

Abstract
Phosphatidylinositol 3-kinases (PI3Ks) play important roles in human tumorigenesis. Activation of the PI3K target AKT is frequent in neuroblastoma (NB) and correlates with poor prognosis. PI3K pan-inhibitors reduce NB tumor formation but present severe toxicity, which limits their therapeutic potential. Therefore, defining the importance of specific PI3K isoforms may aid in developing more effective therapeutic strategies. We previously demonstrated that PI3K Class IIβ (PI3KC2β) and its regulator intersectin 1 (ITSN1) are highly expressed in primary NB tumors and cell lines. Silencing ITSN1 dramatically reduced the tumorigenic potential of NB cells. Interestingly, overexpression of PI3KC2β rescued the anchorage-independent growth of ITSN1-silenced cells suggesting that PI3KC2β mediates ITSN1's function in NB cells. To address the importance of PI3KC2β in NBs, we generated PI3KC2β-silenced lines and examined their biologic activity. Herein, we demonstrate that PI3KC2β-silencing inhibits early stages of NB tumorigenic growth. We also show that loss of endogenous PI3KC2β or ITSN1 reduces AKT activation but does not impact ERK-MAPK activation. These data reveal a novel role for PI3KC2β in human NB tumorigenesis.
Keywords: PIK3C2β, ITSN1, AKT, oncogenesis, pediatric tumors, neuroblastoma
1. Introduction
Neuroblastoma (NB) is the most common extracranial solid tumor in children and is responsible for 15% of pediatric cancer-related deaths [21]. Amplification or overexpression of the myelocytomatosis viral oncogene (MYCN) is present in approximately 20% of NB patients and is a poor prognostic marker [17]. MYCN overexpression has been reported as a cause of increased tumor cell proliferation and invasiveness. Proteins of the phosphoinositide 3-kinase (PI3K) family play a central role in NB tumorigenesis through stabilization of MYCN [7]. In addition, the target of PI3K, protein kinase B (PKB or AKT), is also a marker for poor prognosis in NBs [13]. Activation of the PI3K-AKT pathway leads to phosphorylation and inhibition of GSK3β which normally phosphorylates MYCN leading to its degradation by the proteasome [7]. Pharmacological inhibition of PI3Ks with pan-inhibitors blocks malignant progression in vivo; however, these inhibitors have serious toxicity due to their promiscuity that limits their usefulness in NB treatment [5, 27]. Thus, defining the specific PI3K isoforms important for NB tumorigenesis represents an important step towards the development of better therapeutic treatments for this disease.
Accumulating evidence reveals that PI3KC2β contributes to oncogenic transformation. PI3KC2β expression is upregulated in several cancers and PI3KC2β overexpression leads to transformation of colonic epithelial cells [15, 24]. Dominant-negative PI3KC2β inhibits human small cell lung cancer growth as well as growth factor-stimulated AKT activation [2]. We have previously shown that silencing expression of the ITSN1 scaffold, which activates PI3KC2β [11], reduced NB cell growth in anchorage-independent conditions in vitro as well as tumor growth in vivo [23]. Furthermore, PI3KC2β overexpression restored the anchorage-independent growth of ITSN1-depleted NB cells suggesting that PI3KC2β mediates the tumorigenic effect of ITSN1 in NBs [23]. Specific pharmacological inhibition of PI3KC2β demonstrates a role for this isoform in additional human cancers as well [6]. Thus, we sought to determine the importance of PI3KC2β for NB tumorigenesis. Herein, we report that PI3KC2β depletion reduces anchorage-independent growth in vitro as well as tumor growth in vivo of NB cells. We also demonstrate that loss of either ITSN1 or PI3KC2β in MYCN-amplified IMR-5 NB cells results in reduced EGF-induced AKT activation. These studies reveal an important role for PI3KC2β in human tumorigenesis and highlight it's role in regulating the AKT pathway in MYCN-amplified tumor cell lines.
2. Materials and Methods
2.1. Reagents and cell culture
All NB cells used in this study were maintained in RPMI with 10% fetal bovine serum at 37°C in 5% CO2 and were the kind gifts of Drs. Bernard Weissman (University of North Carolina at Chapel Hill) and Naohiko Ikegaki (University of Illinois at Chicago). Puromycin (GIBCO) was used at 1μg/ml.
2.2 Stable silencing of PI3KC2β
Phoenix-GP cells were used to generate retrovirus for infection of NB cells as previously described [23]. These packaging cells were transiently transfected using calcium phosphate method with 20 μg of vector alone (pSUPER.retro.puro; pSR) or pSR expressing shRNAs to PI3KC2β (βsh2 or βsh5) along with a plasmid encoding the VSV-G envelope glycoprotein to generate viral particles. On the following day, the media was replaced with fresh media, and NB cells were seeded for infection. On day 2 post-transfection, conditioned media from the Phoenix-GP cells was collected, filtered, and used to infect NB cells followed by selection in puromycin. Following selection, colonies were pooled to generate a polyclonal cell line, which was used for all subsequent analyses. Western Blot analyses of polyclonal cell lines were performed as previously described [1]. The sequences of oligonucleotides used to construct these vectors are as follows: βsh2-F-5'-GATCCCCGACATCAACACTTTCTCTTTGTTCAAGAGACAAAGAGAAAGTGTTGATGTCTTTTTA3’(15); βsh2-R-5'-AGCTTAAAAAGACATCAACACTTTCTCTTTGTCTCTTGAACAAAGAGAAAGTGTTGATGTCGGG 3'; βsh5-F-5'-GATCCCCCCAGAAGGCAAGAGAGGAATTCAAGAGATTCCTCTCTTGCCTTCTGGTTTTTA 3'; βsh5-R-3' AGCTTAAAAACCAGAAGGCAAGAGAGGAATCTCTTGAATTCCTCTCTTGCCTTCTGGGGG 3'; pSCR-A-5’-GATCCCCGGTACTAAAGCGAATATTATTCAAGAGATAATATTCGCTTTAGTACCTTTTT and pSCR-B 5’-AAAAAGGTACTAAAGCGAATATTATCTCTTGAATAATATTCGCTTTAGTACCGGGGATC. The βsh5 shRNA targets the 3‘UTR of PI3KC2β.
