

Abstract
Angelman syndrome (AS) is a single gene disorder characterized by intellectual disability, developmental delay, behavioral uniqueness, speech impairment, seizures, and ataxia1,2. It is caused by maternal deficiency of the imprinted gene UBE3A, encoding an E3 ubiquitin ligase3-5. All patients carry at least one copy of paternal UBE3A, which is intact but silenced by a nuclear-localized long non-coding RNA, UBE3A antisense transcript (UBE3A-ATS)6-8. Murine Ube3a-ATS reduction by either transcription termination or topoisomerase I inhibition increased paternal Ube3a expression9,10. Despite a clear understanding of the disease-causing event in AS and the potential to harness the intact paternal allele to correct disease, no gene-specific treatment exists for patients. Here we developed a potential therapeutic intervention for AS by reducing Ube3a-ATS with antisense oligonucleotides (ASOs). ASO treatment achieved specific reduction of Ube3a-ATS and sustained unsilencing of paternal Ube3a in neurons in vitro and in vivo. Partial restoration of UBE3A protein in an AS mouse model ameliorated some cognitive deficits associated with the disease. Although additional studies of phenotypic correction are needed, for the first time we developed a sequence-specific and clinically feasible method to activate expression of the paternal Ube3a allele.
Phosphorothioate modified chimeric 2′-O-methoxyethyl (2′-MOE) DNA ASOs (n=240) were designed complementary to a 113-kb region of mouse Ube3a-ATS downstream of the Snord115 cluster of snoRNAs (Fig. 1a). Following nuclear hybridization of the ASO to the target RNA, RNase H cleaves the RNA strand of the ASO-RNA heteroduplex resulting in subsequent RNA degradation by exonucleases11. A high-throughput imaging screen identified ASOs that unsilenced the Ube3a paternal allele. Primary neurons from Ube3a+/YFP (PatYFP) knock-in mice12 were cultured and treated with ASO (15 μM, 72 h), and we determined the fold increase of paternal UBE3AYFP signal in NeuN-positive cells (Fig. 1b). The negative control non-targeting ASO had no effect on fluorescence (0.96 ± 0.01) whereas the positive control topoisomerase I inhibitor (topotecan, 300 nM) increased fluorescence (3.61 ± 0.00). ASO A and ASO B had an increase in paternal UBE3AYFP fluorescence of 2.11 ± 0.02 and 2.47 ± 0.03, respectively (Fig. 1c). ASOs modulated RNA expression in a dose-dependent manner with greater than 90% reduction of Ube3aYFP-ATS (Fig. 1d, upper) within 48 h of treatment (Fig. 1d, lower).
Figure 1. Unsilencing of the Ube3a paternal allele by Ube3a-ATS targeted ASOs in cultured mouse neurons.

a, Schematic mouse Ube3a genomic locus. IC, imprinting center. b, UBE3AYFP fluorescence (arbitrary units, a.u.) in ASO-treated primary neurons relative to untreated control. Ctl ASO, non-targeting control ASO. c, YFP fluorescent imaging of treated PatYFP neurons. d, Normalized mRNA levels in PatYFP neurons treated with increasing dose (upper panel) or for increasing time (lower panel). e, Northern blot of Snord116 expression. Snord116 intensity relative to 5.8S rRNA is quantified. f, Normalized mRNA levels of long genes. g, Western blot (upper) and qRT-PCR (lower) from PatYFP neurons. ASO, inactive is a sequence-matched RNase H inactive ASO. *P<0.05, two-tailed t-test, n=2 per group, mean ± absolute deviation. h, Western blot from WT or AS primary neurons. UBE3A signal intensity was quantified relative to α-Tubulin. i, DNA methylation analysis of the PWS imprinting center. The paternal allele was distinguished by the conversion of a CpG dinucleotide (CG > AA) in CAST.Chr7 mice.
Snrpn, Snord116, and Snord115 are processed from the same precursor transcript as Ube3a-ATS (Fig. 1a) and are critical genes in Prader-Willi Syndrome (PWS)13. Their expression was not affected by increasing dose or time of ASO treatment (Fig. 1d and Fig. 1e). The ability to down-regulate Ube3a-ATS without affecting Snord116 expression can be attributed to a fast rate of Snord116 splicing (approximately 30 min) relative to the length of time required for transcription of the 332 kb region between Snord116 and the ASO binding site (approximately 80 min) (Extended Data Fig. 1). While Ube3a-ATS ASOs did not affect expression of mature Snord116 or its precursor, ASOs designed directly to Snord116 strongly reduced Snord116 and the entire Ube3a-ATS precursor transcript (Extended Data Fig. 1).
ASO treatment (10 μM, 24 h) specifically reduced Ube3a-ATS (1,000 kb) without affecting expression of five other long genes (Nrxn3, 1,612 kb; Astn2, 1,024 kb; Pchd15, 828 kb; Csmd1, 1,643 kb; Il1rapl1, 1,368 kb), whereas topotecan (300 nM, 24 h), which acts by impairing transcription elongation14, strongly inhibited their expression (Fig. 1f).
