

Ketenes have intrigued chemists with their unusual physical properties and their unique spectrum of chemical reactivity.[1] Ketenes are usually prepared by thermolysis, pyrolysis or in a stoichiometric manner from acyl chlorides using base-promoted elimination reactions.[2] Synthesis of ketenes from α-diazoketones by menas of the Wolff rearrangement[2d] is also noteworthy. However, all these synthetic methods have severe limitations and involve specialised equipment and/or highly unstable starting materials.
Carbonylation of metal–carbene species[3–6] is an interesting alternative method to produce such highly reactive ketenes, which find synthetic applications in various organic transformations.[1, 4, 5] The synthesis of medicinally important β-lactams by [2+2] ketene–imine cycloaddition reactions is especially important in this perspective.[5] Carbonylation reactions of Fischer carbene complexes have indeed been applied successfully to synthesise β-lactams.[5a, b] However, these are stoichiometric transformations, which hamper efficiency. Following the discovery of stoichiometric carbonylation reactions of Fischer-type carbene complexes, thus far only a few transition-metal-catalysed processes have been developed.[5, 6] While interesting results have recently been obtained with a noble (palladium) metal catalyst,[5c] carbene carbonylation by base-metal catalysts reported thus far were associated with low efficiencies and required quite harsh reaction conditions (elevated temperatures and high CO pressures).[6] Therefore, we focused on the development of new catalytic carbene carbonylation processes by catalysts based on abundant first-row transition metals that operate under comparably mild reaction conditions.
As stable metalloradicals with well-defined open-shell doublet d7-electronic configuration, cobalt(II) complexes of porphyrins [CoII(Por)] have emerged as a new class of catalysts capable of carbene-transfer reactions[7] proceeding through radical mechanisms involving discrete CoIII–carbene radical intermediates C (Scheme 1).[8]
Scheme 1.

[CoII(Por)]-catalysed carbene carbonylation leading to ketene formation in one-pot tandem transformations producing amides, esters and β-lactams. DFT calculated relative free energies (ΔGo) in kcalmol−1 between brackets (Turbomole, BP86, def2-TZVP, employing Grimme’s disp3 dispersion corrections).
In comparison with classic electrophilic Fischer-type carbenes, the increased nucleophilicity of the carbene radical intermediate C slows-down unwanted carbene dimerisation while allowing for catalytic cyclopropanation through radical addition to olefinic substrates, including electron-deficient olefins.[7, 8] In this respect, we envisioned that similar intermediates might well be effective in carbene carbonylation reactions, which can be considered as an attack of a nucleophilic cobalt–carbene radical (C) at the π-accepting CO substrate. At the same time, both the [Co(Por)] catalyst and the carbenoid intermediates are expected to interact only weakly with amines, alcohols, imines and the ketene reaction products. Hence, we expected smooth catalytic ketene formation under comparatively mild conditions and their subsequent in situ reactions with nucleophiles in one-pot tandem procedures (see Scheme 1).
Here in we report an efficient one-pot tandem protocol of the carbonylation of α-diazocarbonyl compounds and a variety of N-tosylhydrazones catalysed by CoII–porphyrin metalloradicals leading to the formation of ketenes, which subsequently react with a variety of nucleophiles and imines to form esters, amides and β-lactams. This system has a broad substrate scope and can be applied to various combinations of carbene precursors, nucleophiles and imines. The use of N-tosylhydrazones as precursors of diazo compounds in cobalt–porphyrin-based carbene-transfer reactions is unprecedented, and represents an efficient and convenient way to prepare the key carbenoid intermediates (C in Scheme 1) responsible for ketene formation. The key ketene formation steps were further investigated computationally (DFT).