2.3. Proliferation assay
NB cells (700 per well) were plated on 24-well plates in complete media (RPMI +10%FBS plus puromycin) for the indicated number of days. On the indicated day, media was removed and replaced with 100μl of complete media to which 100μl of CellTiter Glow (Promega) was added to the cells. Luminescence was quantified on a Dynex 96-well microtiter plate luminometer according to the manufacture's instructions.
2.4. Analysis of apoptosis by Annexin V staining
Cells were seeded on Ultra-Low attachment plates (Corning) at a density of 0.5×106 cells per well. After 24hrs cells were collected, washed once with PBS and trypsinized (0.25%) for 10 min @ 37°C, washed again with PBS, centrifuged and resuspended in 1x binding buffer provided by manufacturer (Invitrogen) at a concentration of 1×106 cells/ml. Suspension of cells was transferred into a 5 ml tube to which was added 5ul of FITC conjugated-Annexin V and/or 1ul of PI (Invitrogen). The cells were then incubated for 15 min at RT in the dark. Binding buffer (400 ul) was added into each tube and then apoptosis quantified by flow cytometry within one hour.
2.5. Soft agar assay
Assays were performed essentially as described [9]. Briefly, 5% (w/v) agar (DIFCO, Detroit MI) was prepared in distilled water then diluted to 0.5% final concentration with complete media and kept @ 60°C in water bath. A bottom layer of 0.5% agar in complete media was plated in each well of six-well plates to which 10,000 cells were subsequently plated in triplicate in a top layer of 0.37% agar in complete media and cooled at 37°C. Samples were placed in humidified cell culture incubators at 37°C for 2-3 weeks after which colonies were stained using 100 μl solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; 2 mg/ml in H2O). Colonies were counted using the Image Quant LAS4010 (sensitivity set at 9498; size operator at 17; noise at 1) (GE Healthcare).
2.6. Transferrin internalization assay
Cells (2×105 per well) were plated in 6-well plates. The following the day, cells were equilibrated in the presence of 2% BSA for 1h @ 37°C then incubated in the presence of Biotinylated transferrin for 15 min @ 37°C. Cells were washed with ice cold PBS, followed by an acid wash (0.2M NaCl/0.2M acetic acid) on ice for 8 min then lysed and analyzed by Western blot using horseradish peroxidase linked to streptavidin as previously described [11].
2.7. EGFR internalization assay
Cells (0.7×105 per well) were plated in triplicate in 24-well plates. The following the day, cells were equilibrated in the presence RPMI, BSA 0.1%, 25mM HEPES for 1h @ 37°C. Cells were incubated on ice in the presence of RPMI, BSA 0.1%, 25 mM HEPES and 100 ng/mL EGF for 1hour. Cells were then incubated at 37°C for indicated times, washed with ice cold PBS and then fixed with 4% paraformaldehyde for 5 min on ice. Following a wash with ice cold PBS, cells were incubate with anti-EGFR antibody AB12 (1:200) for 1h @ RT in RPMI/HEPES/BSA, washed 3x with PBS, and incubated with anti-mouse-HRP for 1h @ RT. Cells were then washed 3X (10min each) with PBS and incubated with 200ul of TMB (tetramethylbenzidine) diluted in PBS 1:10 from Sigma and incubated RT for 5-20 min depending on color appearance. Samples (150 ul) were transferred to 96-well flat bottom microtiter dish and absorbance read at 655nm.
2.8. Xenograft tumor assays
Approximately 1×107 cells for each stable NB line were collected and resuspended in Matrigel (BD bioscience) at a 1:1 volume ratio in a total volume of 200 μl. This cell-Matrigel mixture was kept on ice then injected subcutaneously into the flanks of athymic nude mice using a 25G syringe. Tumor growth was monitored by caliper every other day for 4 weeks or until the tumors reached a diameter of 1 cm. Tumor volume was calculated using the formula (d1 × d2 × d3) × π/6. Animals were then sacrificed and tumors extracted and analyzed by Western blot. Tumors were fixed in formalin, processed and embedded in paraffin wax, and H&E stained sections were prepared for histological evaluation.
3. Results 3.1. Silencing PI3KC2β does not alter proliferation of NB cells in adherent conditions
We previously showed that ITSN1 regulates tumorigenic properties of NBs and that PI3KC2β overexpression rescued the tumorigenic properties of ITSN1 silenced cells [23]. In addition, we showed that ITSN1 binds and activates PI3KC2β [11]. These results suggested that PI3KC2β acts downstream of ITSN1 to mediate tumorigenesis. PI3KC2β is expressed in NB cell lines as well as primary tumor samples consistent with a role in NB tumorigenesis [23]. To determine the importance of PI3KC2β in NB, we stably silenced endogenous PI3KC2β expression in both MYCN-amplified IMR-5 cells and MYCN-non-amplified SK-N-AS cells using retrovirally delivered shRNAs specific for PI3KC2β (Fig. 1A-B). Western blot analysis of the resulting cell lines demonstrates significant reduction in PI3KC2β levels in the PI3KC2β shRNA infected lines (βsh2 and βsh5) but not in the control lines (pSCR and pSR). Validity of shRNAs targeting ITSN1 (sh#1 and sh#2) has been previously demonstrated [23]. Given the role of Class I PI3Ks in proliferation, we tested whether loss of PI3KC2β affected the growth of NB cell lines in culture. As shown in Figs 1C & D, the proliferation of PI3KC2β-silenced cell lines was not significantly different from the control lines.
Figure 1. PI3KC2β silencing does not alter adherent growth of NB cells.