Primary neurons from PatYFP mice treated with ASO (10 μM, 72 h) or topotecan (300 nM, 72 h) resulted in biallelic UBE3A protein expression due to unsilencing of the paternal allele (Fig. 1g). Additionally, ASO treatment of primary neurons from Ube3aKO/+ (AS) mice15 achieved 66-90% wild-type (WT) levels of UBE3A protein (Fig. 1h). ASO treatment (10 μM) did not affect DNA methylation at the PWS imprinting center (Fig. 1i). A sequence-matched ASO that was rendered unresponsive to RNase H by complete modification with 2′-MOE nucleotides (ASO, inactive) did not affect paternal UBE3A expression, indicating reduction of the antisense transcript is required for paternal Ube3a unsilencing (Fig. 1g).
While reduction of the antisense transcript was required, additional studies indicated it was not sufficient for paternal Ube3a unsilencing. ASOs complementary to the region of Ube3aYFP-ATS upstream of Ube3a (non-overlapping ASOs, n=15) up-regulated Ube3aYFP RNA 7.4 ± 0.6 fold relative to untreated control neurons (Extended Data Fig. 2). ASOs complementary to the region of Ube3aYFP-ATS located within the Ube3a gene body (overlapping ASOs, n=12) only up-regulated Ube3aYFP RNA 1.7 ± 0.2 fold. Because both non-overlapping and overlapping ASOs reduced Ube3aYFP-ATS to a similar level, a mechanism independent of the presence of the long non-coding RNA may play a role in Ube3a silencing.
Next, we tested if central nervous system (CNS) administration of Ube3a-ATS ASOs unsilenced paternal Ube3a in vivo. A single intracerebroventricular (ICV) injection of ASO was administered into the lateral ventricle of adult PatYFP mice. The ASO treatment was generally well tolerated, despite transient sedation following surgery. No significant changes in body weight, microglial activation marker (AIF1) expression, or astrocyte marker (GFAP) expression were observed one month post-treatment (Extended Data Fig. 3). Four weeks post-treatment, ASO A and ASO B reduced Ube3a-ATS RNA by 60-70% and up-regulated paternal Ube3aYFP RNA 2 to 5-fold in the brain and spinal cord (Fig. 2a). However, compared to Ube3aYFP/+ (MatYFP) mice, ASO treatment did not fully unsilence the paternal allele. Ube3aYFP RNA in ASO-treated PatYFP mice was 30-40% the level in MatYFP mice (Fig. 2a). Western blot quantification showed that UBE3AYFP protein was up-regulated in the cortex (82 ± 7%), hippocampus (33 ± 3%), and thoracic spinal cord (73 ± 33%) in ASO A-treated PatYFP mice compared to MatYFP mice (Fig. 2b). No significant down-regulation of Snrpn, Snord116, Snord115, or the sentinel long genes was observed, including any Snord116 reduction in the hypothalamus (Fig. 2a, Fig. 2c, and Extended Data Fig. 4).
Figure 2. A single administration of Ube3a-ATS ASOs resulted in paternal UBE3A unsilencing for 4 months.

a and b, mRNA levels (a), and UBE3AYFP protein (b) in cortex, hippocampus, and spinal cord 4 wk after ICV injection of PBS or ASO to PatYFP mice. MatYFP mice are included for comparison. c, Normalized mRNA levels of long genes in the cortex. d and e, RNA levels (d) and UBE3AYFP protein (e) in ASO-treated PatYFP mice 2 to 20 wk post-treatment. *P<0.05, **P<0.005, two-tailed t-test, n=3-4 per group, mean ± s.e.m. For Western blot quantification, YFP signal intensity was calculated relative to α-Tubulin.
Following a single ASO dose, Ube3a-ATS reduction was sustained for 16 wk in the CNS, and returned to basal expression by 20 wk post-treatment (Fig. 2d). Both the RNA and protein levels of paternal UBE3AYFP were significantly higher than PBS-treated mice at 2 to 16 wk post-treatment, and returned to the silenced state 20 wk post-treatment (Fig. 2d, e). No significant changes in Snrpn, Snord115, or Snord116 expression were observed (Fig. 2d). Immunostaining on brain sections 16 wk post-treatment further confirmed the long stability of the ASO and duration of paternal UBE3A protein expression (Extended Data Fig. 5). This result is consistent with the long stability of other centrally administered ASOs that are chemically modified to resist intracellular nuclease degradation16,17.