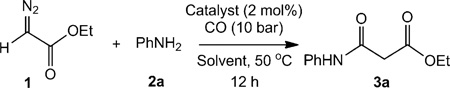
Since ketenes are highly reactive[5, 6] and can generally only be trapped in the presence of a strong nucleophile, we first evaluated the activity of [CoII(P1)] (P1 = tetraphenylporphyrin; Figure 1) in the catalytic carbonylation of ethyl diazoacetate (EDA; 1) in presence of aniline (2a) under 10 bar CO at 50 °C. Under these conditions, (ethoxycarbonyl) ketene was formed and trapped by aniline to produce ethyl 2-(phenylcarbamoyl)acetate (3a) in 57% isolated yield (Table 1, entry 1). Further initial experiments focused on the evaluation of ligand and solvent effects on the carbonylation of EDA in the presence of aniline. Four different CoII–porphyrin catalysts, including two chiral ones, were employed in an attempt to improve the catalytic process and to investigate potential asymmetric induction of the reaction (Figure 1). The catalysts [CoII(P1)], [CoII(P2)], and [CoII(P3)] (see Supporting Information for experimental details) showed more or less similar activity, while the catalyst [Co(P4)] was found to be ineffective for this reaction (Table 1). No enantioselectivity was obtained using the chiral catalysts [CoII(P3)] and [CoII(P4)] under the various reaction conditions applied. This suggests that the ketene intermediate is liberated from the catalyst to react subsequently with the amine, freely in solution.
Figure 1.

Structures of cobalt(II) complexes of porphyrins. P1 = Tetraphenylporphyrin; P2 = 3,5-DitBu-IbuPhyrin; P3=3,5-DitBu-ChenPhyrin; P4 = 21H,23H-porphine-5,10,15,20-tetrakis[(1R,4S,5S,8R)-1,2,3,4,5,6,7,8-octahydro-1,4:5,8-dimethanoanthracen-9-yl].
Table 1.
Conditions of [CoII(Por)]-catalysed β-ketoester synthesis using CO, EDA and aniline.[a]
 | |||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yield [%][b] |
| 1 | [CoII(P1)] | PhMe | 57 |
| 2 | [CoII(P1)] | THF | 25 |
| 3 | [CoII(P1)] | dioxane | 50 |
| 4 | [CoII(P1)] | DCE | 10–15 |
| 5 | [CoII(P1)] | MeCN | <5 |
| 6 | [CoII(P1)] | DMF | – |
| 7 | [CoII(P1)] | PhCl | 55 |
| 8 | [CoII(P1)] | PhMe/K3PO4 | 69 |
| 9 | [CoII(P1)] | PhMe/K2CO3 | 65 |
| 10 | [CoII(P1)] | PhMe/KHCO3 | 58 |
| 11 | [CoII(P1)] | PhMe/NEt3 | 63 |
| 12 | [CoII(P2)] | PhMe/K3PO4 | 70 |
| 13 | [CoII(P3)] | PhMe/K3PO4 | 72 |
| 14 | [CoII(P4)] | PhMe/K3PO4 | 35 |
Stoichiometry EDA:aniline = 1:2.
Isolated yields after column chromatography.
Optimisation of the reaction conditions revealed that the reaction proceeded most efficiently in nonpolar solvents, such as toluene and chlorobenzene, whereas reactions in solvents of high polarities, such as THF, dioxane, MeCN and DMF, afforded poor yields (Table 1, entries 2–6). The use of inorganic bases K2CO3 or K3PO4 further improved the yields (Table 1, entries 8–11).
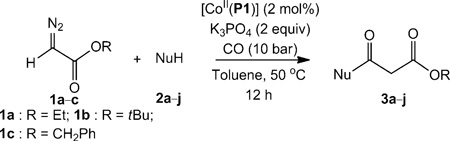
Reactions with other nucleophiles were evaluated to investigate the versatility of the reaction. The experiments indeed showed that the ketenes generated by the CoII–porphyrin-catalysed carbene carbonylation could be trapped by a wide range of nucleophiles (Table 2). The reaction occurred smoothly with substituted aromatic amines (Table 2, entries 1–5), primary and secondary aliphatic amines (Table 2, entries 6–8) and alcohols (Table 2, entries 9 and 10).
Table 2.