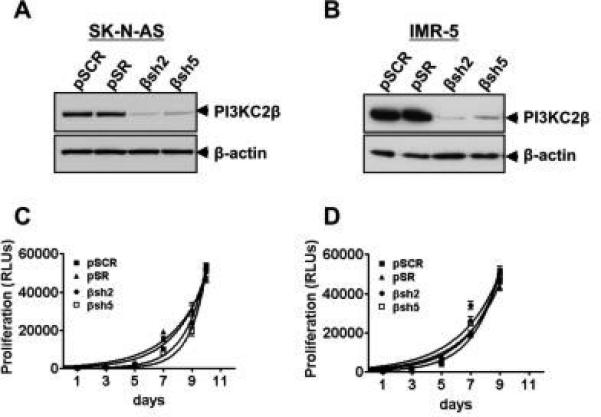
(A&B) PI3KC2β expression levels in shRNA-expressing NB cell lines. SK-N-AS (A) and IMR-5 (B) NB cells were infected with retrovirus containing the indicated empty vector control (pSR), scrambled shRNA (pSCR), or PI3KC2β shRNAs (βsh2 and βsh5). Stable polyclonal cell lines were generated, lysed, and analyzed by Western blot to assess PI3KC2β levels. Actin is shown as a control for loading. Growth curves of shRNA-expressing SK-N-AS (C) and IMR-5 (D) NB cells from above were determined using Cell TiterGlo (Promega). Each point is the average of three independent wells of cells +/− standard deviation.
3.2. PI3KC2β contributes to NB tumorigenesis
Our previous results demonstrated that ITSN1 silencing reduced the tumorigenic properties of MYCN-amplified and non-MYCN-amplified NB cells both in vitro and in vivo and that overexpression of PI3KC2β in ITSN1-silenced IMR-5 cells rescued the soft agar growth of this cell line [23]. To address whether PI3KC2β contributes to the tumorigenic properties of NB cells, we assessed the effects of silencing PI3KC2β on the anchorage-independent growth of NB cells in soft agar. Although PI3KC2β silencing reduced the number of soft agar colonies formed by non-MYCN-amplified SK-N-AS cells, reduction of PI3KC2β in MYCN-amplified IMR-5 cells did not reduce colony formation in an anchorage-independent growth assay in vitro (Fig. 2A-B). However, reduction of PI3KC2β in MYCN-amplified IMR-5 cells reduced colony formation following EGF stimulation (Fig. 2C-D) suggesting a differential role of PI3KC2β in regulating tumorigenesis in different NB lines.
Figure 2. PI3KC2β-silencing decreases anchorage-independent growth of IMR-5 NB cells.
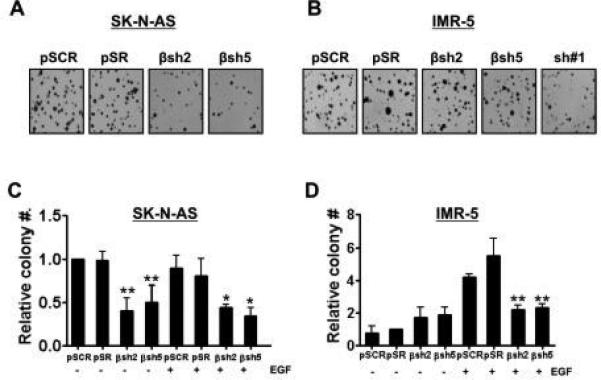
SK-N-AS (A) and IMR-5 (B) NB cells expressing the indicated shRNAs were plated in triplicate in soft agar. Colonies were stained with MTT after 3 weeks of growth and then photographed using a digital scanner. SK-N-AS (C) and IMR-5 (D) cells infected with indicated shRNAs or controls were plated in triplicate in soft agar with or without 100 ng/ml of EGF. Colonies were stained with MTT after 3 weeks of growth and then photographed using a digital scanner. Colonies were counted using the Image Quant LAS4010. Data represent the mean +/− standard error of three independent experiments. For SK-N-AS, βsh2 p=0.0026** and βsh5 p=0.0072** compared to control pSCR without EGF; for EGF-treatment, βsh2 p=0.021* and βsh5 p=0.029* compared to control pSCR. For IMR-5 in absence of EGF stimulation, differences observed with βsh2 and βsh5 were not significant compared to control pSCR. For EGF-treated samples, βsh2 p=0.00029** and βsh5 p=0.00022** compared to control pSCR.
To further examine PI3KC2β's potential role in tumorigenesis, we tested whether PI3KC2β-silenced cells were capable of forming tumors in vivo. Although injection of control NB cells into the flanks of athymic nude mice resulted in tumor development, silencing PI3KC2β in both IMR-5 and SK-N-AS cells significantly reduced their ability to form tumors in athymic nude mice (Fig. 3A-B). PI3KC2β-silenced cells formed smaller tumors with slower kinetics (Fig. 3C-D). These results parallel our findings with ITSN1 in which loss of ITSN1 expression resulted in decreased tumor formation in vivo (12). However, tumor growth of PI3KC2β-silenced cells recovered over time while ITSN1-silenced tumors (sh#1) did not suggesting that ITSN1's role in NB tumorigenesis is more pronounced (Fig. 3C-D). Despite the differences in growth kinetics, the histomorphology of the tumors was similar in all groups and the tumor cell morphology was consistent with that of neuroblastoma cells, the SK-N-AS tumors showing a whorl-like growth pattern while the IMR-5 tumors tended to grow in solid nests and sheets (data not shown). There was evidence of central necrosis only in some of the largest tumors. All tumors for which adequate margins were available for examination appeared to be invasive.
Figure 3. PI3KC2β-depletion results in reduced tumor growth in vivo.
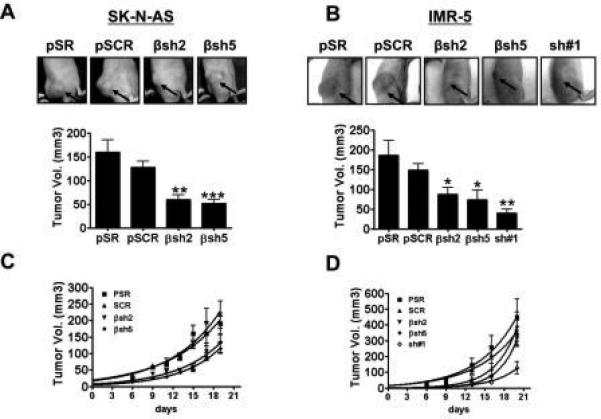
Xenograft assays. SK-N-AS (A) or IMR-5 (B) cells were injected into nude mice (eight injections per condition). Shown are tumors that formed in mice at 15 days post injection. Graphs below pictures represent the average tumor volume +/− standard error at day 10 post-injection. A two-tailed Student's T test was applied to determine significance compared to pSCR. For IMR-5 βsh2 has p=0.02* and βsh5 p=0.016*; sh#1 has p=0.0038**; for SK-N-AS βsh2 has p=0.002** and βsh5 p=0.0005***. Differences between pSR and pSCR were not significant. Tumor growth for SK-N-AS cells (C) and IMR-5 cells (D) were monitored for the indicated times.