Following ICV delivery the ASO displayed widespread bilateral distribution throughout the brain, as demonstrated by immunostaining (Fig. 3a), and in situ hybridization confirmed the in vivo down-regulation of Ube3a-ATS (Fig. 3b). UBE3AYFP protein was expressed in ASO positive cells (Fig. 3c). Increased UBE3AYFP signal was detected in NeuN positive cells throughout the brain (Fig. 3d and Extended Data Fig. 6 and 7). However, paternal unsilencing was not complete compared to the maternal UBE3AYFP level, consistent with the Western blot analysis. To further increase the concentration of ASO in the brain, intrahippocampal delivery of ASO A was performed in PatYFP mice and complete unsilencing of UBE3AYFP was observed near the injection site (Extended Data Fig. 8).
Figure 3. Widespread distribution of paternal UBE3A unsilencing throughout the brain.

Imaging of brain sections 4 wk post-treatment of non-targeting control ASO (Ctl ASO) or Ube3a-ATS ASOs (ASO A and/or B) in PatYFP mice, as labelled. a, ASO immunofluorescence on whole brain coronal sections. b, In situ hybridization of Ube3a-ATS. c, High magnification staining of UBE3AYFP and ASO. d, Immunofluorescence for ASO, UBE3AYFP, and NeuN in critical brain regions. MatYFP mice treated with PBS were included for expression comparison.
Based on the ability of ASO A to up-regulate UBE3A, it was chosen for assessment of phenotypic correction in AS mice. AS mice phenocopy the impaired motor coordination and memory deficit observed in AS patients15. They have additional phenotypes including obesity, hypoactivity, and decreased marble burying behavior18-20. Sex-matched AS littermates at two to four months of age were treated with ASO A or non-targeting control ASO (Ctl ASO). To determine the ability of ASO A to correct expression and behaviors relative to WT levels, a group of PBS-treated WT mice was included. Following a single ICV injection, AS mice treated with ASO A showed reduction of Ube3a-ATS and partial restoration of UBE3A protein in the cortex (35 ± 19%), hippocampus (35 ± 15%) and cerebellum (47 ± 7%) compared to WT mice (Fig. 4b and Extended Data Fig. 9). UBE3A immunofluorescence also showed partial restoration of UBE3A protein in these brain regions (Fig. 4c and Extended Data Fig. 9). Four weeks after treatment, the mice were subjected to behavioral tests. A reversal of contextual freezing comparable to normal behavior was observed in ASO A-treated AS mice [ANOVA, F(2,39)=5.242, P<0.01], indicating the memory impairment was reversed (Fig. 4d and Extended Data Fig. 9). However, there was no difference between mice treated with ASO A or Ctl ASO in open field, marble burying, and accelerating rotarod tests (Extended Data Fig. 9). Complete phenotypic reversal may require treatment before a critical developmental window, a longer recovery time for rewiring of neural circuits, or a higher UBE3A induction level. Body weight was measured in a set of female mice that were injected at three months of age and followed for five months (Fig. 4e and Extended Data Fig. 9). The obesity phenotype in AS mice was corrected one month after treatment, and body weight remained significantly decreased compared to control ASO-treated mice for five months.
Figure 4. ASO administration in adult AS mice unsilenced paternal UBE3A and ameliorated abnormal phenotypes.

a, Experimental schedule. b, Western blot with anti-UBE3A in brain regions of treated mice. Quantification of UBE3A normalized to α-Tubulin is indicated below the images. c, UBE3A immunofluorescence in WT or AS mice. d, Contextual fear measured during the fear conditioning assay. *P<0.05, one-way ANOVA with Newman–Keuls post-hoc, n=13-15 per group. e, Growth curve of age-matched female mice, *P<0.05, **P<0.01 (ASO A versus Ctl ASO), two-way ANOVA of repeated measurements with Newman–Keuls post-hoc), n=5 per group. Ctl or Ctl ASO, non-targeting control ASO.
The genomic organization and regulation at the imprinting control center is highly conserved between mouse and human. Therefore, ASO-mediated reduction of UBE3A-ATS is expected to restore UBE3A mRNA and protein in AS patient neurons. It is believed that maternal deficiency of UBE3A causes the majority of phenotypic findings in AS, and it is reasonable to expect that all AS patients, regardless of exact genotype, would benefit enormously from restored UBE3A expression. ASO therapy has been tested for neurological diseases in non-human primates and human clinical trials via intrathecal administration, with no serious adverse events16,21-23. Well tolerated delivery, broad tissue distribution, and long duration of action sets a framework for ASOs as a viable therapeutic strategy for CNS diseases and builds enthusiasm toward further development of an ASO drug for AS.
Methods
Animals
All the animals of WT, Ube3aKO/+ [15], and Ube3a+/YFP [12] were kept on C57BL/6 background and housed under the standard conditions in a pathogen-free mouse facility. All animal procedures were performed in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine and Isis Pharmaceuticals, Inc. To generate AS mice, Ube3aKO/+ mice were born to mothers who carry the mutation on their paternal chromosome. WT and AS littermates were housed in the same cage whenever possible.
