

Abstract
Purpose
Epidermal growth factor receptor (EGFR) and cyclooxygenase 2 inhibitors synergistically inhibit head and neck squamous cell carcinoma tumorigenesis in preclinical studies. We conducted a phase I and pharmacokinetic study with the erlotinib and celecoxib combination in patients with advanced premalignant lesions.
Patients and Methods
36 subjects with oral leukoplakia, mild, moderate, or severe dysplasia, or carcinoma in situ were screened for study participation; 12 consented and received therapy for a median of 5.38 months. Erlotinib was escalated following a standard 3+3 design at 50, 75, and 100mg orally daily and celecoxib was fixed at 400mg twice daily for 6 months. Biopsy of lesions and cytobrush of normal mucosa were performed at baseline, 3, 6 and 12 months. Erlotinib pharmacokinetics were analyzed in 10 subjects.
Results
The maximum tolerated dose of erlotinib with celecoxib 400mg BID was 50mg per day with skin rash being the main observed toxicity. Overall histologic response rate was 63% (complete response 43%, partial response 14%, stable disease 29%, disease progression 14%). With median follow-up of 36 months, mean time to progression to higher-grade dysplasia or carcinoma was 25.4 months. Downregulation of EGFR and p-ERK in follow-up biopsies correlated with response to treatment. Larger average erlotinib V/F (∼308L) and CL/F (8.3L/hr) compared to previous studies may be related to relatively large average bodyweights. Average erlotinib t1/2 was 25.6hr.
Conclusion
Encouraging responses to the celecoxib and erlotinib combination correlated with EGFR pathway inhibition. Although erlotinib-related rash was the main limitation to dose escalation, the intervention was well tolerated.
Introduction
The epithelial growth factor receptor (EGFR) is expressed in a wide variety of malignant tumors including head and neck, colon, pancreatic, non-small cell lung, breast, kidney, ovarian, bladder carcinomas and gliomas (1-3). The incidence of EGFR expression in head and neck squamous cell carcinoma (HNSCC) is over 90%, suggesting that EGFR inhibition may be effective in HNSCC (4-6). One approach to block EGFR activity involves the use of small molecule tyrosine kinase inhibitors (TKIs) that target the intracellular domain of EGFR. This ability to specifically inhibit intracellular tyrosine kinase activity has been observed over precise dose ranges (7, 8).
A broad range of laboratory investigations, animal models, and epidemiological studies provide evidence that inhibition of cyclooxygenase-2 (COX-2) pathways may contribute to cancer treatment in general (9-11) and HNSCC in particular (12, 13). In HNSCC, COX-2 is expressed in both tumor tissue and adjacent epithelium, with increased expression in invasive carcinoma compared to normal epithelium. COX-2 inhibition has been shown to result in cell growth inhibition in HNSCC cell lines (14). Furthermore, we have previously elucidated the differential expression pattern of COX-2 in stages of head and neck premalignant lesions and invasive carcinoma (15). Our findings indicated that COX-2 is involved in early and intermediate stages of carcinogenesis in HNSCC. COX-2 levels increased progressively throughout all stages of carcinogenesis. This may reflect a role for COX-2 in this process, further supporting the rationale for COX-2 inhibition as a valid strategy for cancer chemoprevention. Based on this evidence, a number of chemoprevention and therapeutic trials in HNSCC using COX-2 inhibitors (COX-2I) are underway (16). Evidence suggests that COX inhibitors, including non-steroidal anti-inflammatory drugs (NSAIDs), protect against a variety of tumors (9, 17, 18). In patients with familial adenomatous polyposis, celecoxib, a selective COX-2I, caused a 28% reduction in the number of colorectal polyps compared with 4.5% reduction for placebo (19). In HNSCC, Western blot analysis showed expression of COX-2 in mucosa of subjects at different stages of carcinogenesis and not in normal mucosa, suggesting a possible role for COX-2 inhibition in HNSCC chemoprevention (20). Previous studies suggested that NSAIDs might have a similar effect in delaying the growth of head and neck tumor cell lines (21-23). Furthermore, prior studies using the COX-2I as a single agent in oral premalignant lesions revealed evidence of improvement in the degree of dysplasia (24).
Preclinical and clinical studies have described the interaction between EGFR and COX-2 and shown that targeting these two pathways can synergistically or additively block progression of HNSCC growth in vitro and in vivo (14, 25, 26), providing a scientific basis for using the combination of EGFR-TKI and COX-2I as a chemopreventive approach in HNSCC. We report here the results of a phase I clinical trial and pharmacokinetic studies of this combination in subjects with premalignant lesions (i.e. leukoplakia, erythroplakia, and/or erythroleukoplakia) of the oral cavity oropharynx or larynx. The correlation of response with biomarker modulation was reported in a separate publication (27).