[CoII(Por)]-catalysed β-ketoester synthesis using CO, α-diazocarbonyl compounds and different nucleophiles.[a]
 | |||
|---|---|---|---|
| Entry | R | NuH | Yield[%][b] |
| 1 | Et | PhNH2 | 69 (3a) |
| 2 | Et | p-MeOC6H4NH2 | 64 (3b) |
| 3 | Et | p-NO2C6H4NH2 | 61 (3c) |
| 4 | Et | 3,4-Cl2C6H3NH2 | 64 (3d) |
| 5 | Et | Ph2NH | 63 (3e) |
| 6 | Et | PhCH2NH2 | 67 (3f) |
| 7 | Et | nBuNH2 | 57 (3g) |
| 8 | Et | Morpholine | 53 (3h) |
| 9 | Et | Ph(CH 2)3OH | 63 (3i) |
| 10 | Et | EtOH | 62 (3j) |
| 11 | tBu | PhNH2 | 75 (3k) |
| 12 | CH2Ph | PhNH2 | 72 (3l) |
Stoichiometry EDA:NuH = 1:2.
Isolated yields after column chromatography
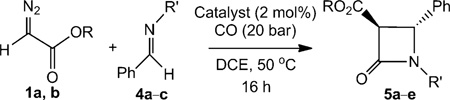
To further investigate the scope of the reaction, imines were introduced into the reaction medium in an attempt to produce β-lactams in a one-pot tandem procedure involving [2+2] cycloaddition of the intermediate ketene with the imine. Indeed, a 1:2 mixture of ethyl diazoacetate (EDA; 1) and N-methylbenzaldimine (4a) in dichloroethane (DCE) under a carbon monoxide atmosphere (20 bar) at 50°C in the presence of a catalytic amount of [CoII(P1)] (2 mol%) led to formation of trans-N-methyl-α-ethoxycarbonyl-βphenyl-β-lactam (5a) in 60% isolated yield (Table 3). Similar reactions using other imines PhCH=NR (R=CH2Ph (4b) and tBu (4c)) also produced the desired β-lactams (Table 3). Notably, the (ethoxycarbonyl)ketene generated by [CoII(P1)]-catalysed carbene carbonylation participates selectively in [2+2] cycloaddition reactions with imines to produce the desired β-lactams (Table 3).
Table 3.
[CoII(Por)]-catalysed trans-selective β-lactam synthesis from αdiazocarbonyl compounds.[a]
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Diazo | Imine | Yield [%][b] (trans:cis) |
| 1 | [CoII(P1)] | R = Et | R′ = Me | 65 (5a) (>95:5) |
| 2 | [CoII(P2)] | R = Et | R′ = Me | 65 (5a) (>95:5) |
| 3 | [CoII(P3)] | R = Et | R′ = Me | 67 (5a) (>95:5) |
| 4 | [CoII(P1)] | R = Et | R′ = CH2Ph | 50 (5b) (>90:5) |
| 5 | [CoII(P1)] | R = Et | R′ = tBu | 55 (5c) (>95:5) |
| 6 | [CoII(P1)] | R = tBu | R′ = Me | 66 (5d) (>95:5) |
Stoichiometry EDA:imine = 1:2.
Isolated yields after column chromatography.
Reactions of other diazoacetates with imines also produced the desired β-lactams (Table 3). This behaviour is markedly different from the [4+2] cycloaddition reactions producing 1,3-dioxin-4-ones reported by Wang and co-workers in related palladium-catalysed reactions (see Scheme 2).[5c]
Scheme 2.

Markedly different behaviour of [CoII(Por)] compared to [Pd2-(dba)3] in reactions with α-diazo carbonyl compounds and imines.
Again, the use of chiral catalyst [CoII(P3)] and [CoII(P4)] did not lead to any enantioselectivity, pointing to liberation of free ketene in solution reacting subsequently with the imine. Introduction of Cinchona alkaloid, quinidine and its derivatives into the reaction mixture in an attempt to trap the liberated ketene with a chiral organocatalysts[9] for chirality transfer in a one-pot cascade manner produced the βlactams with a low ee of only 5% at 30 °C. This temperature is probably much too high for efficient chirality transfer with these organocatalysts (typically operating at −78°C), but this is the lowest possible temperature at which still some conversion can be achieved with these cobalt(II)-based catalysts. Nonetheless, this result reveals the potential of combining metallo and organo catalysts in cascade processes, which may become a useful concept to achieve enantioselective ketene reactivity when combined with more active cobalt catalysts working at lower temperatures in the future.
