even after the discovery of the mitochondrial DNA methyltransferase 1 (mtDNMT1) enzyme by Shock et al. For instance, Hsieh et al. reported no evidence of CpG methylation in human mitochondrial DNA by adopting both regionally-specific and genome-wide analyses (Hong et al. 2013).
Although a recent report suggested the existence of several histone family members in the mitochondria (Choi et al. 2011), the general consensus is that of an absence of histone complexes in mitochondria. Such a lack of histone complexes would further suggest an important role for DNA methylation in mtDNA stability and mitochondrial function. Mitochondrial DNA methylation, by means of 5-methylcytosine and 5-hydroxymethylcytosine, has been increasingly well established and has led to several disease related studies (Iacobazzi et al. 2013), such as those into neuroepigenetics (Chestnut et al. 2011; Manev et al. 2012), aging (Dzitoyeva et al. 2012), and cervical cancer (Sun et al. 2011). While Kanno et al. indicated that mtDNA methylation is not useful as a marker for the detection of cancer due to the lack of methylated DNA (Maekawa et al. 2004), Yang et al. found that decreased methylation on the D-loop promoter area plays a key role in regulating NADH dehydrogenase 2 (ND2) expression during the initiation and/or progression of colorectal cancer (Feng et al. 2012). Control regions in the D-loop in mitochondrial DNA show unusual CpG and non-CpG methylation patterns (Bellizzi et al. 2013). Studies using mouse embryonic stem cells have revealed that non-CpG hydroxymethylation is prevalent in the mitochondrial genome (Sun et al. 2013). However, epigenetic regulation of mtDNA is not limited to DNA methylation, as pre-microRNAs and mature microRNAs have been found in human mitochondria (Barrey et al. 2011). Their role in regulating the expression of mitochondrial genes is yet to be established.
The role of mitochondrial DNA methylation has been suspected due to the dependency of mitochondria upon the nuclear genome (Iacobazzi et al. 2013; Leigh-Brown et al. 2010). Recent studies have shown that mtDNA methylation, in a fashion similar to nuclear DNA (ncDNA) methylation, may have a role in controlling the expression of mitochondrially-encoded genes. Feng et al. observed an inverse correlation between mitochondrial MT-ND2 expression and methylation of its promoter sequence in mtDNA in cases of colorectal cancer (Feng et al. 2012). Similarly, methylation of the NADH dehydrogenase 6 (MT-ND6) that is encoded in the mitochondrial genome has also been demonstrated to be inversely correlated with expression of the gene in nonalcoholic fatty liver disease patients (Pirola et al. 2013). However, there is a need for functional studies to elucidate how mtDNA methylation may be implicated in controlling gene expression.
Mitochondrial activity varies by tissue type. For example, the brain and heart are tissues with high demands for biological energy (Chan 2006), and subsequently they are expected to have different levels of mitochondrial activity and differential expression of mitochondrial genes. Therefore, it would be informative to assess the tissue-differential methylation patterns in mtDNA. It has been reported that mtDNA methylation differs by developmental stage in mice, where mtDNA is unmethylated in blastocysts and embryonic stem cells (ESCs) but is partially methylated in germ cells (Kobayashi et al. 2012), suggesting a possible role for mtDNA methylation in early developmental processes.
Common diseases associated with mitochondria
Mitochondria can be found in every cell type in the human body, with the exception of red blood cells. Most nucleated cells in tissue, and even some anucleated cells, contain 500 to 2000 mitochondria. The size of a mitochondrion is only 0.5 to 1.0 micrometer (μm) in diameter, but the number of mitochondria in particular cell types is sufficiently high that they can fill up to 40% of the cytoplasmic volume in heart muscle, 60% of the volume in lateral rectus, and up to 80% in the cone cell photoreceptors of the eye.
Mitochondrial science is one of the fastest growing fields in genetics and clinical studies, connecting research areas ranging from early embryo development to aging (Dumollard et al. 2007; Lee and Wei 2012). Alterations in mitochondrial metabolism are known to play a role in rare childhood diseases (Borchert et al. 2002; Shrikhande et al. 2010), but they have has also been implicated in many other more common diseases (Vafai and Mootha 2012). In particular, there has been much research into the role of mitochondrial function in neuronal diseases, such as Alzheimer’s disease (Sultana et al. 2013), Parkinson’s disease (Mortiboys et al. 2013), Huntington’s disease (Stuwe et al. 2013), amyotrophic lateral sclerosis (ALS) (Pickles et al. 2013), and mental retardation (Haddad et al. 2013). It is also well known that mitochondrial dysfunction is associated with metabolic syndrome (James et al. 2012), although the underlying molecular mechanisms are not fully elucidated. Risk factors and components related to metabolic syndrome such as insulin resistance (Kim et al. 2008), hypertension (Puddu et al. 2007), abnormal lipid levels (Vamecq et al. 2012), impaired glucose tolerance (Mathews et al. 1999), age (Conley et al. 2007), and diabetes (Newsholme et al. 2012) are all also related to mitochondrial dysfunction. Mitochondrial dysfunction has also been implicated in vascular inflammation (Yu et al. 2012), atherosclerosis (Madamanchi and Runge 2007), renal and liver diseases (Grattagliano et al. 2011; Sharma et al. 2013), cardiovascular disease (Griffiths 2012), stroke (Martinez-Fernandez et al. 2001), autoimmune diseases – such as multiple sclerosis, Sjogrens syndrome, lupus and rheumatoid arthritis – as well as deafness (Yelverton et al. 2013), blindness (Schrier and Falk 2011) and a wide range of solid tumors (Kroemer 2006) and the aging process (Seo et al. 2010). The impact of mitochondrial dysfunction is more severe in tissues that demand high cellular energy (Chan 2006), such as the brain, heart, and muscle. The disease phenotypes do not occur until the proportion of mtDNA mutations reaches a threshold that varies by each mutation (Taylor and Turnbull 2005). However, many mitochondrial diseases do not follow Mendelian inheritance and so cannot be completely explained by genomic study. Indeed, only approximately 15 % of known mitochondrial diseases are thought to occur through mitochondrial DNA sequence (Dimauro and Davidzon 2005). There is therefore increasing interest in the role of mitochondrial epigenetics in common diseases associated with mitochondrial dysfunction.
