

Anterior pituitary cell types express distinct hormone gene products. These include ACTH, GH, PRL, TSH, FSH and LH. Pituitary adenomas are distinguished by both excess proliferation of one of these differentiated cell types, as well as dysregulated specific hormone hypersecretion. Physiologically, the six pituitary trophic hormones regulate respective target hormone production and endocrine gland function in tightly controlled regulatory loops (Figure 1). Adenoma hypersecretion is usually a reflection of dysregulated hormone synthesis and or secretion.
Figure 1.
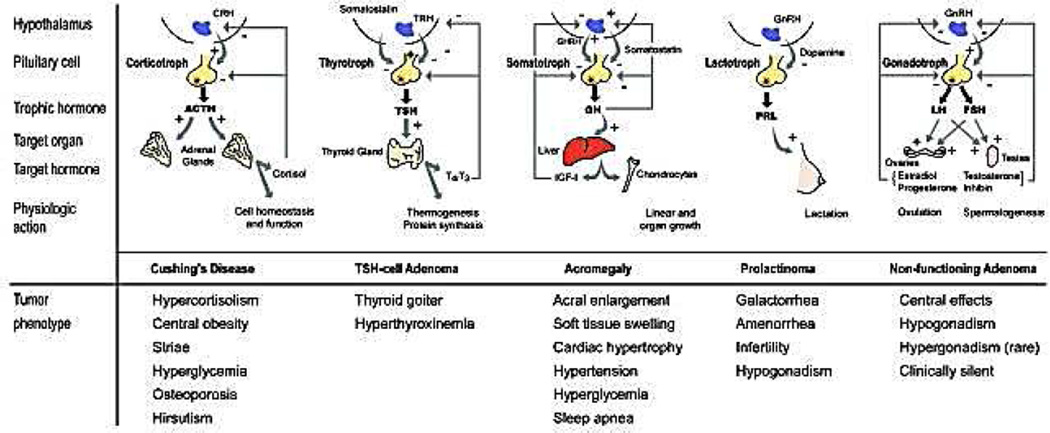
Hormone secretion from pituitary tumors, although excessive and associated with unique phenotypic features, often retains intact trophic control. For example, dopaminergic agents appropriately suppress PRL secretion by prolactinomas, and dexamethasone may suppress ACTH secretion in patients with pituitary Cushing disease.
From Melmed S 2003 Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest 112:1603-1618, with permission.
Each of these cell-specific adenomas are associated with a benign sellar mass, as well as a unique clinical syndrome associated with the hormone being hypersecreted (12). Thus, lactotroph adenomas result in hyperprolactinemia, with features of hypogonadanism accompanied by galactorrhea. Corticotroph adenomas hypersecrete ACTH leading to features of hypercortisolism associated with central obesity, hypertension, hyperglycemia, infections and psychologic disturbances. Somatotroph adenomas hypersecrete GH, leading to features of acromegaly including acral changes, arthritis, hypertension, headache, soft tissue swelling and hyperglycemia. Thyrotroph adenomas hypersecreting TSH are extremely rare, and may be associated with mild hyperthyroidisms and goiter. Tumors arising from gonadotroph cells rarely hypersecrete intact FSH or LH, and more commonly express either glycoprotein subunits, or no excess hormone. These adenomas are often discovered incidentally, and are usually associated with hypogonadism and pituitary failure, because of compressive effects of the expanding mass (Table 1).
Table 1.