To expand the scope of the catalytic ketene synthesis methodology, carbonylation reactions were carried out with N-tosylhydrazones,[10] which are widely used as precursors for in-situ generation of non-stabilised diazo compounds. The use of N-tosylhydrazones as precursors of diazo compounds in cobalt–porphyrin-based carbene-transfer reactions was thus far unprecedented. The easy synthesis of these N-tosylhydrazones from ketones and aldehydes combined with the above described [Co(Por)]-catalysed carbene carbonylation methodology offers a convenient method to convert an organic carbonyl moiety into a ketene functionality, applicable in one-pot three-component tandem transformations.
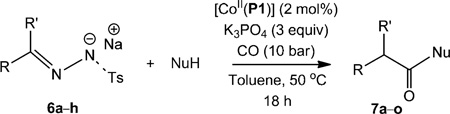
Reaction of the benzaldehyde tosylhydrazone sodium salt (6a) and aniline (2a) was catalysed by [CoII(P1)] under similar reaction conditions as mentioned above (50 °C, 10 bar CO). The benzyl ketene, thus generated in-situ, reacted with aniline to produce N-2-diphenylacetamide (7a) in 70% isolated yield (Table 4, entry 1). Further optimisation of the reaction conditions revealed that the yield could be improved by adding a phase transfer agent (Aliquat 336, trioctyl-methylammonium chloride; Table S1 in the Supporting Information). Both polar and nonpolar solvents were suitable for this reaction (Table S1 in the Supporting Information), and again toluene was the best solvent. The use of inorganic bases K2CO3 or K3PO4 further improved the yields (Table S1 in the Supporting Information).
Table 4.
[CoII(Por)]-catalysed amide/ester synthesis using N-tosylhydrazone sodium salt and different nucleophiles.[a]
 | ||||
|---|---|---|---|---|
| Entry | R | R′ | NuH | Yield [%][b] |
| 1 | Ph | H | PhNH2 | 75 (7a) |
| 2 | Ph | H | p-MeOC6H4NH2 | 79 (7b) |
| 3 | Ph | H | p-NO2C6H4NH2 | 72 (7c) |
| 4 | Ph | H | PhCH2NH2 | 75 (7d) |
| 5 | Ph | H | Ph2NH | 79 (7e) |
| 6 | Ph | H | nBuNH2 | 75 (7f) |
| 7 | Ph | H | Morpholine | 77 (7g) |
| 8 | Ph | H | Ph(CH 2)3OH | 65 (7h) |
| 9 | p-MeC6H4 | H | PhNH2 | 82 (7i) |
| 10 | o-MeC6H4 | H | PhNH2 | 75 (7j) |
| 11 | p-ClC6H4 | H | PhNH2 | 72 (7k) |
| 12 | p-MeOC6H4 | H | PhNH2 | 80 (7l) |
| 13 | 2-naph | H | PhNH2 | 77 (7m) |
| 14 | Ph | CH3 | PhNH2 | 56 (7n) |
| 15 | PhCH=CH | H | PhNH2 | 65 (7o) |
Stoichiometry N-Tosylhydrazone:NuH = 1:3, PTA used.
Isolated yields after column chromatography.
Reactions with nucleophiles other than aniline were also investigated. The experiments showed that the ketenes generated from the tosylhydrazone sodium salts by means of [CoII(P1)]-catalysed carbene carbonylation method could also be trapped by different nucleophiles. Expected products were obtained with several amines and alcohols (Table 4, entries 1–8).
These results prompted us to explore the scope of the catalytic carbene carbonylation reactions using a series of N-tosylhydrazone sodium salts under the above optimised reaction conditions. The reactions produced the expected acetamide derivatives in good to high yields (Table 4, entries 9– 13). The reaction also worked well with disubstituted N-tosylhydrazones, although requiring longer reaction times (Table 4, entry 14). Also α,β-unsaturated N-tosylhydrazones could be converted to amides, albeit in modest yields (Table 4, entry 15).