Environmental toxicants and the mitochondria
There are an abundance of sources of environmental and cellular oxidative stress. Oxidative stress occurs in a condition when the production of reactive oxygen species is greater than the intrinsic antioxidant defenses (Burton and Jauniaux 2011). Mitochondria are implicated in such oxidative stress, and therefore environmental toxicants have often been studied directly or indirectly with mitochondrial function in relation to the human diseases that they are associated with. Oxidative stress in mitochondria can be generated from environmental pollutants, smoke, xenobiotics, temperature, and many other factors, and the sensitivity or toxicity of these pollutants varies in mitochondria and also between people. Furthermore, reactive oxygen species (ROS) are produced in the mitochondria as part of the normal cellular metabolism process involved in the production of cellular energy (ATP), and the accumulating ROS can damage mitochondria structure and function. Mitochondria are particularly vulnerable to oxidative stress, being more than five times more sensitive to such stresses than the nucleus in terms of accumulating DNA damage.
There are many reports regarding the impact of exposure to environmental toxicants upon mitochondrial function. Cadmium (Cd) is a toxic metal and an important environmental pollutant that can cause mitochondrial malfunction, particularly in reducing mitochondrial efficiency and ATP production (Kurochkin et al. 2011). Dibenzofuran (DBF) is a common environmental pollutant that bioaccumulates and is toxic to humans, and exposure to DBF has been demonstrated to alter mitochondrial permeability and may cause mitochondrial dysfunction (Duarte et al. 2013). Bisphenol A (BPA) is widely used in plastic products, and BPA-induced mitochondrial defects observed in β cells include the depletion of ATP, the release of cytochrome c, loss of mitochondrial mass, and loss of the regulation of membrane potential (Lin et al. 2013). Exposure to ambient particle matter (PM) has been association with changes in mitochondrial DNA copy number in steel workers (Byun et al. 2013), while exposure of cells to ionizing radiation has been shown to cause decreased activity of NADH dehydrogenase (Complex I), which is an important enzyme for regulating the release of ROS.
Oxidative stress from exposure to environmental toxicants has been widely studied in regard to DNA damage, such as DNA mutations and lesions in both the nucleus and mitochondria. Epigenetic factors such as DNA methylation, histone modifications and expression of micro RNAs (miRNAs) have also been extensively studied in the nucleus, but not yet in mitochondria. Since oxidative stresses in the nucleus have been shown to modify nuclear DNA methylation, many environmental toxicants might be potential candidates for study with relevance to mitochondrial epigenetics. Recent work has demonstrated an effect of air pollutants on mtDNA methylation, which was hypothesized to occur through oxidative stress (Byun et al. 2013). However, much further work is required to elucidate the effects of environmental exposures upon mitochondrial epigenetics.
Mitochondrial epigenetics and Epidemiology study design
1) Define the question
We have thus far described how the growing field of mitochondrial epigenetics may help to explain the role of mitochondrial dysfunction in a range of human diseases, and how environmental exposures may be implicated in this process. So, what issues must be addressed when designing an epidemiological study to investigate the role of mitochondrial epigenetics in human health and disease? There are perhaps four major questions that must be answered as part of the design process. Firstly, a clear hypothesis must be in place regarding the role of mitochondria in the etiology of the disease, and so we must answer the question of how mitochondria might be implicated in cell death or disease etiology, or how they contribute to a particular metabolic pathway. For example, it may be hypothesized that altered mitochondrial function may lead to increased oxidative stress and subsequently to neuronal cell death that could be implicated in the development of neurodegenerative disease. Secondly, we must ask whether environmental exposures or other risk factors could lead to the epigenetic modification of mtDNA. Is there a clear biological process by which the exposures could impact upon the methylation of mtDNA? This could be through direct or indirect mechanisms, such as increased oxidative stress inducing mutations that eliminate CpG sites or increasing DNMT activity with oxidative stress. Thirdly, could such epigenetic modifications be implicated in the development of disease? This could be through aberrant regulation of mitochondrial gene expression, which may in turn lead to altered mitochondrial function by means of reduced capacity to deal with oxidative stress. Finally, the study must be performed in a tissue that is relevant to the disease or to the risk of its development. We must ask whether blood can act as a suitable surrogate tissue. The use of blood is obviously advantageous for epidemiological studies, and it is not always possible to obtain the primary tissue of interest, such as when studying neurodegenerative diseases, but changes in mitochondrial function may not necessarily be seen in blood or other readily available tissues.
Once the hypothesis has been built based upon the principal issues above, a study design can be developed for the questions to be addressed (Figure 3).
Figure 3.

Epidemiology Design of Mitochondrial Epigenetic Studies
Cross-sectional study
Measurement of mitochondrial epigenetic levels at a single time point in a well-defined population to study the association between risk factors and disease mediated by mitochondrial epigenetic level.