Classification of pituitary adenomas
| Cell tvDe Adenoma Type | Population Prevalence ftotal/lO5) | Tumor transcription factor expression | Unregulated differentiated gene expression | Clinical features |
|---|---|---|---|---|
| Lactotroph Sparsely or densely granulated | 45–50 | Pit-1 | Prolactin | Hypogonadism and/or galactorhea |
| Gonadotroph | 15–20 | SF-l,GATA-2 | FSH and/or LH and/or glycoprotein subunit. (depending on type, null or oncocytic) | Silent or pituitary failure Ovarian hyperstimulation (reproductive age women) Testicular enlargement (prepubertal) |
| Somatotroph | 10 | Pit-1 | ||
| Sparsely granulated | GH | Acromegaly or gigantism | ||
| Densely granulated | GH | Acromegaly | ||
| Combined GH and | GHand | Acromegaly or | ||
| PRL cells | prolactin | gigantism | ||
| Mixed GH and | GHand | Hypogonadism and | ||
| PRL | prolactin GH and | acromegaly Acromegaly or | ||
| Mammos omatotroph | prolactin | gigantism | ||
| Acidophil Stem | Prolactin and | Hypogonadism and | ||
| Cell | GH | acromegaly | ||
| Silent | GH | Hypopituitarism | ||
| Corticotroph | 5 | T-Pit | ACTH | |
| Cushing | Cushing Disease | |||
| Silent | Hypopituitarism | |||
| Nelson | Pituitary hyperplasia | |||
| Thyrotroph | <1 | Pit-1 | TSH | Hyperthyroidism |
| Plurihormonal | Unknown | All | GH, prolactin, ACTH, glycoprotein | Mixed |
All tumor types exhibit features of a pituitary mass, visible on MRI. Glycoprotein refers to intact FSH or LH, or respective α or β glycoprotein subunits. Although null-cell tumors do not express hormone genes, they are classified as gonadotroph in origin.
Abbreviations: ACTH, adrenocorticotropic hormone; FSH, follicle-stimulating hormone; GATA2, endothelial transcription factor GATA-2; GH, growth hormone; LH, luteinizing hormone; PIT1, pituitary-specific positive transcription factor 1; STF1, steroidogenic factor 1; TBX19, T-box transcription factor TBX19 (also known as TPIT).
From Melmed S 2011 Pathogenesis of pituitary tumors. Nat Rev Endocrinol 7:257-266, with permission.
All these adenomas, regardless of cell type, are invariably benign. Although often locally aggressive and invasive, they very rarely progress to true malignancy with documented extracranial metastases (1). Mechanisms underlying the constraints buffering pituitary adenomas against malignant transformation include development of DNA damage and premature proliferative arrest, i.e. cell senescence. Senescent pituitary cells are growth-constrained by CDK inhibitors including p21 for somatotroph tumors. p27 for corticotroph tumors and p15/p165 for non-functioning adenomas (3). These CDK inhibitors lead to cell cycle arrest, while maintaining differentiated hormone secretion and preventing the malignant transformation of respective adenoma cell types.
Another pathogenetic feature of these tumors is their monoclonality (7). As pituitary tumors appear to arise from a single cell, it would be unlikely that broad, generalized signals such as hypothalamic hormone excess, or estrogen excess, lead to polyclonal hyperplasia with resultant hormone hypersecretion. In support of this postulate, activating mutations of hypothalamic hormone receptors in pituitary adenomas (GHRHR; CRHR; GnRHR; TRHR) have not been reported. Similarly, inactivating mutations of hypothalamic inhibitory factor receptors (SSTR1-5; D2R) have not been reported. Consistent with these observations, adenoma monoclonality is also suggested by the fact that surgical resection of small (<10 mm) discrete hormone-secreting adenomas (especially those expressing GH or ACTH) may result in long-term remission with sustained hormonal control. Interestingly, pituitary tissue surrounding the adenoma does not exhibit features of hyperplasia, further suggesting the discreet monocellular pathogenesis of these adenomas.
Multiple extracellular and intracellular signals determine pituitary cell proliferation and specific hormone synthesis and secretion (Figure 2). Mitogenic hormones (e.g. hypothalamic GHRH, CRH or GnRH) or growth factors (e.g. EGF) or steroids (e.g. estrogen) may induce pituitary cell proliferation as well as transcription of pituitary hormone genes. These mitogenic inputs are balanced by important constraining factors of pituitary cell proliferation and hormone synthesis including hypothalamic somatostatin and dopamine. Hormones and growth factors also drive the cell cycle mediated by CDK-cyclin complexes and Rb phosphorylation. Release of E2F by phosphorylated Rb acts to drive cell cycle progression. This complex process is disrupted in adenomas, leading to chromosomal instability and DNA damage (2). In fact, pituitary adenomas are remarkably aneuploidy despite their benign phenotypes. Importantly, dysregulated hormone gene transcription occurs pari passa with cell proliferative changes (11). It is apparent that disruption or activation of multiple sites within the hormone-secreting adenoma cell leads to the dual outcome of adenoma formation with subsequent proliferation, associated with a unique hormone hypersecretory phenotype.