Oligonucleotide synthesis
Synthesis and purification of all chemically modified oligonucleotides was performed as previously described24. The 2′ MOE gapmer ASOs are 20 nucleosides in length, wherein the central gap segment comprising ten 2′-deoxynucleosides is flanked on the 5′ and 3′ wings by five 2′-MOE modified nucleosides. All internucleoside linkages are phosphorothioate linkages, and all cytosine residues are 5′-methylcytosines. The RNase H inactive ASO consists of twenty 2′-MOE modified nucleosides. The sequences of the ASOs are as follows: Ctl ASO, 5′-CTCAGTAACATTGACACCAC -3′25; ASO A, 5′- GATCCATTTGTGTTAAGCTG-3′; ASO B, 5′-CCAGCCTTGTTGGATATCAT-3′; ASO116, 5′-CAGAGTTTTCACTCATTTTG-3′.
Primary neuron culture and ASO treatment
Primary cultures of hippocampal and cortical neurons were established as previously described8 from P0-P2 offspring of WT C57BL/6 or Ube3a+/YFP mice. Four days after plating, half of the medium was replaced and the ASO (10 μM) or topotecan (300 nM) was added to the culture medium for 72 h, unless otherwise noted. Arabinofuranosyl cytidine (Sigma) was used to inhibit glial proliferation.
Immunofluorescence
Primary neurons were fixed with 4% paraformaldehyde (PFA) for 1 h washed in phosphate buffered saline (PBS). For in vivo samples, mice were anesthetized and perfused with PBS and 4% PFA. Brain tissue was fixed with PFA overnight and dehydrated in 30% sucrose. Coronal sections of 35 μm were prepared and stained as previously described9. The following antibodies were used: anti-GFP (ab13970, Abcam, 1:1000), anti-NeuN (MAB377, Millipore, 1:1000 dilution), anti-UBE3A (A300-352A, Bethyl Laboratories,1:500), and anti-ASO (Isis, 1:10,000)26. For the high throughput in vitro ASO screen, the plates were imaged with ImageXpressUltra confocal system (Molecular Device) and then further processed with the MetaXpress software (Molecular Device). Typically 200-800 cells were scored per well and the signal intensities were averaged, and normalized to untreated control cells. For tissue sections, images were taken using a confocal microscope (Leica).
Quantitative RT-PCR (qRT-PCR)
Total cellular RNA was isolated from cultured neurons and mouse tissue using the RNeasy kit (Qiagen). For preparation of mouse tissue, samples were first lysed using FastPrep Lysing Matrix Tubes (MP-Biomedicals) in RLT buffer containing 1% β-mercaptoethanol. On-column DNase digestion was performed for all samples. For qRT-PCR, approximately 10 ng RNA was added to EXPRESS One-Step SuperScript qRT-PCR Kit (Life Technologies) with Taqman primer and probe sets or EXPRESS One-Step SYBR GreenER Kit (Life Technologies) with SYBR primer sets (see Extended Data Table 1 for sequences). All quantification was performed by the relative standard curve method and normalized to total RNA by Ribogreen or to the housekeeping genes Gapdh.
DNA methylation analysis
Primary neuron cultures were derived from the F1 hybrid of CAST.chr7 male and C57BL/6 female mice and treated with ASO (10 μM, 72 h). Genomic DNA was then extracted and processed for bisulfite sequencing of the PWS imprinting center at the Snrpn DMR1 region (Snrpn promoter and exon 1) as previously described8.
Northern blot
Total RNA was isolated from ASO-treated primary neurons (10 μM, 72 h) by TRIzol (Life Technologies) according to the manufacturer’s protocol. 3 μg total RNA was separated on an 8% polyacrylamide-7M urea gel, and then transferred by semi-dry transfer (12 V, 30 min) to GeneScreen plus hybridization transfer membrane (Perkin Elmer). The Northern probes were 5′ end labelled with ATP Gamma 32P (Perkin Elmer) using T4 polynucleotide kinase (New England Biolabs), and then hybridized to the membrane at 42°C for 30 min. After washing membrane in wash buffer (2X SSC containing 0.1% SDS), the membrane was exposed to a PhosphorImager and quantified. The oligonucleotide probe sequences used were Snord116 5′-TTCCGATGAGAGTGGCGGTACAGA-3′ and 5.8S rRNA 5′-TCCTGCAATTCACATTAATTCTCGCAGCTAGC-3′.