Cohort study
The use of cohorts is particularly suitable for examining environmental exposures and risk factors in relation to mitochondrial epigenetic changes. When follow-ups are performed at different time points, longitudinal mitochondrial epigenetic changes can be studied. This is of particular relevance to studies into aging, response to chronic environmental exposure, and biomarkers of disease for screening purposes.
Nested case-control study
Similarly to cohort studies, this study design is ideal for mitochondrial epigenetic marks that give predisposition to disease or that could serve as biomarkers for disease risk, as data on exposure and predisease state are more likely to have been collected in advance to the diagnosis of disease than in conventional case-control studies. This design provides cost saving compared to cohort studies by concentrating resources on individuals that are most informative, i.e. the cases with the disease of interest and a set of appropriate controls.
Intervention study
The epigenetic modification of mtDNA occurs in a dynamic faction and is most likely to be reversible with environmental stimuli. Thus, they are susceptible to pharmacological therapies. In vitro study has demonstrated the effect of treatment of cultured cells with Valproic acid (valproate) on mtDNA methylation levels after three days of treatment (Chen et al. 2012). The use of DNMT enzyme inhibitors, such as Azacytidine or Decitabine, is clinically approved for the treatment of leukemias and some solid cancers. It could be beneficial if in vitro systems could demonstrate alterations in mtDNA methylation in response to treatment with DNMT inhibitors, as it may suggest that such therapies could be of use in mitochondrial diseases. There are currently no pharmaceutical treatments for any mitochondrial disease (Nunnari and Suomalainen 2012).
Birth cohort
Studies of this type are required to understand how preconceptional and prenatal exposures may affect mitochondrial epigenetics. Mitochondria are inherited only from the maternal side, but comparisons between maternal and fetal mtDNA methylation have not yet been made. It could potentially be very interesting, and a source of much further research and discussion, should differential mitochondrial epigenetic patterns be observed from mother to fetus, and to infant (Figure 1).
Twin study
Monozygotic twins have the same genetic background in nuclear, but epigenetic patterns show variation (Fraga et al. 2005). It could therefore be of interest to study differential mitochondrial epigenetic patterns between discordant disease pairs, especially in neuronal cells with regard to cognitive activity.
Migrant study
Mitochondrial DNA is considered as the ‘genetic Eve’, originating in Africa and following human migration (Lewin 1987). Evidence supporting this theory includes the presence of unique mitochondrial haplotypes in each ethnicity (van Oven and Kayser 2009). Therefore, it is reasonable to think that these different mitochondrial epigenetic patterns may play a role in explaining the discrepancies in disease susceptibility and phenotypes between ethnicities.
2) Relevant tissues for studying mitochondrial epigenetics
Most epigenetic epidemiological studies have used DNA from blood, nasal epithelial cells or buccal swabs for studying epigenetic patterns in response to environmental exposures. This is predominantly due to the non-invasive nature and accessibility of these tissues from healthy individuals. While leukocytes commonly contain about 20~30 mitochondria per cell, erythrocytes do not contain mitochondria (Zhang et al. 2011) and other cellular organelles, and nasal epithelial cells similarly contain few mitochondria (Coene et al. 2005; Hansen 2007). Buccal cell mitochondrial DNA has been utilized for the study of mitochondrial dysfunction in children and has shown the potential for use with screening for mitochondrial disease (Yorns et al. 2012).
Indeed, the number of mitochondria varies greatly among different cell types, depending upon how much cellular energy is required for the metabolism of the cell. Human sperm contains 100 to 1000 mitochondria per cell (which degrade in the fertilized egg), fibroblasts contain a few hundred, neurons may contain thousands, and cardiomyocytes tens of thousands. Epigenetic patterns in nuclear DNA are highly tissue-specific and therefore reflect cell-type specific signatures, and it is easy to presume that there will be even more substantial tissue-differential mitochondrial epigenetic patterns on account of the differing numbers of mitochondria and rates of cellular metabolism. The use of peripheral blood, which contains a heterogeneous mix of cell types, could therefore give rise to confounding as the proportions of the different cell types may differ between individuals and time points. Therefore, careful analytical approaches are required to avoid this bias. A method has been established to adjust for the proportions of the most prevalent cell types in blood, but this requires counts of cell compositions from each individual in fresh blood samples, which might be an issue for epidemiological studies. Although, Houseman et al. developed an analytical method for inferring changes in the distribution of white blood cells between different subpopulations using DNA methylation signatures (Houseman et al. 2012), the mitochondrial epigenome is more problematic in this regard, as each cell type may contain different numbers of mitochondria. It is hard to adjust for the number of each cell type and also for the number of mitochondria per cell. It is therefore necessary to obtain a homogenous cell population for use as a surrogate marker to make for a more sophisticated epigenetic epidemiological study.
3) Measurement of Mitochondrial Epigenetics
Study by molecular type: mtDNA methylation
Current mitochondrial epigenetic studies are limited to measuring 5-methylcytosine and 5-hydroxymethycytosine at CpG or non-CpG sites in mtDNA, by techniques that are very similar to those used with studies of nuclear DNA. In fact, many techniques for studying methylation of ncDNA have been adapted for use with mtDNA, such as immunoprecipitation (Shock et al. 2011), mass spectrometry (Infantino et al. 2011; Song et al. 2005), pyrosequencing (Byun et al. 2013), and bisulfite genomic sequencing (Hong et al. 2013).