To assess whether tumor growth of PI3KC2β-silenced cells recovered due to re-expression of PI3KC2β, we analyzed emergent tumors by Western blot. PI3KC2β was not re-expressed in tumors from PI3KC2β-silenced cells (Sup. Fig. 1A-B). In addition, silencing PI3KC2β did not lead to increased expression of other PI3K isoforms (Sup. Fig. 1C-D). These results suggest that the stronger effect of ITSN1 in NB tumorigenesis is due to regulation of multiple signaling pathways and that PI3KC2β may regulate a more limited set of these pathways important in the early stage of NB tumor growth.
3.3. Silencing PI3KC2β did not alter apoptotic or endocytic pathways
Our previous findings suggest that the ITSN1-PI3KC2β pathway activates AKT (11). Given the role of AKT in inhibiting apoptosis, we next tested whether the observed decrease in soft agar growth and tumor formation in PI3KC2β-silenced cells was due to an increase in apoptosis. We cultured control or PI3KC2β-depleted IMR-5 and SK-N-AS lines in low-attachment plates and quantified apoptosis by Annexin V staining. Loss of PI3KC2β did not lead to increased apoptosis in either IMR-5 or SK-N-AS cells (Sup. Fig. 2A-B) consistent with our previous results with ITSN1 [23].
Given ITSN1's role in regulating endocytosis [12] and the association of PI3KC2β with ITSN1 on endocytic vesicles [11], we examined whether loss of PI3KC2β resulted in defects in receptor-mediated endocytosis. Depletion of PI3KC2β had no effect on transferrin internalization in IMR-5 or SK-N-AS cells (Sup. Fig. 3A-B). Thus, stable loss of either ITSN1 or PI3KC2β does not adversely affect constitutive endocytosis. In addition we assessed whether loss of ITSN affects EGF-induced EGFR internalization. We found that ITSN1-depletion did not have effect on EGFR internalization in SK-N-AS cells (Sup. Fig. 4).
3.4. Silencing ITSN1 or PI3KC2β decreased AKT activation in NB cells
Our results suggest that ITSN1 and PI3KC2β regulate a pro-tumorigenic signaling pathway in NB cells. We previously reported that ITSN1 stimulates a PI3K-dependent activation of AKT [11, 28] and that ITSN1 and PI3KC2β regulate an AKT-dependent cell survival signaling pathway in neurons [11]. Furthermore, expression of a PI3KC2β dominant-negative inhibits growth factor activation of AKT as well as growth of human lung cancer cell lines [2]. Given that AKT activation is a poor prognostic marker in NB, we next examined whether the ITSN1-PI3KC2β pathway contributes to AKT activation in NB cells. Silencing PI3KC2β did not impair AKT activation in non-MYCN amplified SK-N-AS cells (Fig. 4A). However, depletion of PI3KC2β resulted in reduced growth factor stimulated AKT activation in MYCN amplified IMR-5 cells (Figs. 4B). As a control, depletion of ITSN1 resulted in >50% reduction in AKT activation following EGF stimulation in these cells as well (Figs. 4B). However, this reduction in AKT activation in PI3KC2β-silenced cells did not result in increased apoptosis (Sup. Fig. 2).
Figure 4. Silencing PI3KC2β or ITSN1 results in reduced AKT activation in IMR-5 cells.
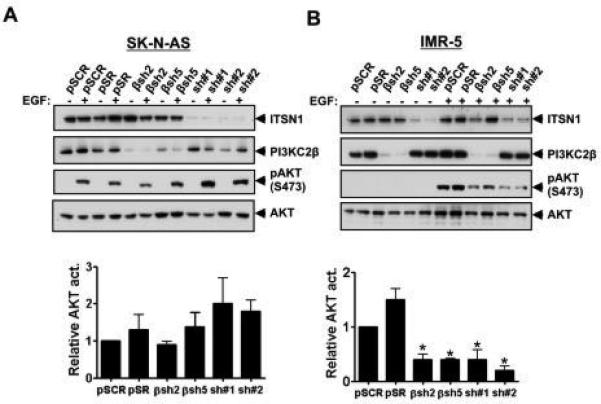
SK-N-AS (A) and IMR-5 (B) cells stably infected with virus expressing control (pSCR or pSR), ITSN1 (sh#1 or sh#2), or PI3KC2β (βsh2 or βsh5) shRNAs and grown in complete media containing 10% serum, then stimulated with or with EGF (100 ng/ml for 10 min), lysed and analyzed by Western blot to assess phosphorylation of AKT at Ser473 as well as total AKT levels. Levels of ITSN1 and PI3KC2β are also shown. Relative AKT phosphorylation was calculated by comparing the level of AKT phosphorylation in samples in the presence of EGF and normalizing to AKT phosphorylation levels in pSCR stimulated with EGF. Graphs are the average +/− standard error of three independent experiments. For IMR-5 βsh2, βsh5, sh#1 and sh#2, p<0.05 compared to control pSCR. For SK-N-AS, differences in phosphorylation between samples were not significant.
Given reports of ITSN1's contribution to ERK1/2 activation [30], we also tested whether loss of ITSN1 or PI3KC2β altered EGF activation of ERK1/2. As illustrated in Fig. 5, we did not observe any differences in phosphoERK1/2 levels in ITSN1-silenced IMR-5 or SK-N-AS cells following EGF stimulation consistent with our previous findings [1, 19].
Figure 5. Silencing PI3KC2β or ITSN1 did not affect ERK1/2 activation in either IMR-5 or SK-N-AS.