Western blot
Cultured neurons and mouse tissue were homogenized and lysed in RIPA buffer (Sigma-Aldrich) containing EDTA-free cOmplete Protease Inhibitor Cocktail (Roche). Protein concentration of the supernatant was determined by the DC protein assay (Bio-Rad). 10-40 μg protein lysate was separated on a precast 4-20% Bis-Tris gel (Life Technologies) and transferred by iBlot (Life Technologies). The following primary antibodies were diluted in Odyssey blocking buffer: anti-UBE3A (611416, BD Biosciences, 1:500), anti-GFP (NB600-308, Novus Biologicals, 1:500), anti-β-Tubulin (T9026, Sigma, 1:20,000), and anti-α-Tubulin (T5168, Sigma, 1:8,000). Following primary antibody incubation, membranes were probed with goat anti-rabbit IRDye 680LT (LiCor) or goat anti-mouse IRDye 800CW (LiCor) and imaged and quantified using the LiCor Odyssey system.
ASO in vivo administration
Lyophilized ASOs were dissolved in sterile PBS without calcium or magnesium and quantified by ultraviolet (UV) spectrometry. The ASOs were then diluted to the desired concentration required for dosing mice and sterilized through a 0.2 μm filter. Mice were anesthetized with 2% isoflurane and placed in a stereotaxic frame (David Kopf Instruments). After exposing the skull, a needle (Hamilton, 1701 RN 10 μl micro syringe, needle 26s/2"/2) was used to penetrate the skull at 0.2 mm posterior and 1.0 mm lateral to the bregma, and lowered to a depth of 3.0 mm, to deliver PBS or ASO (ASO A, 700 μg; ASO B, 500 μg) at a rate of approximately 1 μl /30 s. The needle was left in place for 5 min, slowly withdrawn and the incision was sutured. For intrahippocampal injection, the coordinate of −2.0 mm anterior, 1.5 mm lateral, and −2.0 mm dorsal to the bregma was used.
Fluorescence in-situ hybridization (FISH)
Tissue preparation and RNA FISH was carried out by the RNA In-Situ Hybridization Core at Baylor College of Medicine, as previously described9. Primers for DNA template synthesis are 5′-ATTTAGGTGACACTATAGAAGCGAAGATGAGTCAGTTTGGTTTT-3′ and 5′-TAATACGACTCACTATAGGGAGATTCTGAGTCTTCTTCCATAGC-3′. The T7 promoter was used to generate the Ube3a-ATS probe.
Behavioral tests
Three groups of age and sex-matched littermates were generated, and mice were randomly assigned to each treatment group. At 2 to 4 months of age, AS mice received a single 700 μg dose of non-targeting control ASO or ASO A. WT mice injected with an equal volume of PBS were included as controls. Four weeks post-treatment, a battery of behavioral tests was performed by an experimenter blind to the genotype and treatment group using a protocol previously described9 in the Neurobehavioral Core at Baylor College of Medicine. The open field and marble burying tests were performed on day 1, the accelerating rotarod test was performed on day 2 and 3, and the fear conditioning test was performed on day 4 and 5. Mice were acclimated to the test room for 30 min before each behavior test.
Open field assay
Each mouse was placed in the center of a clear Plexiglas (40 x 40 x 30 cm) open-field arena (Versamax Animal Activity Monitor, AccuScan Instruments, Columbus, OH) and allowed 30 min to explore. Overhead lighting was ~800 lux inside the field, and the white noise was at ~60 dB. Mouse activity was recorded and quantified.
Marble burying test
Each mouse was placed in a standard mouse cage containing 20 small (1.5-2 cm) clean black marbles on top of 4 inches of corn cob bedding, forming 4 rows of 5 columns. After a period of 30 min exploration, the mouse was removed from the cage and the number of marbles buried at least 50% was recorded.
Accelerating rotarod
The test was performed with a rotating rod system that rotates from 4 to 40 rpm within 5 min (model 7650 Rota-rod, Ugo Basile, Collegeville, PA). Mice were placed on the rotating rod and the time until falling off or losing balance (mice not walking on the rod for two consecutive turns) was recorded. For two consecutive days, four trials were performed per day with at least a 30 min interval between trials.
Contextual fear conditioning
On the training day, each mouse was placed in a test chamber. After two minutes of free exploration (baseline/pre-shock freezing), the mouse received an auditory tone (2800 Hz, 85 db, 30 s) followed by a foot-shock (0.7 mA, 2 s). The training was repeated once. The mouse remained in the chamber for one additional min (post-shock freezing) and then was returned to the home cage. Twenty-four hours after training, mice were returned to the same test chamber for 5 min and tested for freezing in response to the training context (contextual freezing). Afterwards, the environmental settings of the test chamber were drastically altered and the mice were placed back in the modified context. They were allowed 3 min of free exploration, and then the auditory tone was presented for 3 min to test the fear response to the cue (cued freezing). Freezing frequency was analyzed with FreezeFrame software (San Diego Instruments, San Diego, CA).
Isolation of nascent RNA
Nascent RNA was isolated using the Click-iT Nascent RNA Capture Kit (Life Technologies), according to the manufacturer’s protocol. In brief, WT primary neurons were incubated with 5-ethynyl uridine (EU, 0.5 mM) for 0 to 150 minutes at which time total RNA was isolated by TRIzol. 5 μg total RNA was biotinylated with 0.5 mM biotin azide, and 500 ng biotinylated RNA was precipitated on streptavidin beads. Nascent EU-containing RNA captured on the beads was used for SuperScript VILO cDNA synthesis (Life Technologies) followed by qPCR.