Figure 2.
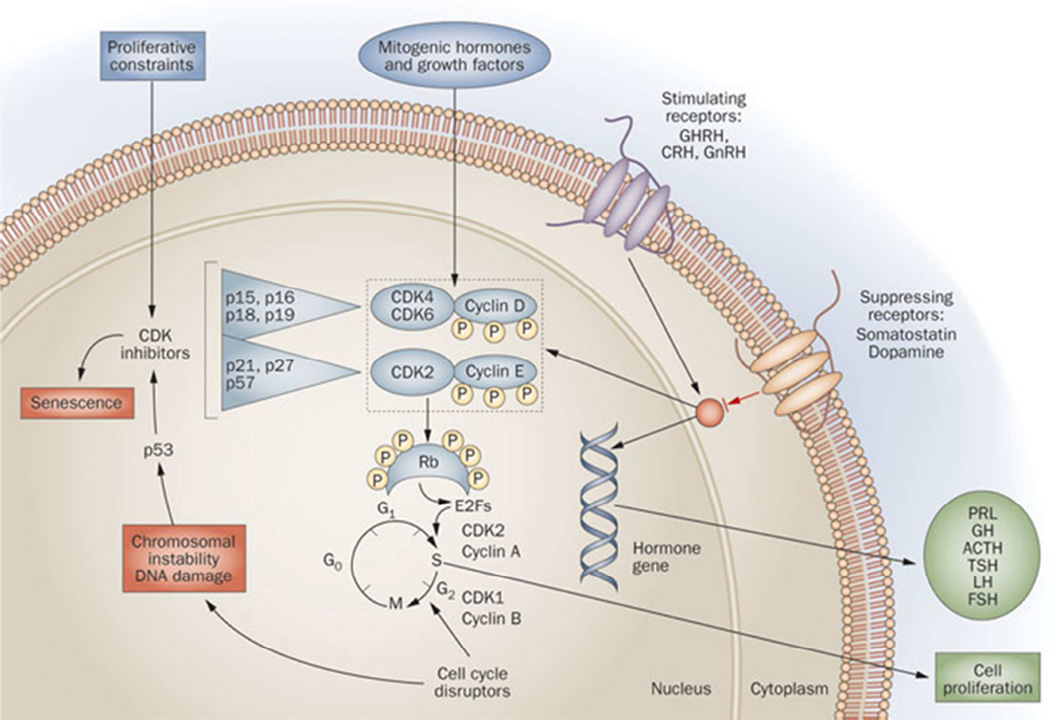
Pituitary adenoma signaling. Transcription of pituitary hormone genes and cell proliferation are induced by pituitary mitogenic factors including hypothalamic hormones and transcription factors, as well as peripheral hormones. Proliferative constraints include somastostatin and dopamine, as well as tumor suppressor genes. Cell cycle progression is mediated by CDK–cyclin complexes that phosphorylate Rb and cause it to release E2F, which drives cell proliferation. CDK inhibitors block kinase phosphorylation, thereby restraining cell cycle progression. Chromosomal instability, DNA damage and senescence may act to constrain malignant transformation of pituitary tumors.
From Melmed S 2009 Acromegaly pathogenesis and treatment. J Clin Invest 119:3189-3202, with permission.
Animal models
Although classic oncogene mutations are not encountered in sporadic tumors, several growth factor, hormone and cell cycle genes are reported to determine development of sporadic pituitary adenomas (13). Many have been tested in genetically modified mice which either exhibit gain of function or loss of function (Table 2). For example, heterozygote Rb+/− mutant mice develop pituitary adenomas with high penetrance, usually arising from the intermediate lobe with overexpressed POMC (8). Although several reported transgenic knock-in, universal knockout, and conditional knockout mice may faithfully recapitulate human phenotypes with specific cell type adenoma development and hormone hypersecretion, few such models have yielded a viable therapeutic drug discovery.
Table 2.