Patients and Methods
Patient accrual
Between October 24, 2006 and June 28, 2012, 36 subjects with documented premalignant lesions including mild (mild-D), moderate (MD) or severe (SD) oral leukoplakia and carcinoma in situ (CIS) were screened. Lesion sites included oral cavity oropharynx, and the larynx. Participants were eligible regardless of their smoking status. Eligibility requirements included: ECOG performance status (PS) of 0-1, age ≥18 years, adequate bone marrow, liver and renal function, signed written informed consent, negative serum pregnancy test (β-HCG) within 72 hours of receiving treatment, willingness to use appropriate contraception during study participation for women of child-bearing potential, adequate pulmonary function (FEV1 and FVC ≥60% by spirometry), adequate cardiac function (echocardiogram with normal LV ejection fraction).
Subjects were excluded from participation if they had documented hyperplasia only, acute inter-current illness or recent major surgery, history of previous malignancies excluding stage I or II cancers rendered disease-free more than 1 year from time of consent, documented pregnancy, were breast feeding, had active cardiovascular events including angina, unstable angina, palpitation, tachycardia, arrhythmia, a recent cerebrovascular accident or myocardial infarction within 6 months from enrollment, documented history of coagulopathy and/or were taking warfarin or warfarin-derivative anticoagulants, hypertension not adequately controlled by medication, history of congestive heart failure greater than NYHA Grade II, confusion, disorientation, or a history of major psychiatric illness impairing their understanding of the informed consent, history of intake of COX-2I or EGFR-TKI within 3 months of study entry, documented history of interstitial lung disease or known connective tissue disease, history of NSAID-induced ulcers or participation in a clinical trial using an investigational drug within 12 months of enrollment.
Study participants were required to have a complete inspection of the oral cavity oropharynx and larynx. A baseline biopsy for initial diagnosis and grading was mandatory. All evaluable patients were required to have repeated biopsies at 3, 6 and 12 months from initiation of therapy. Biopsies of suspicions lesions in the oral cavity or laryngeal lesions were performed using standard “punch biopsy” procedure with approximately a 3 mm punch. If a follow-up biopsy was deemed difficult to obtain at the suspicious site, a brushing or wash was performed instead. Histologic assessments were performed by a head and neck pathologist. Buccal scrapings (cytobrush) for target lesions and normal buccal mucosa were performed at baseline, 3- 6- and 12-months as surrogates for biomarker modulation.
Drug treatment
Participants received a fixed dose of celecoxib 400mg orally BID continuously for 6 months. Erlotinib was dose escalated at 3 dose levels of 50, 75 and 100mg orally QD for 6 months. Dose escalation followed a standard 3+3 escalation design.
Definition of response
Response evaluation was based on pathologic examination of the degree of dysplasia observed and recorded by an expert head and neck pathologist. Pathologic complete response (pCR) was defined as complete disappearance of dysplasia from the epithelium. Pathologic partial response (pPR) was defined as improvement of dysplasia by at least one degree (i.e., severe dysplasia becomes moderate dysplasia). Pathologic minor response or stable disease (pSD) was defined as minor focal improvement without change of degree of dysplasia (i.e., focal improvement from moderate to mild dysplasia with still moderate dysplasia overall) or no pathologic changes after treatment. Pathologic progressive disease (pPD) was defined as worsening by at least one degree of dysplasia (i.e., mild to moderate dysplasia) or development of invasive cancer on or following treatment.
Pharmacokinetic (PK) studies
Blood sampling for measurement of erlotinib PK was performed prior to drug administration (t = 0), and 0.5, 1, 3, 6, 8, 24, and 48 hr post-administration. Blood was centrifuged using a refrigerated centrifuge and plasma collected and frozen prior to being assayed for erlotinib.
Assays of erlotinib and active metabolites
Erlotinib was extracted from plasma samples and plasma concentrations were measured by LC-MS, as detailed in Supplemental Materials. A standard curve was constructed using weighted (1/X2) linear regressions of the peak area (Y) of erlotinib against the corresponding nominal concentrations of erlotinib (X, ng/mL) in blank plasma. The LC-MS/MS assay was validated with specificity, precision (coefficients of variation <15%), accuracy (>85%), matrix effects, and linearity (0.1 to 500ng/mL, r>0.99). The limits of detection and quantitation for erlotinib were 0.5 and 2pg on the column. Recoveries of erlotinib from human plasma were 80-82%.