The first human studies on the effects of environmental pollutants on mtDNA methylation showed that exposure to air pollutants significantly affected mtDNA methylation levels (Byun et al. 2013). The study was performed using DNA from buffy coat cells, extracted by a conventional method for isolating DNA that resulted in a mixture of ncDNA and mtDNA from a variety of cell types. There are a number of ongoing epidemiological studies that have taken the same approach and therefore face the same issue. The problem of using a mixture of ncDNA and mtDNA is that there are many homologous DNA sequences in the mitochondrial genome and in ncDNA, due to the evolutionary origins of mitochondria (Hazkani-Covo et al. 2010). These nuclear mitochondrial DNA (numt) sequences are often confused with those originating from mtDNA and may therefore affect studies investigating the methylation of mitochondrial DNA. Therefore, future studies may need to consider the issue of contamination with numts. This analytical challenge will be discussed in more detail in the next section. However, Shock et al. have recently reported on mtDNA methylation and hydroxymethylation by using an extract of pure mtDNA following the isolation of mitochondria from a human colon cancer cell line. The study reported a low but considerable level of methylation in mtDNA (Shock et al. 2011).
Study by molecular type: RNA modification
The mammalian mitochondrial genome encodes 13 mRNAs for genes involved in oxidative phosphorylation, 2 ribosomal RNAs and 22 transfer RNAs (Table 1). Mitochondrial RNA exists as 5 % of total cellular RNA in most tissues but can be as high as 30% in myocardial cells that have an enrichment of mitochondria (Mercer et al. 2011). The post-transcriptional mechanisms that control mRNA maturation, stability and translation are critical in controlling gene expression (Rorbach and Minczuk 2012). Unlike DNA methylation, RNA methylation varies in terms of the atoms, nucleotides, sequences and tertiary structures involved. Most RNA species, including transfer RNA (tRNA), ribosomal RNA (rRNA), messenger RNA (mRNA), transfer-messenger RNA (tmRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), microRNA (miRNA), and viral RNA, can be methylated (Motorin and Helm 2011). Methylation of mitochondrial ribosomal RNAs has been observed in fungi (Kuriyama and Luck 1974) and mammals (Baer and Dubin 1981; Dubin et al. 1978). Environmental stressors, such as for instance heat shock, induce methylation of ribosomal RNA, which subsequently controls RNA metabolism (Bugl et al. 2000). RNA methylation is considered a new player in epigenetics and may deserve more attention in mitochondrial epigenetics in the future.
Table 1.
List of mitochondrial genes
| Function | Mitochondrial gene | |
|---|---|---|
| NADH dehydrogenase (complex I) | Essential for the assembly of the membrane arm and the respiratory function of the enzyme | MT-ND1 |
| MT-ND2 | ||
| MT-ND3 | ||
| MT-ND4 | ||
| MT-ND4L | ||
| MT-ND5 | ||
| MT-ND6 | ||
| Coenzyme Q-cytochrome c reductase/Cytochrome b (complex III) | Plays an important role in the production of ATP | MT-CYB |
| Cytochrome c oxidase (complex IV) | Helps to establish a transmembrane difference of proton electrochemical potential that the ATP synthase then uses to synthesize | MT-CO1 |
| MT-CO2 | ||
| MT-CO3 | ||
| ATP synthase | Provides energy for the cell to use through the synthesis of ATP | MT-ATP6 |
| MT-ATP8 | ||
| tRNAs | Carries an amino acid to ribosome | MT-TA, MT-TR, MT-TN, MT-TD, MT-TC, MT-TE, MT-TQ, MT-TG, MT-TH, MT-TI, MT-TL1, MT- TL2, MT-TK, MT-TM, MT-TF, MT-TP, MT-TS1, MT-TS2, MT- TT, MT-TW, MT-TY, MT-TV |
| rRNAs | Essential for protein synthesis | MT-RNR1 (12S) and MT-RNR2 (16S) |
Study by molecular type: Nucleoid remodeling and miRNAs
Histone structures have not been identified in the mitochondria, however studies have suggested the presence of nucleoid which is the nucleoprotein complex of mtDNA replication and gene expression machinery. Nucleoid remodeling is essential for the maintenance of the mitochondrial genome and the control of mitochondrial copy number (Kucej et al. 2008). Any form of remodeling of the nucleoid might be directly or indirectly related to mitochondrial gene expression. Manipulation of the protein-mtDNA interactions have been shown to cause decreased in vivo methylation (Rebelo et al. 2009), which is due to the altered accessibility of regions of mtDNA to DNA methyltransferases based upon the levels of protein occupancy.
Mitochondrial gene expression may also be regulated by miRNAs. Pre-miRNAs and miRNAs are present in human mitochondria for post-transcriptomic regulation by RNA interference (Barrey et al. 2011; Bienertova-Vasku et al. 2013), but whether these miRNAs are transported from cytosol or processed in the mitochondrion is not known. Further work is required into the profiling of mitochondrial miRNAs and establishing their function in regulating mitochondrial gene expression.
4) Analytical challenges in the study of mitochondrial epigenetics
Since the advent of the study of mitochondrial epigenetics in 1971, there have of course been substantial improvements in the methodological approaches available and the understanding of the nature of mtDNA. However there are still numerous challenges that the researcher must be aware of and understand when studying mitochondrial epigenetics.
Heteroplasmy
Many mtDNA copies contain mutations. Since there are multiple copies of mtDNA in every mitochondrion and mutations will not be present in all of these at once, the mutation rate varies between 1% and 100% (heteroplasmy). The mutation rate can vary from person to person and even between cells within the same individual. Once this mutation rate reaches a certain threshold, the heteroplasmy of mtDNA mutation can be linked to many human disease phenotypes, including neurological, gastrointestinal, cardiac, endocrinal etc. (Taylor and Turnbull 2005). It is very possible that every mitochondrial DNA copy has a different methylation pattern that can be dependent or independent of the mutations present, and therefore studies into mtDNA methylation may need to take into account the percentage of heteroplasmy as well. Further studies are required to investigate the possible link between these two factors, and how this can be addressed within study design.