Animal models of pituitary tumors include spontaneous tumors
| Model type | Genetic modification | Type of tumor |
|---|---|---|
|
TRANSGENIC | ||
| PyLT | Polyoma large T antigen overexpression | Corticotropinoma |
| POMC-SV40 large T antigen | SV40 large T antigen overexpression (POMC promoter) | Intermediate lobe |
| AVP-SV40 large T antigen | SV40 large T antigen overexpression (AVP promoter) | GH-producing adenomas |
| FSHβ-SV40 large T antigen | SV40 large T antigen overexpression (FSHβ promoter | Null cell adenomas |
| hGRHH | GHRH overexpression | Somatotrophs and mammosomatotrophs |
| PRL-TGFα | TGFα overexpression | Prolactinomas |
| PRL-ptd-FGFR4 | Overexpression of N-terminally truncated isoform of FGFR4 | Prolactinomas |
| αGSU-PTTG1 | PTTG1 overexpression | Gonadotroph hyperplasia |
| CMV-HMGA1 | HMGA1 overexpression | Mixed growth hormone/prolactin adenomas |
| CMV-HMGA2 | HMGA2 overexpression | Somatotrophs, lactotrophs and mammosomatotrophs |
|
CONVENTIONAL KNOCK OUT | ||
| Men1 heterozygous | Men1 inactivation | Prolactinomas |
| Men1 heterozygous | Men1 inactivation | Prolactinomas, NFPA |
| Men1 heterozygous | Men1 inactivation | Prolactinomas, GH-producing adenomas |
| Men1 heterozygous | Men1 inactivation | Mammosomatotrophs (90%). ACTH-producing intermediate lobe (10%) |
| AIP heterozygous | AIP inactivation | GH-producing adenomas (90%), prolactinomas |
| Drd2 homozygous | Drd2 inactivation | Prolactinomas |
| Prl homozygous | Prl inactivation | Prolactinomas |
| Prlr homozygous | Prl receptor inactivation | Prolactinomas |
| Rb haterozygous | Rb inactivation | ACTH-producing intermediate lobe |
| P27Kip1 homozygous | p27Kip1 inactivation | Intermediate lobe |
| p18Ink4c homozygous | p18Ink4c inactivation | intermediate and anterior lobe |
| Compound p18Ink4c p27Kip1 homozygous | Double p18Ink4c and p27Kip1 inactivation | Intermediate and anterior lobe |
| Cdk4R24C/R24C mutant mice | Mutation rendering the Cdk4 protein insensitive to INK4 inhibitors | Anterior lobe |
| Compound Cdk4R24C/R24C p27Kip1 homozygous mutant mice | Mutation rendering the Cdk4 protein insensitive to INK4 inhibitors and and p27Kip1 inactivation | Poorly differentiated adenomas |
|
CONDITIONAL KNOCK OUT | ||
| rGhrhr-Cre PRKAR1A | Pituitary-specific (somatotrophs, lactotrophs, and thyrotrophs) PRKAR1A inactivation | Somatotrophs, lactotrophs, and thyrotrophs |
| POMC-specific Rb heterozygous | Rb inactivation in POMC-expressing cells | Intermediate lobe |
| POMC-specific Rb homozygous | Rb inactivation in POMC-expressing cells | Intermediate lobe |
| POMC-specific Rb homozygous | Rb inactivation in POMC-expressing cells | Not reported |
| Ins-Cre Men1 | Men1 homozygous inactivation in pituitary due to ectopic Cre expression | Prolactinomas |
| GFAP-Cre Bmi1 | Bmi1 homozygous pituitary-specific inactivation | Intermediate lobe |
| Hesx1-Cre; Ctnnb1lox(ex3)+ Sox2-CreERT2;Ctnnb1lox(ex3)+ | Pituitary-specific overexpression of constitutively active form of β-catenin | Adamantinomatous, craniopharyngioma |
Animal models of pituitary tumors include spontaneous tumors, subcutaneous xenografted implantation, and genetically engineered mouse models may (transgenic and knockout mice). Adapted from Cano DA, Soto-Moreno A, Leal-Cerro A 2014 Front Oncol Aug 1;4:203, with permission.
Transgenic overexpression or disruption of several cell cycle-associated genes have been observed to uniquely lead to pituitary tumor formation in several animal models (Table 3). These experimental tumors exhibit varying degrees of penetrance, may be located in the anterior or intermediate lobes, and may or may not hypersecrete specific trophic hormones with resultant peripheral hormonal phenotypes. Two examples are presented of how further unraveling pituitary tumor signaling pathways in transgenic animal models may lead to translational discovery of novel subcellular therapeutic targets:
Table 3.