Extended Data
Extended Data Figure 1. ASOs targeting Snord116 reduced Ube3a-ATS pre-mRNA.

a, Upper panel, schematic of the ASO binding sites and location of qRT-PCR primer and probe sets. Lower panel, qRT-PCR from WT primary neurons treated with ASO A or ASO116 (72 h) using primer and probe sets to the indicated regions of Ube3a-ATS pre-mRNA and mRNA. b, Nascent transcripts were isolated from WT primary neurons incubated with 5-ethynyl uridine (see Methods) for the indicated time. qRT-PCR for pre-mRNA and mature mRNA (HG, host gene) within the Snord116 region. The red line indicates the 30 min delay between the appearance of pre-mRNA and mature mRNA. Assuming a transcription elongation rate of 4 kb/min, it would take RNAPII 80 min to transcribe the 332 kb distance from the last copy of Snord116 to the ASO binding site. n=2 per group, mean ± absolute deviation.
Extended Data Figure 2. ASOs complementary to two regions of Ube3a-ATS differed in their ability to unsilence paternal Ube3a.
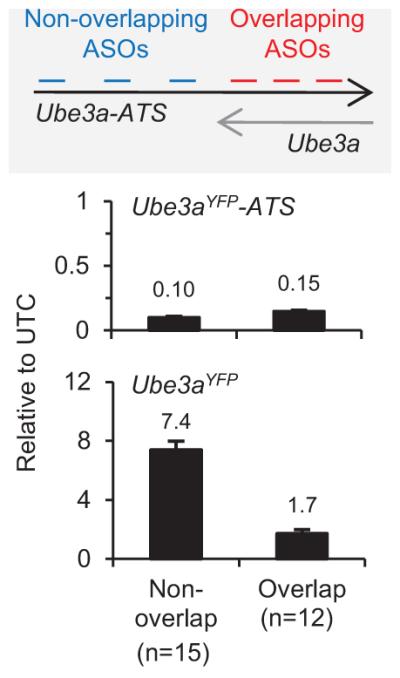
PatYFP primary neurons were treated with ASOs that bind Ube3a-ATS 5′ of Ube3a (non-overlap ASOs, n=15) or that bind to the gene body region (overlap ASOs, n=12) for 72 h. The level of Ube3aYFP-ATS reduction and Ube3aYFP up-regulation was analyzed by qRT-PCR and normalized to untreated control (UTC) neurons. Mean ± s.e.m.
Extended Data Figure 3. In vivo ASO administration was well tolerated.

a, Left, body weight of individual WT C57BL/6 female mice (2 months old) treated with PBS or ASO measured weekly for 4 wk post-treatment. Right, Change in body weight at each time point relative to body weight at time of treatment. n=4 per group, mean ± s.e.m. b, Percent change in body weight of PatYFP mice 4 wk post-treatment relative to pre-treatment. n=3-4, mean ± s.e.m. c, Microglial activation was measured by Aif1 qRT-PCR 4 wk post-treatment. CTX, cortex; HIP, hippocampus; SC, thoracic spinal cord. *P<0.05, two-tailed t-test, n=3-4 per group, mean ± s.e.m. d, Immunohistochemistry for AIF1 and GFAP on sagittal brain sections from WT C57BL/6 female mice treated with PBS or ASO for 2 wk.
Extended Data Figure 4. Snord116 was not reduced in the hypothalamus.
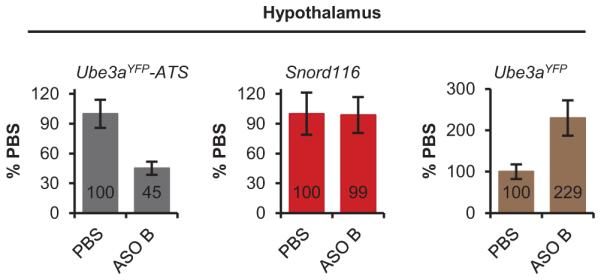
qRT-PCR on RNA isolated from PatYFP mice 4 wk post-treatment of PBS or ASO B.
Extended Data Figure 5. UBE3A unsilencing persisted 4 months post-treatment.