Upon introduction of imines to the reaction medium, the ketenes generated from these N-tosylhydrazone salts also reacted smoothly to produce the desired β-lactams. The use of a phase-transfer agent in this case did not have any influence. A series of N-tosylhydrazone salts were subjected to the reaction conditions with different benzaldimines (Table 5). In all cases, the corresponding β-lactams were obtained in good yields. Interestingly, for most of the substrates, the reactions afforded trans products with excellent diastereoselectivity. However, again no enantioselectivity was obtained using either chiral catalysts or by adding quinidine-based chiral organocatalysts into the reaction mixture.
Table 5.
[CoII(Por)]-catalysed trans-selective β-lactam synthesis using different N-tosylhydrazone sodium salts and imines.[a]
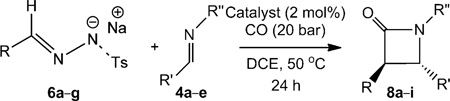 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | R | R′ | R″ | Yield (%)[b] (trans:cis) |
| 1 | [CoII(P1)] | Ph | Ph | Me | 64 (8a) (>95:5) |
| 2 | [CoII(P2)] | Ph | Ph | Me | 65 (8a) (>95:5) |
| 3 | [CoII(P3)] | Ph | Ph | Me | 65 (8a) (>95:5) |
| 4 | [CoII(P1)] | Ph | Ph | PhCH2 | 62 (8b) (>90:10) |
| 5 | [CoII(P1)] | Ph | pClC6H4 | Me | 63 (8c) (>95:5) |
| 6 | [CoII(P1)] | Ph | pOMeC6H4 | Me | 73 (8d)(> 90:10) |
| 7 | [CoII(P1)] | pMeC6H4 | Ph | Me | 77 (8e) (>95:5) |
| 8 | [CoII(P1)] | pClC6H4 | Ph | Me | 62 (8f) (>95:5) |
| 9 | [CoII(P1)] | pMeOC6H4 | Ph | Me | 73 (8g) (>85:15) |
| 10 | [CoII(P1)] | 2-napthyl | Ph | Me | 67 (8h) (>95:5) |
| 11 | [CoII(P1)] | PhCH = CH | Ph | Me | 52 (8i) (>95:5) |
Stoichiometry: N-tosylhydrazone:imine=1:2.
Isolated yields after column chromatography.
To obtain more insight into the mechanistic aspects of [Co(Por)]-catalysed ketene formation, several controlled infrared studies of the reaction mixtures were carried out. Since, diphenylketene is known to be more stable than the corresponding monosubstituted ketenes, diphenyl tosylhydrazone sodium salt was intentionally chosen as the carbene source for IR analysis of the carbene carbonylation reaction mixtures (see Supporting Information Figure S1–S4). Indeed, carbonylation of the diphenyl tosylhydrazone sodium salt with 20 bar CO produces free diphenyl ketene, as indicated by observation of a characteristic IR stretch frequency at 2102 cm−1 directly after the reaction.[11] Formation of the diphenyl diazo compound[12] from the diphenyl tosylhydrazone sodium salt was also detected by IR spectroscopy, as revealed by observation of a characteristic stretch frequency at 2042 cm−1. These data are in excellent agreement with the proposed reaction mechanism shown in Scheme 1.