Selected cell cycle-associated genes associated with pathogenesis of pituitary adenomas
| Gene | Function | Mode of activation/inactivation |
|---|---|---|
| CCNB2 | Cyclin | Induced by HMGA |
| CCND1 | Oncogene | Overexpression |
| CDKNIB | CDK inhibitor | Inactivating |
| HMGA2 | Oncogene | Overexpression |
| FGFR4 | Oncogene | Alternative transcription |
| PTTG | Securin | Overexpression |
| Rb | Tumor suppressor | Epigenetic silencing |
| CDKN2A | Cyclin-dependent kinase inhibitor | Epigenetic silencing |
Select genes that contribute to the molecular pathogenesis of pituitary adenomas
Adapted from Melmed S 2009 Acromegaly pathogenesis and treatment. J Clin Invest 119:3189-3202, with permission.
TARGETING CDK SIGNALLING
PTTG, pituitary tumor transforming gene, isolated from rat pituitary tumor cells is the index mammalian securin, and drives pituitary tumor formation in murine and zebrafish models (15). The gene is also induced by estrogen in experimental prolactinomas (6) and is overexpressed in human pituitary tumor cells (16). In an attempt to recreate an ACTH-secreting pituitary adenoma, the zebrafish Pttg gene was linked to the POMC promoter to create a corticotroph targeted transgene. Transgenic zebrafish expressing pituitary corticotroph-directed zpttg develop phenotypic features which recapitulate human Cushing disease. These include corticotroph hyperplasia, hypercortisolism, hyperglycemia and fatty liver and cardiac effusions and hypertrophy (9). Furthermore, partial glucocorticoid resistance was also demonstrated. Using this animal model as a drug therapy, screening target, the transgenic zebrafish model was utilized for screening of small molecules exhibiting specific CDK inhibitory activity in the hyperplastic transgenic pituitary. Accordingly, R-Roscovitine, a selective CKD inhibitor, was shown to suppress tumorous pituitary POMC gene expression and ACTH secretion in both zebrafish and murine models of ACTH-secreting corticotroph cell adenomas. The drug also reversed phenotypic features in tumor-bearing mice. Suppressive effects of R-Roscovitine on ACTH secretion were also recapitulated in cell cultures derived from resected human pituitary tumor specimens (9). Based on these translational studies elucidating mechanisms for pituitary tumorigenesis, R-Roscovitine has now been approved to enter clinical trials to test for safety and efficacy in patients with ACTH-secreting pituitary adenomas and Cushing disease.
TARGETING Erb SIGNALLING
Mechanisms underlying prolactinoma pathogenesis are challenging to elucidate due to the unavailability of functional prolactin-secreting human pituitary tumor cell lines, as well as to the relative sparseness of available pituitary tumor tissue specimens. Both ErbB family receptors and ligands are expressed in normal anterior pituitary cells. Activation of this signaling pathway leads to induced PRL production by regulating PRL gene expression and lactotroph differentiation (14). To further elucidate mechanisms underlying prolactinoma tumorigenesis, transgenic mice expressing EGFR or HER2 driven by the tissue-specific rPRL enhancer/promoter to achieve pituitary over-expression were generated lactotroph-targeted murine transgenic hEGFR/HER2 expression induced murine prolactinomas secreting high PRL levels. When EGFR/HER2 activity was inhibited by treating mice with lapatinib, a dual tyrosine kinase inhibitor (TKI) for both EGFR and HER2, hyperprolactinemia and tumor MAPK and Akt pathways were attenuated (10).
Furthermore, in both ACTH-secreting murine AtT20 cells, as well as in primary human cell cultures derived from resected ACTH-secreting tumors were suppressed by TKI treatment. In ATCH-secreting AtT20 cells, overexpression of EGFR led to activation of MAPK-Erk signaling, which was inhibited by gefitinib, an EGFR TKI (5).
These observations provide a compelling rationale for the feasibility of employing ErbB targeted therapy for patients with prolactinomas and those with Cushing disease. Indeed, in a pilot clinical study, lapatanib was shown to attenuate PRL secretion in two patients harboring aggressive prolactinoma found to be tumors resistant to high dose dopamine agonist therapy (4).