PK data analysis and modeling
Plasma concentrations of erlotinib were utilized for PK modeling. Assay results (confirmed by patient drug diaries), indicated variations with respect to timings of erlotinib administration, and certain individuals were missing blood samples at critical times, making the data unsuitable for compartmental modeling on a per individual basis. However, modeling was feasible using population PK methods with the NONMEM modeling program (28). The population PK of erlotinib was previously modeled in cancer patients enrolled in Phase 2 studies, using an open 1-compartmental model with first order absorption into plasma (29-31). Here, a similar modeling approach was used to study the PK of erlotinib in comparatively healthy subjects with premalignant lesions, using NONMEM software (ver 7.1, ICON Development Solutions, Ellicott City, MD) (32) run with PLT Tools (version 2.6, PLTsoft, San Francisco, CA). Goodness of fit was analyzed using commonly-used graphical methods for population PK, and the influence of body weight on V/F and CL/F was explored using previously reported covariate model structures (29-31). Methods are detailed in Supplemental Materials.
Results
Patient characteristics
A total of 36 subjects with pathologic dysplasia (Mild-D, MD, SD) or CIS were screened. Nineteen screened subjects were not enrolled due to social or personal reasons mostly related to reasons of convenience such as transportation or other personal commitments, in addition to concerns about toxicity, co-morbidities or ineligibility. One patient with Mild-D was enrolled after the protocol was amended to include Mild-D. Seventeen subjects were enrolled on the study, 3 of whom withdrew consent (one male 69 yrs of age, and two females 43 and 42 yrs of age). Two patients who signed informed consent were deemed to be screen failures, one secondary to prior history of oral squamous cell carcinoma and the other secondary to a history of cardiac arrhythmias. A total of 12 subjects received therapy. None of the 12 patients who received therapy had a history of prior treated malignancy. Their characteristics with baseline pathology, response, and duration of response are described in Table 1. Two subjects were taken off treatment prior to response evaluation due to the following reasons: grade 3 rash, elevated serum creatinine, or urosepsis. Two subjects chose to withdraw from therapy prior to first response evaluation. One patient was excluded from the efficacy analysis as she was found to have microinvasive carcinoma on her resected pre-therapy tissue biopsy. A total of 7 subjects were evaluable for response.
Table 1. Evaluable patient characteristics, baseline pathology, pathologic response, and time to progression.
| Patient | Age (Yrs) | Gender | Site | Lesion | Pathologic Response | Progression/Discontinuation | Time to progression or last evaluation (Months) / cause of withdrawal |
|---|---|---|---|---|---|---|---|
| 1 | 50 | F | Tongue | MD | CR | Y | 4 |
| 2 | 42 | M | Tongue | MD | PR | Y | 26 |
| 3 | 41 | F | Tongue | SD | SDi | Y | 26 |
| 4 | 65 | F | Tongue | SD | PD | Y | 6 |
| 5 | 62 | M | Buccal mucosa | SD | CR | Y | 55 |
| 6 | 64 | M | Larynx | Mild-D | SDi | Y | 6 |
| 7 | 40 | M | Larynx | MD | CR | N | 36 |
| 8 | 69 | F | Tongue | HG | NE | D (elevated creatinine) | Renal insufficiency |
| 9 | 74 | F | Tongue | MD | NE | D (grade 3 rash) | Rash |
| 10 | 87 | F | BOT | SD | NE | D (withdrew) | Patient chose to withdraw |
| 11 | 46 | F | Tongue | HG | NE | D (withdrew) | Withdrew after signing consent |
| 12 | 57 | F | Tongue | HG | N/A | N/A | Excluded from efficacy analysis |
Legend: M=Male, F=Female; MD=Moderate Dysplasia, SD=Severe Dysplasia, Mild-D=Mild Dysplasia; BOT=Base of tongue; HG=High Grade Dysplasia
SDi=Stable disease, PD=Progression of disease; CR=Complete remission; PR=Partial Remission; NE=Non evaluable
N=No Progression as of last follow up; Y= Documented Progression; D=Discontinued therapy before first evaluation of response.