Repeated attempts to detect the more reactive benzyl ketene directly after reaction of the corresponding tosylhydrazone sodium salt with 20 bar CO were unsuccessful. This is likely due to too rapid dimerisation/oligomerisation and/or other side-reactions of the ketene in absence of a nucleophile or imine. The reaction mixture obtained directly after reacting the benzaldehyde tosylhydrazone salt with 20 bar CO in the presence of N-methylbenzaldimine does show characteristic C=O stretch frequency of the β-lactam at 1752 cm−1.[5c]
The mechanism of the above carbene carbonylation reactions was further investigated computationally by using DFT methods (see Supporting Information for details). In these studies, in contrast to previous investigations reported by our groups, we decided to include dispersion (VdW) corrections in the geometry optimisations. Remarkably, this modification had a profound influence on the affinity of the cobalt center for the diazo compound (bound through carbon), shifting the association equilibrium from A to B and resulting in a somewhat lower transition state barrier TS1 (from B) for formation of the key CoIII–carbene radical intermediate C (Scheme 1) than reported previously. It is also worth noting that with dispersion forces, the carbonbound methyl diazoacetate (MDA) adduct B was calculated to be more stable than the nitrogen-bound MDA adduct B′, while this was reversed in previous calculations without dispersion corrections.[8] Apart from this interesting effect of inclusion of dispersion forces in the calculations, this part of the catalytic mechanism is in fact identical to carbenoid formation in the calculated mechanism for olefin cyclopropanation.[8] Carbene radicals C are in equilibrium with “bridging carbenes” C′ according to these calculations (nearly thermoneutral), and both C- and C′-type species were previously detected by EPR spectroscopy after reacting [CoII(P3)] with ethyl diazoacetate.[8a]
Subsequent carbonylation of terminal carbenoid C proceeds by means of a concerted one-step CO addition to the carbene moiety with simultaneous homolysis of the Co–C bond to produce the free ketene, according to DFT. The ketene has little to no affinity for cobalt, and spontaneously dissociates from the cobalt center directly after its formation. These calculations are in good agreement with the detection of free ketene in the above-mentioned IR experiments. The computed carbene carbonylation step has a somewhat lower energy barrier (ΔG*(TS2)=8.6 kcalmol−1 from C; ΔG*(TS2)=9.2 kcalmol−1 from C′) than the barrier for formation of C through dinitrogen loss from diazo adduct B (TS1=13.6 kcalmol−1).[8a] The rate-limiting step in the catalytic cycle according to these calculations under standard conditions in the gas phase is therefore the formation of carbene radical C. However, the energy differences between TS1 and TS2 are not large and at relative low CO concentrations and relative high EDA concentrations (i.e., non-standard conditions as in the catalytic experiments) the entropy contributions lower the relative barrier of TS1 compared to TS2, allowing side reactions of C (e.g., carbene dimerisation, reaction with the solvent). This, in combination with the relatively low solubility of CO in common organic solvents,[13] most likely explains why CO pressures >10 bar are required in the experimental catalytic runs.
The ketene, thus generated in-situ, has no affinity for the [Co(Por)] catalyst according to these DFT calculations, and hence most likely reacts subsequently in a noncatalysed manner with nucleophiles or imines present in solution, leading to the final ester, amide or β-lactam products.[5c, 6] Given that carbon monoxide has a weak affinity for the cobalt(II) center,[14] it is unlikely for carbon monoxide to bind to the catalyst and thus influence the carbonylation of diazo compounds under the applied catalytic reaction conditions. The more nucleophilic character of C compared to Fischertype carbenes further reduces its reactivity towards nucleophiles, which is likely an important feature in preventing unwanted side-reactions.
In summary, we have demonstrated [CoII(Por)] are effective metalloradical catalysts for carbene carbonlyation, producing ketenes from CO and diazo compounds or tosylhydrazones under mild conditions. The [CoII(Por)]-catalysed reaction, which involves a low-barrier carbene carbonylation step, provides a valuable synthetic alternative for the production of ketenes, which can be trapped in situ with amines, alcohols or imines to produce esters, amides or βlactams in a one-pot cascade manner. This straightforward methodology has a broad substrate scope and can be applied for various combinations of diazo compounds and nucleophiles or imines. In addition to diazo compounds, [CoII(Por)]-catalysed carbonylation process can also employ tosylhydrazones derived from aldehydes and ketones as carbene sources. Consequently, this procedure offers an efficient way for the homologation of ketones and aldehydes, and also provides a diastereoselective method for the transformation of aldehydes to β-lactam derivatives. Future research in this area will focus on achieving enantioselective one-pot cascade reactions involving ketene intermediates.