SUMMARY
Pituitary tumors are commonly encountered intracranial neoplasms which are invariably benign. Classic oncogene mutations are not encountered in these tumors, and disrupted cell cycle control and growth factor signaling likely contribute to pathogenesis and natural history. They exhibit unique clinical features which are determined by the secreted hormone gene product.
Key Points.
Pituitary tumors are commonly encountered intracranial neoplasms which are invariably benign
Classic oncogene mutations are not encountered in these tumors, and disrupted cell cycle control and growth factor signaling likely contribute to pathogenesis and natural history
Pituitary tumors exhibit unique clinical features which are determined by the secreted hormone gene product.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES:
Dr. Melmed has received research funding from the NIH.
References
- 1.Chesnokova V, Zhou C, Ben-Shlomo A, Zonis S, Tani Y, Ren SG, Melmed S. Growth hormone is a cellular senescence target in pituitary and nonpituitary cells. Proc Natl Acad Sci U S A. 2013;110:E3331–E3339. doi: 10.1073/pnas.1310589110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chesnokova V, Zonis S, Kovacs K, Ben-Shlomo A, Wawrowsky K, Bannykh S, Melmed S. p21(Cip1) restrains pituitary tumor growth. Proc Natl Acad Sci U S A. 2008;105:17498–17503. doi: 10.1073/pnas.0804810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chesnokova V, Zonis S, Zhou C, Ben-Shlomo A, Wawrowsky K, Toledano Y, Tong Y, Kovacs K, Scheithauer B, Melmed S. Lineage-specific restraint of pituitary gonadotroph cell adenoma growth. PLoS One. 2011;6:e17924. doi: 10.1371/journal.pone.0017924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper O, Mamelak A, Bannykh S, Carmichael J, Bonert V, Lim S, Cook-Wiens G, Ben-Shlomo A. Prolactinoma ErbB receptor expression and targeted therapy for aggressive tumors. Endocrine. 2014;46:318–327. doi: 10.1007/s12020-013-0093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, Melmed S. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest. 2011;121:4712–4721. doi: 10.1172/JCI60417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heaney AP, Fernando M, Melmed S. Functional role of estrogen in pituitary tumor pathogenesis. J Clin Invest. 2002;109:277–283. doi: 10.1172/JCI14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S. Clonal origin of pituitary adenomas. J Clin Endocrinol Metab. 1990;71:1427–1433. doi: 10.1210/jcem-71-6-1427. [DOI] [PubMed] [Google Scholar]
- 8.Jacks T. Tumor suppressor gene mutations in mice. Annu Rev Genet. 1996;30:603–636. doi: 10.1146/annurev.genet.30.1.603. [DOI] [PubMed] [Google Scholar]
- 9.Liu NA, Jiang H, Ben-Shlomo A, Wawrowsky K, Fan XM, Lin S, Melmed S. Targeting zebrafish and murine pituitary corticotroph tumors with a cyclin-dependent kinase (CDK) inhibitor. Proc Natl Acad Sci U S A. 2011;108:8414–8419. doi: 10.1073/pnas.1018091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, Kano M, Araki T, Cooper O, Fukuoka H, Tone Y, Tone M, Melmed S. ErbB receptor-driven prolactinomas respond to targeted lapatinib treatment in female transgenic mice. Endocrinology in press. 2014 doi: 10.1210/en.2014-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119:3189–3202. doi: 10.1172/JCI39375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112:1603–1618. doi: 10.1172/JCI20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011;7:257–266. doi: 10.1038/nrendo.2011.40. [DOI] [PubMed] [Google Scholar]
- 14.Murdoch GH, Potter E, Nicolaisen AK, Evans RM, Rosenfeld MG. Epidermal growth factor rapidly stimulates prolactin gene transcription. Nature. 1982;300:192–194. doi: 10.1038/300192a0. [DOI] [PubMed] [Google Scholar]
- 15.Pei L, Melmed S. Isolation and characterization of a pituitary tumor-transforming gene (PTTG) . Mol Endocrinol. 1997;11:433–441. doi: 10.1210/mend.11.4.9911. [DOI] [PubMed] [Google Scholar]
- 16.Vlotides G, Eigler T, Melmed S. Pituitary tumor-transforming gene: physiology and implications for tumorigenesis. Endocr Rev. 2007;28:165–186. doi: 10.1210/er.2006-0042. [DOI] [PubMed] [Google Scholar]