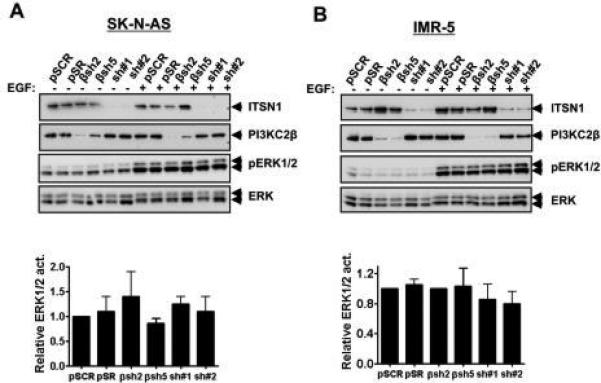
SK-N-AS (A) and IMR-5 (B) cells stably infected with pSCR or pSR, sh#1, sh#2, βsh2 or βsh5, were lysed and analyzed by Western blot to assess phosphorylation of phosphoERK1/2 or ERK1/2 total levels. Levels of ITSN1 and PI3KC2β are also shown. Phosphorylation of ERK1/2 was normalized to total ERK1/2 and relative value calculated normalized to pSCR value. Graphs are the average +/− standard error of three independent experiments. P values were not significant.
A number of receptor tyrosine kinases have been implicated in NB tumorigenesis including ALK, TRK family members, IGF-1, and EGFR [8]. Given PI3KC2β's role in regulating RTK-dependent AKT activation [2, 11, 28], we examined the contribution of PI3KC2β to growth factor stimulated enhancement of anchorage-independent growth. In the case of IMR-5 cells, EGF stimulation increased colony formation when compared to non-stimulated cells; however, SK-N-AS cells did not respond to EGF treatment (Fig. 6 A-B). Furthermore, silencing PI3KC2β attenuated EGF-stimulated colony formation in IMR-5 cells further supporting a role for PI3KC2β in NB tumorigenesis. Inhibition of AKT completely abolished colony formation in SK-N-AS. In IMR-5, AKT inhibition reduced colony formation under basal conditions. In the presence of EGF, the AKT inhibitor abolished EGF-induced increase in colony formation consistent with a pivotal role for AKT in NB tumorigenesis (Fig. 6C-D).
Figure 6. AKT inhibition reduces anchorage-independent growth of NB cell lines.

SK-N-AS (A) and IMR-5 (B) cells infected with indicated shRNAs or controls were plated in triplicate in soft agar with or without 100 ng/ml of EGF and with or without AKT-inhibitor (AKTi, 10 μM) as indicated. Colonies were stained with MTT after 3 weeks of growth and quantified as in Fig. 2. Data represent the mean +/− ± standard error of three independent experiments. (A) For parental SK-N-AS cells, treatment with AKT-inhibitor completely abolished colony formation +/− ± EGF treatment (AKTi p < 0.0001***; AKTi + EGF p = 0.007**). The effect of AKTi on PI3KC2β silenced SK-N-AS cells was not determined given the lack of effect of PI3KC2β loss on AKT activation in these cells (see Fig. 4). (B) IMR-5 derived cells infected with indicated shRNAs or controls (1 = pSCR; 2 = pSR; 3 = βsh2; 4 = βsh5; 5 = sh#1; 6 = sh#2) were treated with or without AKTi in the presence or absence of EGF stimulation. For IMR-5 treated with vehicle in the absence of EGF, differences observed with βsh2 and βsh5 (columns 3 and 4, respectively) were not significant compared to control pSCR; however, silencing ITSN1 (columns 5 and 6) reduced colony formation as previously reported [11]. p value for sh#1 = 0.007 **; for sh#2 = 0.004 **; compared to pSCR. For EGF-treated samples in the absence of AKTi, silencing PI3KC2β or ITSN1 reduced colony numbers. βsh2 p = 0.001*** and βsh5 p = 0.0002***; sh#1 + p = 0.001***, sh#2 p < 0.0001 *** compared to control pSCR. Treatment with AKTi in the absence of EGF stimulation results in significant reduction in colony formation in all conditions compared to pSCR. Treatment of cells with EGF + AKTi results in loss of EGF-induced increase in colony formation in pSCR and pSR controls when compared to EGF alone, whereas in the presence of EGF, AKTi does not affect colony number when either ITSN1 or PI3KC2β are silenced.
4. Discussion
Given our previous findings on the importance of ITSN1 in NB tumorigenesis and the ability of PI3KC2β to rescue the tumorigenic properties of ITSN1-silenced NB cells [23], we examined the role of PI3KC2β in NB tumorigenesis. Herein, we demonstrate the importance of PI3KC2β for NB tumorigenesis. PI3KC2β depletion reduces tumor growth of NB cells in vivo. A recent study with leukemia and NB cells demonstrates that PI3KC2β pharmacological inhibitors have anti-proliferative effects in vitro [6]. However, our results obtained by silencing PI3KC2β (or ITSN1) do not show any effects on proliferation in adherent conditions. This discrepancy in results may be due to the fact that we are depleting PI3KC2β from cells as opposed to just inhibiting the kinase activity of PI3KC2β. It is well known that Class I PI3Ks have both kinase-dependent and kinase-independent activities. While the difference in our results from those of Boller et. al. may involve these kinase-independent functions, it is unclear how the remaining scaffold functions of PI3KC2β may lead to growth inhibitory activity whereas loss of the entire protein or reduction in its level does not. One possibility is that the remaining scaffold function of PI3KC2β complexes with Ras thereby preventing its activation [28]. Alternatively, it is possible that the anti-proliferative activity of the PI3KC2β inhibitors stems from off-target effects. Finally, since we have selected for stably silenced PI3KC2β cells, it is possible that the resulting cell lines have compensated for the loss of the protein. Nevertheless, these studies together suggest that characterizing the role of PI3KC2β may uncover mechanisms involved in NB tumorigenesis.