Supplementary Material
Acknowledgements
This work was financially supported by the European Research Council (ERC Grant Agreement 202886-CatCIR; B.d.B.), the Netherlands Organisation for Scientific Research (NWO-CW VICI grant 016.122.613; B.d.B.), the National Science Foundation (CHE-1152767; X.P.Z.), the National Institutes of Health (R01-GM098777; X.P.Z.), and the University of Amsterdam (UvA). We thank Dr. Matthias Otte for helpful discussions.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201301731.
Contributor Information
X. Peter Zhang, Email: xpzhang@usf.edu.
Bas de Bruin, Email: B.deBruin@uva.nl.
References
- 1.a) Staudinger H. Ber. Dtsch. Chem. Ges. 1905;38:1735. [Google Scholar]; b) Tidwell TT. Ketenes. Wiley; Hoboken: 2006. [Google Scholar]; c) Tidwell TT. Angew. Chem. 2005;117:5926. Angew. Chem. Int. Ed.2005, 44, 5778. [Google Scholar]; d) Paull DH, Weatherwax A, Lectka T. Tetrahedron. 2009;65:6771. doi: 10.1016/j.tet.2009.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Moore HW, Wilbur DS. J. Org. Chem. 1980;45:4483. [Google Scholar]; b) Kresze G, Runge W, Ruch E. Justus Liebigs Ann. Chem. 1972;756:112. [Google Scholar]; c) Newman MS, Arkell A, Fukunaga T. J. Am. Chem. Soc. 1960;82:2498. [Google Scholar]; d) Chiang Y, Kresge AJ, Popik VV. J. Am. Chem. Soc. 1999;121:5930. [Google Scholar]
- 3.a) de Meijere A, Schirmer H, Duetsch M. Angew. Chem. 2000;112:4124. doi: 10.1002/1521-3773(20001117)39:22<3964::aid-anie3964>3.0.co;2-c. Angew. Chem. Int. Ed.2000, 39, 3964. [DOI] [PubMed] [Google Scholar]; b) Grotjahn DB, Bikzhanova GA, Collins LSB, Concolino T, Lam KC, Rheingold AL. J. Am. Chem. Soc. 2000;122:5222. [Google Scholar]
- 4.a) Dötz KH. Angew. Chem. 1975;87:672. Angew. Chem. Int. Ed. Engl.1975, 14, 644. [Google Scholar]; b) Dötz KH, Tomuschat P. Chem. Soc. Rev. 1999;28:187. [Google Scholar]
- 5.a) Hegedus LS. Tetrahedron. 1997;53:4105. [Google Scholar]; b) Hegedus LS. Topics Organomet. Chem. 2004;13:157. [Google Scholar]; c) Zhang Z, Liu Y, Ling L, Li Y, Dong Y, Gong M, Zhao X, Zhang Y, Wang J. J. Am. Chem. Soc. 2011;133:4330. doi: 10.1021/ja107351d. [DOI] [PubMed] [Google Scholar]
- 6.a) Miyashita A, Shitara H, Nohira H. J. Chem. Soc. Chem. Commun. 1985:850. [Google Scholar]; b) Huser M, Youinou M, Osborn JA. Angew. Chem. 1989;101:1427. Angew. Chem. Int. Ed. Engl.1989, 28, 1386. [Google Scholar]; c) Barletta J, Karimi F, Doi H, Långström B. J. Labelled Compd. Radiopharm. 2006;49:801. [Google Scholar]; d) Ungvári N, Fördős E, Balogh J, Kégl T, Párkányi L, Ungváry F. Organometallics. 2010;29:3837. [Google Scholar]