Nuclear sequences of mitochondrial origin (numts)
Sequences of mitochondrial DNA have been transferred and inserted into the nuclear genome over the course of human evolution. This process is known to be continuously ongoing, and such sequences are sparsely dispersed throughout nuclear DNA (Mourier et al. 2001). Nuclear sequences of mitochondrial origin are referred to as numts, and the longest numt in the human genome is 14,654bp, which corresponds to 90% of the length of the human mitochondrial genome (Mourier et al. 2001). Depending upon the sequence-based search parameters used, it has been suggested that there are 286, 452 or to 612 numts in the human genome (Hazkani-Covo and Graur 2007). This ancient DNA is of consequence because of the high sequence homology with mitochondrial DNA, complicating the study of mtDNA unless mitochondria are isolated from the cell prior to the extraction of DNA. Indeed, there have been misidentifications in many mitochondrial studies between numts and bona fide mtDNA. A recent twin study clearly demonstrated that it is critical to differentiate between numts and mtDNA in genomic sequencing due to the large number of heteroplasmy that could influence interpretation (Bouhlal et al. 2013). Using cells depleted of mtDNA (ρ0 cells) or cybrid cell produced by fusing ρ0 cell with enucleated cells could be a useful tool in understanding the degree of contamination with numts (Hashiguchi and Zhang-Akiyama 2009).
Limitations of laboratory techniques
Mitochondrial DNA methylation has not yet been widely studied. Depending upon the method of measurement, mtDNA methylation has been reported to be either at a very low level or entirely absent. In earlier studies using radiolabeled dinucleotides, the level of mtDNA methylation in cytosine was observed to be approximately 5% in mice and hamsters (Nass 1973; Pollack et al. 1984) and at similar levels in human cells (Shmookler Reis and Goldstein 1983). These results were obtained using mtDNA from isolated mitochondria that are subsequently free of numts. By bisulfite-PCR-single-stranded DNA conformation polymorphism (SSCP) analysis, it has been shown that there is a lack of methylated DNA in mitochondria from gastric and colon cancer patients, although it is not clear whether the measurement of methylation in this study is solely from mtDNA or whether it could also originate from numts. Elsewhere, Feng et al. reported levels as high as 80% for mtDNA methylation in mitochondria extracted from non-cancerous colon tissues, using methylation-specific PCR (Feng et al. 2012). With the same technique, mtDNA methylation levels of around 20% to 40% have been observed in liver tissue (Pirola et al. 2013). A recent gene-specific analysis by bisulfite-pyrosequencing showed mtDNA methylation in human buffy coat ranging between 3 to 12%. Analysis using next-generation sequencing showed CpG methylation frequency to be below 0.66% in human mitochondrial DNA from whole blood (Hong et al. 2013), which was therefore interpreted as absence of mtDNA methylation in humans. The low levels of methylation seen in mtDNA can lead to confusion as to what is the product of DNA methylation and what might arise through incomplete conversion of cytosines by the bisulfite-modification process. This can be exacerbated by the presence of ncDNA in the DNA extract used for sequencing, which may reduce the measured DNA methylation levels and make it harder to distinguish from artifacts of the bisulfite-conversion process.
Ethnicity
Human mitochondrial DNA sequences have specific polymorphic variants that differ by geographic origin, and subsequently human mitochondrial DNA haplotypes have developed. It should be noted that the names of the mitochondrial haplogroups, from A to Z, are based upon the order of their discovery and do not reflect their genetic relationships. The effect of mitochondrial haplotype upon mtDNA methylation is not yet known, but it has been shown that methylation of the nuclear genome is affected by mtDNA variants in peripheral blood DNA (Bellizzi et al. 2012). The relationship between mitochondrial haplotype and the mtDNA epigenome needs to be established and could be of relevance to epidemiological population studies.
Perspective and future directions
As we have demonstrated in this review, the field of mitochondrial epigenetics has not been without controversies. Some of these still remain, but they may be explained by factors such as the presence of numts in the analyzed samples, the limitation of detection techniques, and an inappropriate choice of tissue(s) to study. It is critical to be aware of these factors and to carefully consider them in designing epidemiological studies. Functional studies should accompany analysis of mitochondrial epigenetics in order to support the biological relevance of changes in the epigenetic regulation of mitochondrial DNA, whether by analysis of mitochondrial gene expression or mitochondrial function assays. A range of human diseases need to be studied with regards to mitochondrial epigenetics, such as – among others – diabetes, cardiomyopathy, and neuronal development, due to the known role of mitochondrial dysfunction in the disease. Where mitochondrial epigenetics are implicated in the pathogenesis of the disease, this may open up avenues for the development of novel therapies.
Contributor Information
Hyang-Min Byun, Email: hmbyun@hsph.harvard.edu, Laboratory of Environmental Epigenetics, Exposure Epidemiology and Risk Program, Harvard School of Public Health, Boston, MA, USA 02115.