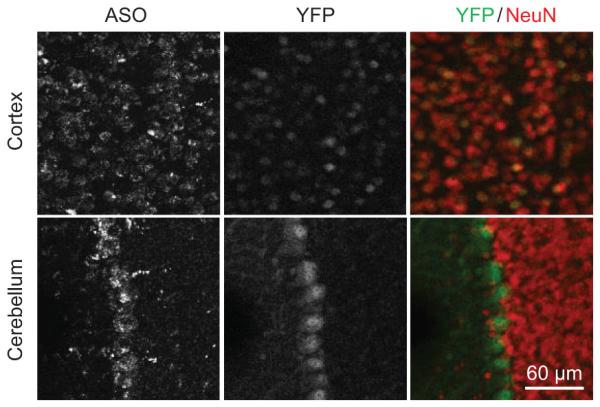
ASO and YFP immunofluorescence on brain sections of cortex and cerebellum in PatYFP mice 4 months post-treatment of ASO A.
Extended Data Figure 6. UBE3AYFP was up-regulated throughout the brain.

Whole brain image of YFP fluorescence in PatYFP mice treated with PBS or ASO 4 wk post-treatment.
Extended Data Figure 7. Imaging of unsilenced UBE3AYFP in specific brain regions.

Immunofluorescence for ASO, UBE3AYFP, and NeuN 4 wk post-treatment in MatYFP or PatYFP mice of the amygdala, hippocampus CA2 and CA3 layers, dentate gyrus, and striatrum (panel 1); thalamus, hypothalamus, medulla, and third ventricle (panel 2); motor cortex, somatosensory cortex, auditory cortex, and visual cortex (panel 3).
Extended Data Figure 8. Intrahippocampal injection of ASO A in PatYFP mice resulted in near complete unsilencing of paternal UBE3AYFP.

YFP immunofluorescence on brain sections from PatYFP mice treated with non-targeting control ASO (Ctl ASO), 100 μg ASO A via intrahippocampal injection, or 700 μg ASO A via ICV injection. A MatYFP mouse treated with PBS was included for comparison.
Extended Data Figure 9. ASO treatment in AS mice up-regulated Ube3a.

a, RNA levels of Ube3a-ATS and Ube3a were determined by qRT-PCR in WT mice treated with PBS and AS mice treated with non-targeting control ASO (ctl ASO), or ASO A. n=2-3 per group, mean ± s.e.m. b, UBE3A immunofluorescence on brain sections was performed 2 to 8 wk post-treatment. c-h, ASO treatment in adult AS mice did not reverse some disease-associated phenotypes. c, Total distance traveled in the open field assay. d, Vertical activity in the open field assay. e, Stereotype activity in the open field assay. f, Marble burying test. The y axis represents the number of marbles at least 50% buried. g, Accelerating rotarod test during eight trials. h, Post-shock and cued response measured during the fear conditioning assay. n=13-15 per group *P<0.05, ***P<0.001 (one way ANOVA with Newman-keuls post-hoc analysis). i, Growth curve of age-matched female mice. Each line represents weight measurements of a single mouse over a 5 month time course post-injection, n=5 per group. Tx age, age of mouse at time of treatment.
Extended Data Table 1.
qRT-PCR primer sequences
| Target | Forward Sequence | Reverse Sequence | Probe Sequence (Taqman) |
|---|---|---|---|
| Ube3aYFP-ATS | CCAATGACTCATGATTGTCCTG | GGTACCCGGGGATCCTCTAG | |
| Ube3aYFP | TGGAGGACTAGGAAAATTGAAGATG | CGCCCTTGCTCACCATG | CCAAAAATGGCCCAGACACAGAAAGG |
| Ube3a-ATS | CCAATGACTCATGATTGTCCTG | GTGATGGCCTTCAACAATCTC | |
| UbeSa | GCACCTGTTGGAGGACTAGG | GTGATGGCCTTCAACAATCTC | |
| Snrpn | TGTGATTGTGATGAGTTCAGGAAGA | ACCAGACCCAAAACCCGTTT | CAAGCCAAAGAATGCAAAACAGCCAGAA |
| Snord116 | GGATCTATGATGATTCCCAG | GGACCTCAGTTCCGATGA | |
| Snord116HG | TGTGCTGACTTGCCCTAG | GTTCGATGGAGACTCAGTTGG | AAACATGCAGAGGAAATGGCCCC |
| Snord116pre | ATTGGTCCCACTGTAATCGG | GTTCGATGGAGACTCAGTTGG | AAACATGCAGAGGAAATGGCCCC |
| Snord115 | CTGGGTCAATGATGACAAC | TTGGGCCTCAGCGTAATCC | |
| Snord115HG | CAGCAATCCCTCTCCAGTTC | AAGGTGGCATGTGAGATGAC | TGTGACCATTCCTACTCTGAGCCAGTT |
| Snord115pre | CCATGTGACCATTCCTACTCTG | AGAATTCGGCTACATCTACTTGG | TGGAAAGGAAGGTAAGTGTTTGGATTAGGT |
| Ipw | GCTGATAACATTCACTCCCAGA | GAATGAGCTGACAACCTACTCC | TTGGACACCCTTGCAGAAGATGACTT |
| Gapdh | GGCAAATTCAACGGCACAGT | GGGTCTCGCTCCTGGAAGAT | AAGGCCGAGAATGGGAAGCTTGTCATC |
| Pgk1 | ATGTCGCTTTCCAACAAGCTG | GCTCCATTGTCCAAGCAGAAT | |
| Nrxn3 | GATGAAGACTTTGTCGAATGTGA | CCGTCTGATTCCTGGCTCCGTG | GACCATCCCTGTTGTACTGC |
| Astn2 | CAGCACCACTACAACTCTCAC | TCACTCCTCCAGACGAAGTCACCA | CGCAGAATCAGATGAGCCTT |
| Pchd15 | CGGCGAAGTCATTGGTGAA | ACCAACTTGATCATTCTTTTTCTTGCCCAC | GTTCTGCTTCTCTGCGACTC |
| Csmd1 | GCTGCCATTCTTGTTCCCT | ACTTTTGGTCTTGTTCTGTGTTTGTAGAGGT | TCAAATGAAGCTTGTCCATTACTG |
| Il1rapl1 | CTTGAAATCCTCCCTGATATGCT | CCAACTGGAACATACATTGAAGATGTGGCT | CCGCTTACTTTGATCTACGCA |
| Aif1 | TGGTCCCCCAGCCAAGA | CCCACCGTGTGACATCCA | AGCTATCTCCGAGCTGCCCTGATTGG |
Acknowledgements
We thank D. Liu and X. Jun for assistance with high throughput imaging, H. Zheng and J. Rosen for surgical equipment, C. Spencer and X. Zhai for phenotyping assistance, and M. Costa-Mattioli and J. Jankowsky for discussions. This research was supported by funding from NIH grant R01 HD037283 (A.L.B.), Angelman Syndrome Foundation (A.L.B. and L.M.), and BCM Intellectual and Developmental Disability Research Core grant P30HD024064.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information