Dose escalation and toxicity
Three subjects were enrolled on cohort 1 (erlotinib 50mg daily) with no dose limiting toxicities (DLT) observed, allowing escalation to cohort 2 (erlotinib 75mg daily). One patient had a grade 3 rash at dose level 2 and cohort 2 was expanded to six subjects. As a second subject had grade 3 rash, the maximum allowable dose (MAD) of erlotinib was deemed to be 75mg. One patient was enrolled at a dose level 3 (erlotinib 100mg daily) prior to toxicity analysis of the patients who received 75mg/day and received continued therapy for the entire duration of the study with no documented grade 3 toxicities. Following dose de-escalation, two additional patients received erlotinib 50mg/day. None of the 5 subjects treated at the 50mg/day dose experienced a DLT. The MTD of erlotinib in combination with celecoxib 400mg BID was therefore determined to be 50mg/day. The observed grade 2-3 toxicities were as follows for the 12 treated patients: rash 2/12 (17%), mucositis 1/12 (17%), mouth sores 3/12 (25%), leukopenia 1/12 (8%), anemia 1/12 (8%), hyperglycemia 2/12 (17%), hypoalbuminemia 1/12 (8%), hypoglycemia 1/12 (8%), throat infection 1/12 (8%), urosepsis 1/12 (8%), elevated creatinine 1/12 (8%) (Table 2).
Table 2. Toxicities including grades 1-4.
| Toxicity | Grade 1 | Grade 2 | Grade 3 | Grade 4 |
|---|---|---|---|---|
| Abdominal cramping | 2 | |||
| Alopecia | 2 | |||
| Anemia | 2 | 1 | ||
| Anxiety | 2 | |||
| Decreased Protein | 2 | |||
| Leukopenia | 1 | 1 | ||
| Depression | 3 | |||
| Diarrhea | 5 | |||
| Dry Eyes | 4 | |||
| Dry Skin | 6 | |||
| Elevated LDH | 3 | |||
| Elevated Serum Creatinine | 4 | 1 | ||
| Elevated Alkaline Phosphatase | 3 | |||
| Elevated ALT | 5 | |||
| Elevated AST | 4 | |||
| Fatigue | 6 | |||
| Hyperbilirubinemia | 2 | |||
| Hypercholesterolemia | 2 | |||
| Hyperglycemia | 7 | 2 | ||
| Hypoalbuminemia | 3 | 1 | ||
| Hypocalcemia | 4 | |||
| Hypoglycemia | 1 | 1 | ||
| Hypokalemia | 2 | |||
| Hyponatremia | 3 | |||
| Mouth Sores | 9 | 3 | ||
| Mucositis | 3 | 1 | ||
| Nausea | 4 | |||
| Neuropathy | 3 | |||
| Pruritis | 2 | |||
| Rash | 8 | 2 | ||
| Shortness of Breath | 3 | |||
| Strep Throat | 1 | |||
| Urosepsis | 0 | 0 | 1 | |
| Vomiting | 2 |
Clinical outcome
Responses were evaluated for 7 subjects using the last documented histologic response at 3, 6 or 12 months. At baseline, 3/7 (43%) had MD, 3/7 had SD (43%) and 1/7 had mild-D (14%). 3/7 achieved a complete remission (CR 43%), 1/7 partial remission (PR 14%), 1/7 progressive disease (PD 14%), and 2/7 had stable disease (SD 29%) with an overall RR of 57% and overall clinical benefit of 86% (histologic responses to therapy are shown in Fig. 1). The median time to achieving a documented response from the time of enrollment was 5.6 months. All three patients who achieved a complete remission also had complete disappearance of their lesions by visual inspection. At the time of the last analysis, 6/7 patients (85%) had documented progression: one to stage I invasive carcinoma 6 months after completion of therapy for MD of the lateral tongue, one to stage II oral cavity carcinoma 4 months after completion of therapy, one to invasive squamous cell carcinoma after having stable SD for 26 months, two with recurrent MD and SD at 26 months and 6 months, respectively, and one patient with recurrent high grade dysplasia 55 months after achieving a complete remission for severe dysplasia of the buccal mucosa. One patient continues to be in complete remission 36 months after treatment of a high grade dysplasia of the vocal cord. The median duration of follow-up for the 7 evaluable subjects was 36 months. The mean time to progression to a higher grade dysplasia or carcinoma was 25.4 months (Fig. 2). No patients had died by the time of data analysis.
Figure 1.