Andrea A. Baccarelli, Email: abaccare@hsph.harvard.edu, Laboratory of Environmental Epigenetics, Exposure Epidemiology and Risk Program, Harvard School of Public Health, Boston, MA, USA 02115
References
- Baer RJ, Dubin DT. Methylated regions of hamster mitochondrial ribosomal RNA: structural and functional correlates. Nucleic Acids Res. 1981;9:323–37. doi: 10.1093/nar/9.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrey E, Saint-Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PLoS One. 2011;6:e20220. doi: 10.1371/journal.pone.0020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi D, D’Aquila P, Giordano M, Montesanto A, Passarino G. Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics. 2012;4:17–27. doi: 10.2217/epi.11.109. [DOI] [PubMed] [Google Scholar]
- Bellizzi D, D’Aquila P, Scafone T, Giordano M, Riso V, Riccio A, Passarino G. The Control Region of Mitochondrial DNA Shows an Unusual CpG and Non-CpG Methylation Pattern. DNA Res. 2013 doi: 10.1093/dnares/dst029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienertova-Vasku J, Sana J, Slaby O. The role of microRNAs in mitochondria in cancer. Cancer Lett. 2013;336:1–7. doi: 10.1016/j.canlet.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Borchert A, Wolf NI, Wilichowski E. Current concepts of mitochondrial disorders in childhood. Semin Pediatr Neurol. 2002;9:151–9. doi: 10.1053/spen.2002.33800. [DOI] [PubMed] [Google Scholar]
- Bouhlal Y, Martinez S, Gong H, Dumas K, Shieh JT. Twin Mitochondrial Sequence Analysis. Mol Genet Genomic Med. 2013;1:174–186. doi: 10.1002/mgg3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugl H, Fauman EB, Staker BL, Zheng F, Kushner SR, Saper MA, Bardwell JC, Jakob U. RNA methylation under heat shock control. Mol Cell. 2000;6:349–60. doi: 10.1016/s1097-2765(00)00035-6. [DOI] [PubMed] [Google Scholar]
- Burton GJ, Jauniaux E. Oxidative stress. Best Pract Res Clin Obstet Gynaecol. 2011;25:287–99. doi: 10.1016/j.bpobgyn.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P, Bertazzi PA, Baccarelli AA. Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. 2013;10:18. doi: 10.1186/1743-8977-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chen H, Dzitoyeva S, Manev H. Effect of valproic acid on mitochondrial epigenetics. Eur J Pharmacol. 2012;690:51–9. doi: 10.1016/j.ejphar.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011;31:16619–36. doi: 10.1523/JNEUROSCI.1639-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Hoon Jeong J, Min HK, Jung HJ, Hwang D, Lee SW, Kim Pak Y. Shot-gun proteomic analysis of mitochondrial D-loop DNA binding proteins: identification of mitochondrial histones. Mol Biosyst. 2011;7:1523–36. doi: 10.1039/c0mb00277a. [DOI] [PubMed] [Google Scholar]
- Coene ED, Hollinshead MS, Waeytens AA, Schelfhout VR, Eechaute WP, Shaw MK, Van Oostveldt PM, Vaux DJ. Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol Biol Cell. 2005;16:997–1010. doi: 10.1091/mbc.E04-10-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley KE, Marcinek DJ, Villarin J. Mitochondrial dysfunction and age. Curr Opin Clin Nutr Metab Care. 2007;10:688–92. doi: 10.1097/MCO.0b013e3282f0dbfb. [DOI] [PubMed] [Google Scholar]
- Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–32. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- Duarte FV, Gomes AP, Teodoro JS, Varela AT, Moreno AJ, Rolo AP, Palmeira CM. Dibenzofuran-induced mitochondrial dysfunction: Interaction with ANT carrier. Toxicol In Vitro. 2013;27:2160–2168. doi: 10.1016/j.tiv.2013.08.009. [DOI] [PubMed] [Google Scholar]
- Dubin DT, Taylor RH, Davenport LW. Methylation status of 13S ribosomal RNA from hamster mitochondria: the presence of a novel riboside, N4-methylcytidine. Nucleic Acids Res. 1978;5:4385–97. doi: 10.1093/nar/5.11.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumollard R, Duchen M, Carroll J. The role of mitochondrial function in the oocyte and embryo. Curr Top Dev Biol. 2007;77:21–49. doi: 10.1016/S0070-2153(06)77002-8. [DOI] [PubMed] [Google Scholar]
- Dzitoyeva S, Chen H, Manev H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol Aging. 2012;33:2881–91. doi: 10.1016/j.neurobiolaging.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Xiong L, Ji Z, Cheng W, Yang H. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol Med Rep. 2012;6:125–30. doi: 10.3892/mmr.2012.870. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grattagliano I, Russmann S, Diogo C, Bonfrate L, Oliveira PJ, Wang DQ, Portincasa P. Mitochondria in chronic liver disease. Curr Drug Targets. 2011;12:879–93. doi: 10.2174/138945011795528877. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ. Mitochondria and heart disease. Adv Exp Med Biol. 2012;942:249–67. doi: 10.1007/978-94-007-2869-1_11. [DOI] [PubMed] [Google Scholar]
- Haddad DM, Vilain S, Vos M, Esposito G, Matta S, Kalscheuer VM, Craessaerts K, Leyssen M, Nascimento RM, Vianna-Morgante AM, De Strooper B, Van Esch H, Morais VA, Verstreken P. Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Mol Cell. 2013;50:831–43. doi: 10.1016/j.molcel.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Hansen A. Olfactory and solitary chemosensory cells: two different chemosensory systems in the nasal cavity of the American alligator, Alligator mississippiensis. BMC Neurosci. 2007;8:64. doi: 10.1186/1471-2202-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi K, Zhang-Akiyama QM. Establishment of human cell lines lacking mitochondrial DNA. Methods Mol Biol. 2009;554:383–91. doi: 10.1007/978-1-59745-521-3_23. [DOI] [PubMed] [Google Scholar]