PI3KC2β also contributes to tumorigenesis in additional cancer types. Over-expression of PI3KC2β in colonic epithelial cells results in oncogenic transformation [18]. Single-nucleotide polymorphisms in the promoter region of PI3KC2β are also associated with increased risk for prostate cancer [15]. Furthermore, PI3KC2β mRNA is elevated in a variety of cancers including pancreatic cancer [24], mixed lineage leukemia [4] and a subset of acute myeloid leukemia [22]. In glioblastoma, ITSN1 has been shown to be required for tumorigenesis [16]; however, the contribution of PI3KC2β to this effect has not been addressed.
Silencing either PI3KC2β or ITSN1 has anti-tumorigenic effects in both MYCN-amplified and non-amplified cells suggesting that this pathway is of broad importance to NB tumorigenesis. In MYCN-amplified IMR-5 cells, depletion of either ITSN1 or PI3KC2β leads to reduced AKT activation following growth factor stimulation, although without a corresponding increase in apoptosis. Interestingly, we observed robust AKT activation in IMR-5 cells only when stimulating cells with growth factor, i.e., EGF, in the presence of serum. Serum starvation of cells prior to EGF treatment abolished AKT activation suggesting that in these cells, EGF was not sufficient for AKT activation but required additional factors present in the serum. The effects of ITSN1 or PI3KC2β on AKT activation are consistent with our previous findings demonstrating that ITSN1 and PI3KC2β regulate an AKT-dependent signaling pathway in neurons [11]. However, in this present case, loss of this pathway does not lead to increased apoptosis in NB cells in contrast to our previous observations. Although ITSN1 silencing increased apoptosis in the N1E-115 mouse NB cell line, this effect was only observed when ITSN1 silenced cells were induced to differentiate into neuron-like cells [11]. ITSN-1 silencing had no effect on the proliferation or survival of N1E-115 grown in the presence of serum. Additional studies demonstrate that expression of a PI3KC2β dominant-negative inhibits growth factor-induced activation of AKT as well as growth of human lung cancer cell lines [2]. These results raise questions regarding the specific phosphoinositide(s) produced by PI3KC2β. It has been previously shown that PI3KC2β produces PtdIns(3)P [10, 25] and PtdIns(3,4)P2 [3]. However, evidence from our lab and others showing that PI3KC2β mediates AKT activation suggests that PI3KC2β may also produce PtdIns(3,4,5)P3 [3, 11]. In contrast to the MYCN-amplified IMR-5 cells, neither ITSN1 nor PI3KC2β depletion affects EGF-induced AKT activation in MYCN non-amplified SK-N-AS cells. These results suggest that PI3KC2β and ITSN1 mediate distinct signaling pathways in MYCN-amplified vs. non-amplified cells despite expression of the receptor in both cell types. In addition, EGF treatment increased colony formation in IMR-5 cells and silencing PI3KC2β and ITSN1 abolished this effect. Using an AKT inhibitor in IMR-5 reduced the EGF-mediated increase in colony formation. In SK-N-AS EGF does not increase colony formation; however, the AKT inhibitor completely abolished colony formation suggesting that AKT is important for SK-N-AS tumorigenesis but EGF may be not as important. These results suggest a differential role for EGF-induced AKT activation in tumorigenesis of diverse NB tumor cell lines. Further experiments are needed to identify the signaling pathways through which PI3KC2β and ITSN1 regulate tumorigenesis in MYCN non-amplified NBs. Interestingly, MYCN downregulates β1integrin resulting in different levels of β1integrin in MYCN-amplified and non-amplified NBs suggesting that β1integrin may play a distinct role in these different NB subtypes [26]. In glioblastoma tumor cells, ITSN1 regulates β1integrin activation [16] suggesting a potentially similar mode of action in NBs.
ITSN1-silencing results in stable reduction of tumor growth whereas PI3KC2β-silencing results in reduction of tumor growth that recovers over time. This difference in effect may be due to a mechanism of compensation in PI3KC2β-silenced cells in which cells adapt to the lack of PI3KC2β by altering additional tumorigenic pathways. Additionally, given ITSN1's ability to couple to multiple biochemical pathways, the more potent effect of ITSN1 silencing may stem from impairment of additional downstream pathways beside PI3KC2β-AKT. We have shown that ITSN1 regulates EGFR trafficking and signaling as well as recruitment of phosphatases such as Shp2 (PTPN11) [20]. PTPN11 activating mutation have been found in NB with a frequency of 3.4% [14] suggesting that regulation of this phosphatase may be important for NB tumorigenesis. In addition, we have identified several ITSN1-binding proteins by yeast two-hybrid screening that may contribute to ITSN1 signaling in NB, such as Rab and Arf family GTPases, both of which regulate receptor endocytosis and recycling [29]. Both Rab and Arf members play a role in cancer [29]. Therefore, our future studies will define the signaling pathways mediated by PI3KC2β and ITSN1 that are important for MYCN+ vs MYCN− NB tumors. Such information may help in the development of new therapeutic strategies for treating NB patients.
Supplementary Material
Supplemental Figure 1. Analysis of tumors from PI3KC2β-silenced cells. (A&B) Tumors from PI3KC2β-depleted (βsh2 and βsh5), ITSN1-depleted (sh#1) as well as control (pSR and pSCR) IMR-5 and SK-N-AS cells were extracted, homogenized, lysed and analyzed by Western blot to assess levels of ITSN1 and PI3KC2β. (C&D) Lysates from IMR-5 and SK-N-AS cells were analyzed by Western blot to assess levels of PI3KC2β and of various PI3K isoforms. Actin is shown as a normalization control for loading.
Supplemental Figure 2. Apoptosis is unaltered in PI3KC2β-silenced cells. (A&B) Annexin V levels were measured by flow cytometry in controls [vector (pSR) or scramble (pSCR)] and in PI3KC2β-silenced (βsh2 and βsh5) polyclonal lines from both SK-N-AS (A) and IMR-5 (B) NB cells. ITSN1 silencing did not increase Annexin V staining in these cells. Results are the average +/− standard deviation of three experiments. Cells were analyzed after one day of growth in suspension.