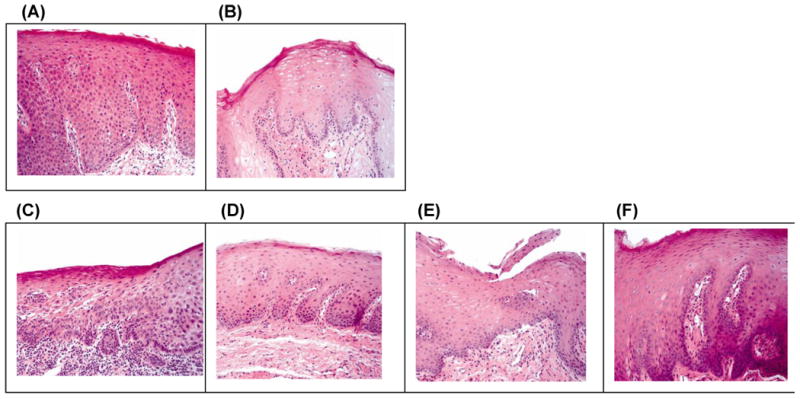
Histologic responses from biopsies of two patients treated with celecoxib and erlotinib. A) Patient 1: baseline severe dysplasia; B) Patient 1: hyperplasia and no dysplasia at 3 months; C) Patient 2: baseline moderate to severe dysplasia; D) Patient 2: mild dysplasia at 3 months; E) Patient 2: hyperplasia and no dysplasia at 6 months; F) Patient 2: hyperkeratosis at 12 months.
Figure 2.
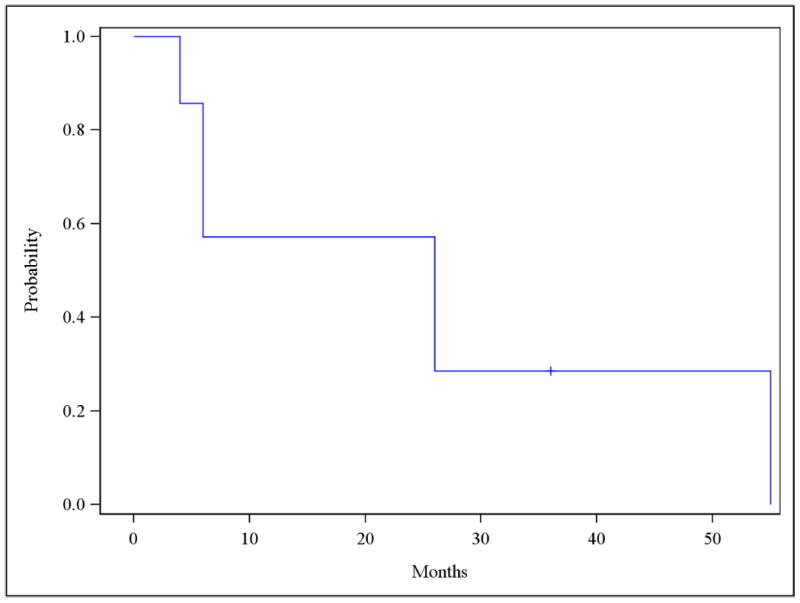
Time to progression to higher grade dysplasia or carcinoma. Mean time to progression was 25.4 months. Median time for follow-up was 36 months.
Correlative biomarker studies
Tissue analysis comparing baseline biopsies to follow-up samples after treatment revealed decreases in EGFR and p-ERK levels in the patients' last biopsies. These changes were correlated significantly with clinical responses to therapy (p=0.019 and p=0.006 for EGFR and p-ERK, respectively). The correlative biomarker studies were described in detail elsewhere (27).
PK studies
Plasma concentrations of erlotinib and the demethyl metabolite (OSI-420) from 10 subjects administered were plotted versus time (Fig. 3A). Erlotinib concentrations only were modeled, as OSI-420 concentrations were <10% of erlotinib and curves were parallel. Four subjects administered erlotinib after the 24 hr blood draw, while the rest did so prior to their 24 hr dose. Also, 6 subjects had plasma concentrations similar to Cmax prior to the 48 hr sampling time. Based on the previously reported erlotinib Tmax (2-3 hr) and long elimination half-life (t1/2 > 24 hr), these undocumented timings were inputted as 3 hr prior to the 48 hr blood draw (31).
Figure 3.