Reprints and permissions information is available at www.nature.com/reprints. A.J.W., C.F.B., and F.R. are employees of Isis Pharmaceuticals. L.M. and A.L.B. declare no competing financial interests.
References
- 1.Dagli A, Buiting K, Williams CA. Molecular and Clinical Aspects of Angelman Syndrome. Molecular syndromology. 2012;2:100–112. doi: 10.1159/000328837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2010;12:385–395. doi: 10.1097/GIM.0b013e3181def138. [DOI] [PubMed] [Google Scholar]
- 3.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nature genetics. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 4.Matsuura T, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nature genetics. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 5.Albrecht U, et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nature genetics. 1997;17:75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- 6.Rougeulle C, Cardoso C, Fontes M, Colleaux L, Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nature genetics. 1998;19:15–16. doi: 10.1038/ng0598-15. [DOI] [PubMed] [Google Scholar]
- 7.Chamberlain SJ, Brannan CI. The Prader-Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics. 2001;73:316–322. doi: 10.1006/geno.2001.6543. [DOI] [PubMed] [Google Scholar]
- 8.Meng L, Person RE, Beaudet AL. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Human molecular genetics. 2012;21:3001–3012. doi: 10.1093/hmg/dds130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meng L, et al. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS genetics. 2013;9:e1004039. doi: 10.1371/journal.pgen.1004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang HS, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481:185–189. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H, et al. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. The Journal of biological chemistry. 2004;279:17181–17189. doi: 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- 12.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Human molecular genetics. 2008;17:111–118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 13.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 14.King IF, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang YH, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 16.Kordasiewicz HB, et al. Sustained Therapeutic Reversal of Huntington's Disease by Transient Repression of Huntingtin Synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigo F, et al. Pharmacology of a central nervous system delivered 2'-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. The Journal of pharmacology and experimental therapeutics. 2014;350:46–55. doi: 10.1124/jpet.113.212407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cattanach BM, et al. A candidate model for Angelman syndrome in the mouse. Mamm Genome. 1997;8:472–478. doi: 10.1007/s003359900479. [DOI] [PubMed] [Google Scholar]
- 19.Allensworth M, Saha A, Reiter LT, Heck DH. Normal social seeking behavior, hypoactivity and reduced exploratory range in a mouse model of Angelman syndrome. BMC genetics. 2011;12:7. doi: 10.1186/1471-2156-12-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang HS, et al. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res. 2013;243:79–90. doi: 10.1016/j.bbr.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith RA, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller TM, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet neurology. 2013;12:435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chirboga C, et al. 65th American Academy of Neurology Annual Meeting; 2013. abstract S36.002. [Google Scholar]
- 24.Swayze EE, et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic acids research. 2007;35:687–700. doi: 10.1093/nar/gkl1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl Acad. Sci. USA. 2013;110:E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler M, Stecker K, Bennett CF. Cellular distribution of phosphorothioate oligodeoxynucleotides in normal rodent tissues. Laboratory investigation; a journal of technical methods and pathology. 1997;77:379–388. [PubMed] [Google Scholar]