Supplemental Figure 3. Transferrin endocytosis is unaltered in PI3KC2β-silenced cells. (A&B) Internalization of biotinylated transferrin (BTfn) in SK-N-AS (A) and IMR-5 (B) NB lines was determined by Western blot of cell lysates using HRP-conjugated streptavidin. Actin is shown as a normalization control for loading. Graphs represent average −/+ standard error of three independent experiments.
Supplemental Figure 4. EGFR endocytosis is unaltered in ITSN-silenced cells. (A&B) SK-N-AS lines were treated with EGF for indicated times and EGFR left at the cell membrane was determined by ELISA. Graphs represent average of three independent experiments.
Supplemental Figure 5. MYCN protein levels are not altered in ITSN and PI3KC2β-silenced. ITSN or PI3KC2β-silenced IMR-5 lines were analyzed by Western blot using MYCN specific antibody. Actin is shown as a normalization control for loading. Graphs represent average −/+ standard error of three independent experiments.
Highlights.
PI3KC2β contributes to neuroblastoma tumorigenesis.
MYCN-amplified vs non-MYCN amplified tumor cells demonstrate a differential dependence on PI3KC2β for their tumorigenic activity.
The ITSN1-PI3KC2β pathway regulates the AKT but not ERK-MAPK pathway activation by growth factor.
Acknowledgements
We wish to thank members of the O'Bryan lab as well as Drs. Graeme Carnegie and Andrei Karginov and members of their laboratories for helpful comments during the course of these studies. These studies were supported by grants to J.P.O from the National Institutes of Health (HL090651 and CA116708), Department of Veteran's Affairs MERIT Award (1I01BX002095), the Foundation Jerome Lejeune, the St. Baldrick's Foundation, and the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. J.P.O also received support from the Center for Clinical and Translational Science at UIC through grant UL1TR000050 from the NIH National Center for Advancing Translational Sciences. The authors wish to dedicate this manuscript to the memory of our colleague Graeme Carnegie who recently passed away.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that there are no competing financial interests in relation to the work described.
Literature Cited
- 1.Adams A, Thorn JM, Yamabhai M, Kay BK, O'Bryan JP. Intersectin, an adaptor protein involved in clathrin-mediated endocytosis, activates mitogenic signaling pathways. J Biol Chem. 2000;275:27414–27420. doi: 10.1074/jbc.M004810200. [DOI] [PubMed] [Google Scholar]
- 2.Arcaro A, Khanzada UK, Vanhaesebroeck B, Tetley TD, Waterfield MD, Seckl MJ. Two distinct phosphoinositide 3-kinases mediate polypeptide growth factor- stimulated PKB activation. EMBO J. 2002;21:5097–5108. doi: 10.1093/emboj/cdf512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arcaro A, Zvelebil MJ, Wallasch C, Ullrich A, Waterfield MD, Domin J. Class II phosphoinositide 3-kinases are downstream targets of activated polypeptide growth factor receptors. Mol Cell Biol. 2000;20:3817–3830. doi: 10.1128/mcb.20.11.3817-3830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 5.Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B, Khwaja A. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene. 2006;25:6648–6659. doi: 10.1038/sj.onc.1209670. [DOI] [PubMed] [Google Scholar]
- 6.Boller D, Doepfner KT, De Laurentiis A, Guerreiro AS, Marinov M, Shalaby T, Depledge P, Robson A, Saghir N, Hayakawa M, Kaizawa H, Koizumi T, Ohishi T, Fattet S, Delattre O, Schweri-Olac A, Holand K, Grotzer MA, Frei K, Spertini O, Waterfield MD, Arcaro A. Targeting PI3KC2beta impairs proliferation and survival in acute leukemia, brain tumours and neuroendocrine tumours. Anticancer Res. 2012;32:3015–3027. [PubMed] [Google Scholar]
- 7.Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, Matthay KK, Rowitch D, Weiss WA. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006;66:8139–8146. doi: 10.1158/0008-5472.CAN-05-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark G, Cox AD, Graham SM, Der CJ. Biological assays for Ras transformation. Methods in Enzymology. 1995;255:395–412. doi: 10.1016/s0076-6879(95)55042-9. [DOI] [PubMed] [Google Scholar]
- 10.Crljen V, Volinia S, Banfic H. Hepatocyte growth factor activates phosphoinositide 3-kinase C2 beta in renal brush-border plasma membranes. Biochem J. 2002;365:791–799. doi: 10.1042/BJ20020316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das M, Scappini E, Martin NP, Wong KA, Dunn S, Chen YJ, Miller SL, Domin J, O'Bryan JP. Regulation of neuron survival through an intersectin phosphoinositide 3′-kinase C2beta-AKT pathway. Molecular and cellular biology. 2007;27:7906–7917. doi: 10.1128/MCB.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter MP, Russo A, O'Bryan JP. Emerging Roles for Intersectin (ITSN) in Regulating Signaling and Disease Pathways. Int J Mol Sci. 2013;14:7829–7852. doi: 10.3390/ijms14047829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izbicka E, Izbicki T. Therapeutic strategies for the treatment of neuroblastoma. Curr Opin Investig Drugs. 2005;6:1200–1214. [PubMed] [Google Scholar]
- 14.Je EM, Choi YJ, Yoo NJ, Lee SH. Oncogenic PTPN11 Mutations are Rare in Solid Tumors. Pathol Oncol Res. 2015;21:225–227. doi: 10.1007/s12253-014-9780-z. [DOI] [PubMed] [Google Scholar]
- 15.Koutros S, Schumacher FR, Hayes RB, Ma J, Huang WY, Albanes D, Canzian F, Chanock SJ, Crawford ED, Diver WR, Feigelson HS, Giovanucci E, Haiman CA, Henderson BE, Hunter DJ, Kaaks R, Kolonel LN, Kraft P, Le Marchand L, Riboli E, Siddiq A, Stampfer MJ, Stram DO, Thomas G, Travis RC, Thun MJ, Yeager M, Berndt SI. Pooled analysis of phosphatidylinositol 3-kinase pathway variants and risk of prostate cancer. Cancer Res. 2010;70:2389–2396. doi: 10.1158/0008-5472.CAN-09-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Y, Wang B, Li W, Liu X, Wang J, Ding T, Zhang J, Ying G, Fu L, Gu F. Intersectin1-s is involved in migration and invasion of human glioma cells. J Neurosci Res. 2012;89:1079–1090. doi: 10.1002/jnr.22616. [DOI] [PubMed] [Google Scholar]