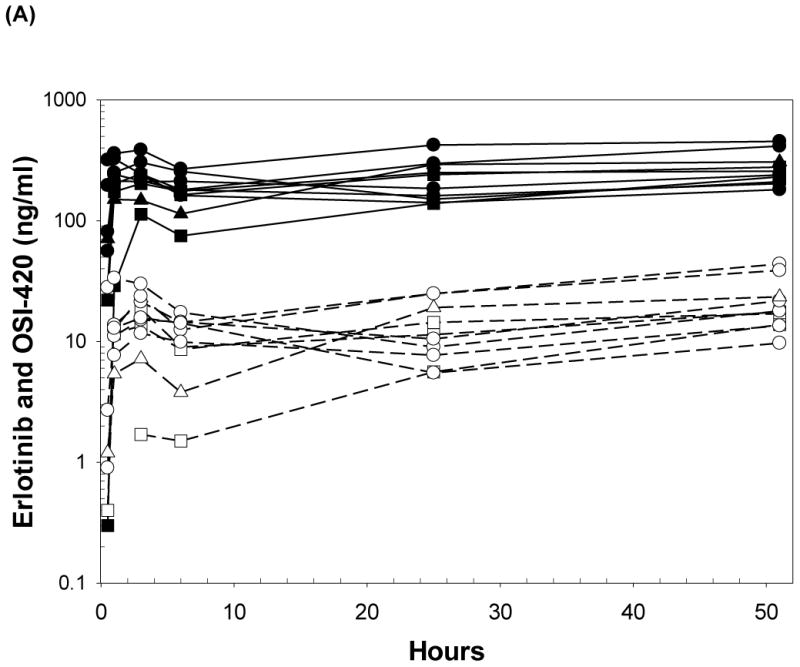
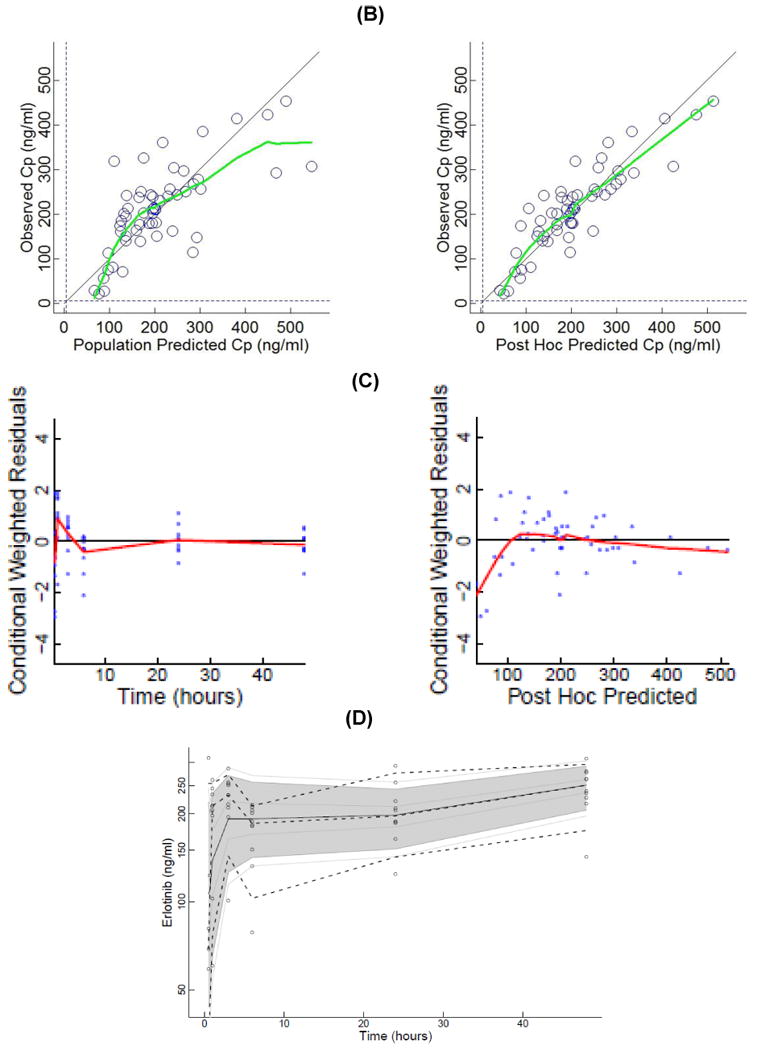
(A) Plasma concentrations of erlotinib and OSI-420 (demethyl erlotinib) versus time. Subjects self-administered 50mg (□, n = 3), 75mg (◯, n = 6) or 100mg erlotinib (Δ, n =1) QD. Closed symbols and solid lines represent erlotinib concentrations, open symbols and dashed lines represent the OSI-420 metabolite. Plasma concentrations of erlotinib were used for PK modeling. (B) Observed versus final model predicted plasma concentrations of erlotinib. The left panel is based on the “typical” prediction without including model variability (IIV), while the right panel is based on the complete model including IIV (post hoc fit). The linear regression coefficients (r2, observed versus predicted) for the respective plots were 0.508 and 0.783, respectively. Inclusion of the error model (post hoc model) decreased the standard errors of prediction of the model from 22 to 15%. (C) Conditioned weighted residuals (CWRES) (39) were plotted versus time and post hoc predicted concentrations. The curvilinear plots depict local regression trend smoother fits to the data. The curvilinear plots in C and D depict local regression trend smoother (R Supersmoother) fits to the data. (D) Model validation by “precision corrected visual predictive check” (40), in which observations are normalized to the median (75mg QD) dosing scheme. One thousand simulations were performed per subject and used to compute percentile ranges (P5, P25, P50, P75 and P95) normalized to median dose and body-weight. Actual observations were also normalized, and superimposed over these ranges for comparison. The shaded area indicates 90% confidence interval; solid lines indicate percentiles: 2.5, 97.5 (red); 5, 95 (blue); 25, 75 (green); 50 (black). Dashed lines indicate percentiles 5, 50, and 95 of observations.