- Hazkani-Covo E, Graur D. A comparative analysis of numt evolution in human and chimpanzee. Mol Biol Evol. 2007;24:13–8. doi: 10.1093/molbev/msl149. [DOI] [PubMed] [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EE, Okitsu CY, Smith AD, Hsieh CL. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol. 2013;33:2683–90. doi: 10.1128/MCB.00220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobazzi V, Castegna A, Infantino V, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab. 2013;110:25–34. doi: 10.1016/j.ymgme.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Infantino V, Castegna A, Iacobazzi F, Spera I, Scala I, Andria G, Iacobazzi V. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol Genet Metab. 2011;102:378–82. doi: 10.1016/j.ymgme.2010.11.166. [DOI] [PubMed] [Google Scholar]
- James AM, Collins Y, Logan A, Murphy MP. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol Metab. 2012;23:429–34. doi: 10.1016/j.tem.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–14. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, Suzuki Y, Kono T. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G. Mitochondria in cancer. Oncogene. 2006;25:4630–2. doi: 10.1038/sj.onc.1209589. [DOI] [PubMed] [Google Scholar]
- Kucej M, Kucejova B, Subramanian R, Chen XJ, Butow RA. Mitochondrial nucleoids undergo remodeling in response to metabolic cues. J Cell Sci. 2008;121:1861–8. doi: 10.1242/jcs.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudriashova IB, Kirnos MD, Vaniushin BF. DNA-methylase activities from animal mitochondria and nuclei: different specificity of DNA methylation. Biokhimiia. 1976;41:1968–77. [PubMed] [Google Scholar]
- Kuriyama Y, Luck DJ. Methylation and processing of mitochondrial ribosomal RNAs in poky and wild-type Neurospora crassa. J Mol Biol. 1974;83:253–66. doi: 10.1016/0022-2836(74)90390-8. [DOI] [PubMed] [Google Scholar]
- Kurochkin IO, Etzkorn M, Buchwalter D, Leamy L, Sokolova IM. Top-down control analysis of the cadmium effects on molluscan mitochondria and the mechanisms of cadmium-induced mitochondrial dysfunction. Am J Physiol Regul Integr Comp Physiol. 2011;300:R21–31. doi: 10.1152/ajpregu.00279.2010. [DOI] [PubMed] [Google Scholar]
- Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol. 2012;942:311–27. doi: 10.1007/978-94-007-2869-1_14. [DOI] [PubMed] [Google Scholar]
- Leigh-Brown S, Enriquez JA, Odom DT. Nuclear transcription factors in mammalian mitochondria. Genome Biol. 2010;11:215. doi: 10.1186/gb-2010-11-7-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin R. The unmasking of mitochondrial Eve. Science. 1987;238:24–6. doi: 10.1126/science.3116666. [DOI] [PubMed] [Google Scholar]
- Lin Y, Sun X, Qiu L, Wei J, Huang Q, Fang C, Ye T, Kang M, Shen H, Dong S. Exposure to bisphenol A induces dysfunction of insulin secretion and apoptosis through the damage of mitochondria in rat insulinoma (INS-1) cells. Cell Death Dis. 2013;4:e460. doi: 10.1038/cddis.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–73. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Taniguchi T, Higashi H, Sugimura H, Sugano K, Kanno T. Methylation of mitochondrial DNA is not a useful marker for cancer detection. Clin Chem. 2004;50:1480–1. doi: 10.1373/clinchem.2004.035139. [DOI] [PubMed] [Google Scholar]
- Manev H, Dzitoyeva S, Chen H. Mitochondrial DNA: A Blind Spot in Neuroepigenetics. Biomol Concepts. 2012;3:107–115. doi: 10.1515/bmc-2011-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab. 2007;18:190–8. doi: 10.1016/j.tem.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Martinez-Fernandez E, Gil-Peralta A, Garcia-Lozano R, Chinchon I, Aguilera I, Fernandez-Lopez O, Arenas J, Campos Y, Bautista J. Mitochondrial disease and stroke. Stroke. 2001;32:2507–10. doi: 10.1161/hs1101.098328. [DOI] [PubMed] [Google Scholar]
- Mathews CE, McGraw RA, Dean R, Berdanier CD. Inheritance of a mitochondrial DNA defect and impaired glucose tolerance in BHE/Cdb rats. Diabetologia. 1999;42:35–40. doi: 10.1007/s001250051109. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS. The human mitochondrial transcriptome. Cell. 2011;146:645–58. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortiboys H, Aasly J, Bandmann O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease. Brain. 2013;136:3038–50. doi: 10.1093/brain/awt224. [DOI] [PubMed] [Google Scholar]
- Motorin Y, Helm M. RNA nucleotide methylation. Wiley Interdiscip Rev RNA. 2011;2:611–31. doi: 10.1002/wrna.79. [DOI] [PubMed] [Google Scholar]
- Mourier T, Hansen AJ, Willerslev E, Arctander P. The Human Genome Project reveals a continuous transfer of large mitochondrial fragments to the nucleus. Mol Biol Evol. 2001;18:1833–7. doi: 10.1093/oxfordjournals.molbev.a003971. [DOI] [PubMed] [Google Scholar]
- Mushkambarov NN, Votrin II, Debov SS. Methylation of preformed DNA in rat liver cell nuclei and mitochondria. Dokl Akad Nauk SSSR. 1976;229:1255–7. [PubMed] [Google Scholar]