- 7.a) Doyle MP. Angew. Chem. 2009;121:864. Angew. Chem. Int. Ed.2009, 48, 850. [Google Scholar]; b) Huang LY, Chen Y, Gao GY, Zhang XP. J. Org. Chem. 2003;68:8179. doi: 10.1021/jo035088o. [DOI] [PubMed] [Google Scholar]; c) Penoni A, Wanke R, Tollari S, Gallo E, Musella D, Ragaini F, Demartin F, Cenini S. Eur. J. Inorg. Chem. 2003:1452. [Google Scholar]; d) Chen Y, Zhang XP. J. Org. Chem. 2007;72:5931. doi: 10.1021/jo070997p. [DOI] [PubMed] [Google Scholar]; e) Chen Y, Ruppel JV, Zhang XP. J. Am. Chem. Soc. 2007;129:12074. doi: 10.1021/ja074613o. [DOI] [PubMed] [Google Scholar]; f) Zhu SF, Ruppel JV, Lu HJ, Wojtas L, Zhang XP. J. Am. Chem. Soc. 2008;130:5042. doi: 10.1021/ja7106838. [DOI] [PubMed] [Google Scholar]; g) Zhu SF, Perman JA, Zhang XP. Angew. Chem. 2008;120:8588. doi: 10.1002/anie.200803857. Angew. Chem. Int. Ed.2008, 47, 8460. [DOI] [PubMed] [Google Scholar]; h) Fantauzzi S, Gallo E, Rose E, Raoul N, Caselli A, Issa S, Ragaini F, Cenini S. Organometallics. 2008;27:6143. [Google Scholar]; i) Ruppel JV, Gauthier TJ, Snyder NL, Perman JA, Zhang XP. Org. Lett. 2009;11:2273. doi: 10.1021/ol9005882. [DOI] [PubMed] [Google Scholar]; j) Zhu SF, Xu X, Perman JA, Zhang XP. J. Am. Chem. Soc. 2010;132:12796. doi: 10.1021/ja1056246. [DOI] [PubMed] [Google Scholar]; k) Chen Y, Zhang XP. J. Org. Chem. 2004;69:2431. doi: 10.1021/jo049870f. [DOI] [PubMed] [Google Scholar]
- 8.a) Dzik WI, Xu X, Zhang XP, Reek JNH, de Bruin B. J. Am. Chem. Soc. 2010;132:10891. doi: 10.1021/ja103768r. [DOI] [PubMed] [Google Scholar]; b) Lu H, Dzik WI, Xu X, Wojtas L, de. Bruin B, Zhang XP. J. Am. Chem. Soc. 2011;133:8518. doi: 10.1021/ja203434c. [DOI] [PubMed] [Google Scholar]; c) Dzik WI, Zhang XP, de Bruin B. Inorg. Chem. 2011;50:9896. doi: 10.1021/ic200043a. [DOI] [PubMed] [Google Scholar]; d) Dzik WI, Reek JNH, de Bruin B. Chem. Eur. J. 2008;14:7594. doi: 10.1002/chem.200800262. [DOI] [PubMed] [Google Scholar]
- 9.Tian S-K, Chen Y, Hang J, Tang L, McDaid P, Deng L. Acc. Chem. Res. 2004;37:621. doi: 10.1021/ar030048s. [DOI] [PubMed] [Google Scholar]
- 10.Fulton JR, Aggarwal VK, de Vicente J. Eur. J. Org. Chem. 2005:1479. [Google Scholar]
- 11.a) Grotjahn DB, Collins LSB, Wolpert M, Bikzhanova GA, Christine Lo H, Combs D, Hubbard JL. J. Am. Chem. Soc. 2001;123:8260. doi: 10.1021/ja004324z. [DOI] [PubMed] [Google Scholar]; b) Wagner BD, Arnold BR, Brown GS, Lusztyk J. J. Am. Chem. Soc. 1998;120:1827. [Google Scholar]
- 12.Bertani R, Biasiolo M, Darini K, Michelin RA, Mozzon M, Visentin F, Zanotto L. J. Organomet. Chem. 2002;642:32. [Google Scholar]
- 13.a) Magee MP, Li HQ, Morgan O, Hersh WH. Dalton Trans. 2003:387. [Google Scholar]; b) Wilhelm E, Battino R. Chem. Rev. 1973;73:1. [Google Scholar]
- 14.a) Mu XH, Kadish KM. Inorg. Chem. 1989;28:3743. [Google Scholar]; b) Wayland BB, Minkiewicz JV, Abd-Elmageed ME. J. Am. Chem. Soc. 1974;96:2795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.