The PK parameters of a basic model with no covariates (model 1), and a model relating V/F as proportional with subject body weight (model 2), are summarized in Table 3. Both estimated V/F, CL/F and Ka with reasonable precision. Including body weight as a covariate of V/F improved the model fit (ΔOF = 5.09), without the need for additional fitted parameters. Unlike previous reports, inclusion of body weight as a covariate of CL/F did not improve model fit further, possibly due to the limited range of body weights in this study (29). There was >20% shrinkage for Ka in model 1 and for Ka and V/F in model 2, indicative of under-estimation of IIV for these parameters. Neither model allowed estimation of the inter-subject variability (IIV) of CL/F. Model 2 was used for all further analyses.
Table 3. Population pharmacokinetic parameters of erlotinib.
| OF (-2LL) | −45.72 | −50.81 |
|---|---|---|
| parameters (RSE, %): | Basic model | kg on V |
| CL/F (L/h) | 8.34 (4.99) | 8.29 (5.10) |
| V1/F (L) | 295.2 (12.69) | 308.35 (10.03) |
| Ka (h-1) | 1.00 (29.55) | 1.02 (31.09) |
| b IIV, σ2 (% shrinkage) | ||
| IIV of V/F | 33.02 (14.92) | 21.19 (28.01) |
| IIV of Ka | 83.87 (20.18) | 66.09 (22.36) |
| σ2 (%) | 31.97 (11.63) | 32.7 (9.48) |
| Number significant digits | 3.8 | 4.9 |
No reference intravenous dose was administered so that the fraction (F) of orally absorbed drug is unknown, as no reference intravenous dose was administered (assumed 1 for calculations). CL/F = oral clearance, V is the volume of distribution (normalized for an 81.7 kg individual in the case of the covariate model), Ka = first-order oral absorption rate constant. IIV are inter-individual variance estimates, σ2 = residual (intra-individual) variance and OF is the value of NONMEM objective function. Relative standard errors of parameters (RSE) were calculated as % CV. IIV, σ2 and RSE were reported as percent coefficient of variations (% CV), noting that the formula for % CV for a log-normal distributed parameter = √(eˆω2 - 1) × 100, where ω2 = variance of a log-normally distributed parameter. % shrinkage estimates were obtained from the NONMEM output.
There was reasonable symmetry of predicted population and post-hoc concentrations versus observed data (Fig. 3B), with a slight tendency to overestimate certain lower concentrations. As expected, the individual post hoc predictions (with IIV) had greater correlations with observations (r2 = 0.508 and 0.783, respectively). CWRES versus time and post-hoc predictions (Fig. 3C), were confirmatory of an adequately fitted model. Internal model validation using a pcVPC plot (Fig. 3D) for model 2 resulted in 12% and 14% of observations greater than or less than the predicted 95th and 5th percentiles, respectively, which could indicate some variance shrinkage. However, the performance may be adequate given only 51 plasma concentrations. Due to data limitations, a study of the influence of celecoxib on the PK of erlotinib was not feasible in this study.
Discussion
The combination of erlotinib and celecoxib has been shown to synergistically inhibit head and neck cancer cell growth in preclinical studies performed by our group (14, 25). Prior phase I trials have examined the clinical activity of this combination with radiation therapy in patients with recurrent HNSCC and advanced lung cancer (33). Unlike our study, the dose of erlotinib was fixed in these studies and the celecoxib dose was escalated. Grade 3 toxicities including dermatitis and mucositis were also reported. Our study focused on primary chemoprevention in healthy subjects with pre-malignancies; therefore, our threshold of defining DLTs was lower, given the chemopreventive nature of our investigation. While both celecoxib and erlotinib have been used as single agents in chemoprevention studies for different tumors including colorectal cancer and HNSCC (19, 34, 35), the combination has not been extensively explored in the chemoprevention setting.