- Nass MM. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells. In vivo and in vitro methylation. J Mol Biol. 1973;80:155–75. doi: 10.1016/0022-2836(73)90239-8. [DOI] [PubMed] [Google Scholar]
- Newsholme P, Gaudel C, Krause M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv Exp Med Biol. 2012;942:235–47. doi: 10.1007/978-94-007-2869-1_10. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–59. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Destroismaisons L, Peyrard SL, Cadot S, Rouleau GA, Brown RH, Jr, Julien JP, Arbour N, Vande Velde C. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum Mol Genet. 2013;22:3947–59. doi: 10.1093/hmg/ddt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola CJ, Gianotti TF, Burgueno AL, Rey-Funes M, Loidl CF, Mallardi P, Martino JS, Castano GO, Sookoian S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2013;62:1356–63. doi: 10.1136/gutjnl-2012-302962. [DOI] [PubMed] [Google Scholar]
- Pollack Y, Kasir J, Shemer R, Metzger S, Szyf M. Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 1984;12:4811–24. doi: 10.1093/nar/12.12.4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu P, Puddu GM, Cravero E, De Pascalis S, Muscari A. The putative role of mitochondrial dysfunction in hypertension. Clin Exp Hypertens. 2007;29:427–34. doi: 10.1080/10641960701613852. [DOI] [PubMed] [Google Scholar]
- Rebelo AP, Williams SL, Moraes CT. In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 2009;37:6701–15. doi: 10.1093/nar/gkp727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorbach J, Minczuk M. The post-transcriptional life of mammalian mitochondrial RNA. Biochem J. 2012;444:357–73. doi: 10.1042/BJ20112208. [DOI] [PubMed] [Google Scholar]
- Schrier SA, Falk MJ. Mitochondrial disorders and the eye. Curr Opin Ophthalmol. 2011;22:325–31. doi: 10.1097/ICU.0b013e328349419d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123:2533–42. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Karl B, Mathew AV, Gangoiti JA, Wassel CL, Saito R, Pu M, Sharma S, You YH, Wang L, Diamond-Stanic M, Lindenmeyer MT, Forsblom C, Wu W, Ix JH, Ideker T, Kopp JB, Nigam SK, Cohen CD, Groop PH, Barshop BA, Natarajan L, Nyhan WL, Naviaux RK. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol. 2013;24:1901–12. doi: 10.1681/ASN.2013020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmookler Reis RJ, Goldstein S. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J Biol Chem. 1983;258:9078–85. [PubMed] [Google Scholar]
- Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011;108:3630–5. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrikhande DY, Kalakoti P, Syed MM, Ahya K, Singh G. A rare mitochondrial disorder: Leigh syndrome--a case report. Ital J Pediatr. 2010;36:62. doi: 10.1186/1824-7288-36-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, James SR, Kazim L, Karpf AR. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:504–10. doi: 10.1021/ac0489420. [DOI] [PubMed] [Google Scholar]
- Stuwe SH, Goetze O, Lukas C, Klotz P, Hoffmann R, Banasch M, Orth M, Schmidt WE, Gold R, Saft C. Hepatic mitochondrial dysfunction in manifest and premanifest Huntington disease. Neurology. 2013;80:743–6. doi: 10.1212/WNL.0b013e318282514e. [DOI] [PubMed] [Google Scholar]
- Sultana R, Baglioni M, Cecchetti R, Cai J, Klein JB, Bastiani P, Ruggiero C, Mecocci P, Butterfield DA. Lymphocyte mitochondria: toward identification of peripheral biomarkers in the progression of Alzheimer disease. Free Radic Biol Med. 2013;65C:595–606. doi: 10.1016/j.freeradbiomed.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol Oncol. 2011;121:59–63. doi: 10.1016/j.ygyno.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Terragni J, Borgaro JG, Liu Y, Yu L, Guan S, Wang H, Sun D, Cheng X, Zhu Z, Pradhan S, Zheng Y. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–76. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–83. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- Vamecq J, Dessein AF, Fontaine M, Briand G, Porchet N, Latruffe N, Andreolotti P, Cherkaoui-Malki M. Mitochondrial dysfunction and lipid homeostasis. Curr Drug Metab. 2012;13:1388–400. doi: 10.2174/138920012803762792. [DOI] [PubMed] [Google Scholar]
- van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30:E386–94. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- Yelverton JC, Arnos K, Xia XJ, Nance WE, Pandya A, Dodson KM. The clinical and audiologic features of hearing loss due to mitochondrial mutations. Otolaryngol Head Neck Surg. 2013;148:1017–22. doi: 10.1177/0194599813482705. [DOI] [PubMed] [Google Scholar]
- Yorns WR, Jr, Valencia I, Jayaraman A, Sheth S, Legido A, Goldenthal MJ. Buccal swab analysis of mitochondrial enzyme deficiency and DNA defects in a child with suspected myoclonic epilepsy and ragged red fibers (MERRF) J Child Neurol. 2012;27:398–401. doi: 10.1177/0883073811420870. [DOI] [PubMed] [Google Scholar]
- Yu E, Mercer J, Bennett M. Mitochondria in vascular disease. Cardiovasc Res. 2012;95:173–82. doi: 10.1093/cvr/cvs111. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Cheng J, Xu F, Chen YE, Du JB, Yuan M, Zhu F, Xu XC, Yuan S. Red blood cell extrudes nucleus and mitochondria against oxidative stress. IUBMB Life. 2011;63:560–5. doi: 10.1002/iub.490. [DOI] [PubMed] [Google Scholar]