- 17.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3- kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Marras E, Concari P, Cortellezzi L, Dondi D, De Eguileor M, Perletti G. Involvement of PI3K in PKCepsilon-mediated oncogenic signal in rat colonic epithelial cells. Int J Oncol. 2001;19:395–399. [PubMed] [Google Scholar]
- 19.Martin NP, Mohney RP, Dunn S, Das M, Scappini E, O'Bryan JP. Intersectin regulates epidermal growth factor receptor endocytosis, ubiquitylation, and signaling. Mol Pharmacol. 2006;70:1643–1653. doi: 10.1124/mol.106.028274. [DOI] [PubMed] [Google Scholar]
- 20.Okur MN, Ooi J, Fong CW, Martinez N, Garcia-Dominguez C, Rojas JM, Guy G, O'Bryan JP. Intersectin 1 enhances Cbl ubiquitylation of epidermal growth factor receptor through regulation of Sprouty2-Cbl interaction. Mol Cell Biol. 2012;32:817–825. doi: 10.1128/MCB.05647-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24:65–86. doi: 10.1016/j.hoc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM. Expression profiling of CD34+ hematopoietic stem/ progenitor cells reveals distinct subtypes of therapy-related acute myeloid leukemia. Proc Natl Acad Sci U S A. 2002;99:14925–14930. doi: 10.1073/pnas.222491799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russo A, O'Bryan JP. Intersectin 1 is required for neuroblastoma tumorigenesis. Oncogene. 2012;31:4828–4834. doi: 10.1038/onc.2011.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato N, Fukushima N, Maitra A, Iacobuzio-Donahue CA, van Heek NT, Cameron JL, Yeo CJ, Hruban RH, Goggins M. Gene expression profiling identifies genes associated with invasive intraductal papillary mucinous neoplasms of the pancreas. Am J Pathol. 2004;164:903–914. doi: 10.1016/S0002-9440(10)63178-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srivastava S, Di L, Zhdanova O, Li Z, Vardhana S, Wan Q, Yan Y, Varma R, Backer J, Wulff H, Dustin ML, Skolnik EY. The class II phosphatidylinositol 3 kinase C2beta is required for the activation of the K+ channel KCa3.1 and CD4 T-cells. Mol Biol Cell. 2009;20:3783–3791. doi: 10.1091/mbc.E09-05-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Golen CM, Soules ME, Grauman AR, Feldman EL. N-Myc overexpression leads to decreased beta1 integrin expression and increased apoptosis in human neuroblastoma cells. Oncogene. 2003;22:2664–2673. doi: 10.1038/sj.onc.1206362. [DOI] [PubMed] [Google Scholar]
- 27.Wetzker R, Rommel C. Phosphoinositide 3-kinases as targets for therapeutic intervention. Curr Pharm Des. 2004;10:1915–1922. doi: 10.2174/1381612043384402. [DOI] [PubMed] [Google Scholar]
- 28.Wong KA, Russo A, Wang X, Chen YJ, Lavie A, O'Bryan JP. A new dimension to Ras function: a novel role for nucleotide-free Ras in Class II phosphatidylinositol 3-kinase beta (PI3KC2beta) regulation. PLoS One. 2012;7:e45360. doi: 10.1371/journal.pone.0045360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong KA, Wilson J, Russo A, Wang L, Okur MN, Wang X, Martin NP, Scappini E, Carnegie GK, O'Bryan JP. Intersectin (ITSN) family of scaffolds function as molecular hubs in protein interaction networks. PLoS One. 2012;7:e36023. doi: 10.1371/journal.pone.0036023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie J, Vandenbroere I, Pirson I. SHIP2 associates with intersectin and recruits it to the plasma membrane in response to EGF. FEBS Lett. 2008;582:3011–3017. doi: 10.1016/j.febslet.2008.07.048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Analysis of tumors from PI3KC2β-silenced cells. (A&B) Tumors from PI3KC2β-depleted (βsh2 and βsh5), ITSN1-depleted (sh#1) as well as control (pSR and pSCR) IMR-5 and SK-N-AS cells were extracted, homogenized, lysed and analyzed by Western blot to assess levels of ITSN1 and PI3KC2β. (C&D) Lysates from IMR-5 and SK-N-AS cells were analyzed by Western blot to assess levels of PI3KC2β and of various PI3K isoforms. Actin is shown as a normalization control for loading.
Supplemental Figure 2. Apoptosis is unaltered in PI3KC2β-silenced cells. (A&B) Annexin V levels were measured by flow cytometry in controls [vector (pSR) or scramble (pSCR)] and in PI3KC2β-silenced (βsh2 and βsh5) polyclonal lines from both SK-N-AS (A) and IMR-5 (B) NB cells. ITSN1 silencing did not increase Annexin V staining in these cells. Results are the average +/− standard deviation of three experiments. Cells were analyzed after one day of growth in suspension.
Supplemental Figure 3. Transferrin endocytosis is unaltered in PI3KC2β-silenced cells. (A&B) Internalization of biotinylated transferrin (BTfn) in SK-N-AS (A) and IMR-5 (B) NB lines was determined by Western blot of cell lysates using HRP-conjugated streptavidin. Actin is shown as a normalization control for loading. Graphs represent average −/+ standard error of three independent experiments.
Supplemental Figure 4. EGFR endocytosis is unaltered in ITSN-silenced cells. (A&B) SK-N-AS lines were treated with EGF for indicated times and EGFR left at the cell membrane was determined by ELISA. Graphs represent average of three independent experiments.
Supplemental Figure 5. MYCN protein levels are not altered in ITSN and PI3KC2β-silenced. ITSN or PI3KC2β-silenced IMR-5 lines were analyzed by Western blot using MYCN specific antibody. Actin is shown as a normalization control for loading. Graphs represent average −/+ standard error of three independent experiments.