The results of this phase I clinical trial support our hypothesis that EGFR-TKI and COX-2I can inhibit tumorigenesis in premalignant lesions of the head and neck. Based on our conservative approach in assessing toxicities we determined the MTD of erlotinib in this combination to be 50mg/day. This regimen was fairly well tolerated with erlotinib-induced rash being the most commonly observed grade 3 toxicity preventing dose escalation. We believe erlotinib at 50mg per day would be a well-tolerated dose using this combination.
A major pitfall of our study is the small number of patients who were evaluable for response. Despite this, our correlative biomarker studies showing a downregulation in EGFR and p-ERK levels which correlated with response to therapy (27), as well as an observed pathologic response rate of 63% with a mean duration of 20.9 months support the clinical benefit of this intervention. This will, however, require further confirmation in a larger phase II study.
There are multiple challenges and obstacles in implementing any chemopreventive intervention in a large population of healthy subjects. These include difficulty in accrual, toxicity and quality of life implications in an otherwise healthy patient population, among others. As chemopreventive agents need to be delivered continuously over a rather extended period of time, it is important to select agents with a very favorable toxicity profile. Erlotinib-induced skin rash has become an accepted consequence in patients with advanced metastatic non-small cell lung cancer when compared to the dangers and side effects of cytotoxic chemotherapy. This is clearly not the case in a chemoprevention patient population such as ours. It could be argued that this is a rather high price to pay in the chemopreventive setting. In our experience, the toxicity of our intervention has been one major limitation to accrual and to dose escalation on the study. Other chemopreventive trials in head and neck cancer are incorporating erlotinib or celecoxib as a single agent or in combination with other agents. In a study using single agent celecoxib in oral premalignant lesions, 12 out of 18 biopsies showed improvement in the degree of dysplasia after 12 weeks (24). Whereas the majority of our treated subjects had severe or high-grade dysplasia, 50% of subjects in the Wirth study had mild dysplasia. Still these results raise the question of whether there is an advantage in combining the two agents versus using single agents. The results of trials using single agent erlotinib are eagerly awaited. Our rationale for combining celecoxib and erlotinib stems from our preclinical data showing a clear synergism between EGFR and COX-2 blockade as far as inhibition of HNSCC progression and growth in vitro and in vivo (14, 25, 26).
The toxicity concern clearly opens the door to exploring better tolerated agents such as natural compounds as future chemopreventive agents, which is currently the focus of several groups including ours (36-38).
Our PK results indicated a larger average V/F (308.35 L) in our study compared to previous studies (∼220 L), which is roughly proportional to the average body weights in the two studies (∼69 and 82 kg, respectively). Furthermore, the average CL/F in the present study (8.29 L/hr) was larger than that reported (∼3.95 L/hr). Since CL/F of erlotinib increases less than proportionally with body weight (29), it is likely that factors in addition to body weight were responsible for the higher CL/F, e.g. the lower dose used in this study (50 to 100mg QD) compared with 150mg QD in the previous phase 2 trials. Furthermore, Thomas et al., reported that erlotinib CL/F was influenced by drug transporter polymorphisms (e.g. ABCB1, CYP3A5, and ABCG2) (29, 31), which were not characterized in this study. The average t1/2 of erlotinib observed in this study (ln2 × V/CL = 25.6 hr) was shorter than in the study with cancer patients (∼37.5 hr).
In summary, despite concerns about toxicity, this phase I study demonstrated clinical responses that correlated with downregulation of activated protein levels of the EGFR pathway. This is further supported by our preclinical findings (27). By the time of the last evaluation most patients had progressed to higher grade dysplasia or invasive carcinoma, however in some instances progression to dysplasia took more than 4 years from the time of documented complete remission and one patient continues to have a durable complete remission after 3 years. The preliminary efficacy signal is therefore encouraging and supports future evaluation of this regimen in a phase II efficacy trial. Our study remains the first to combine an EGFR-TKI and COX2-I with PK analysis for patients with premalignancies of the head and neck. The half-life of erlotinib in our study was shorter compared to other studies (∼37.5 hr). Larger studies are necessary to determine whether coadministration of celecoxib influences the PK of erlotinib.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health (U01 CA101244 and Specialized Programs of Research Excellence in Head and Neck Cancer, P50 CA128613) to DMS and R01CA0983722, P50CA097190 and the American Cancer Society (to JRG). We thank Genentech for providing erlotinib for this study. We also thank Dr. Anthea Hammond for her critical reading of the article and Dana Ray for her administrative support of the study.
Footnotes
Presented at the 2010 American Society of Clinical Oncology (ASCO) meeting: J Clin Oncol 28:15s, 2010 (suppl; abstr 5535)
There are no disclaimers to report
References
- 1.Rusch V, Klimstra D, Venkatraman E, Pisters PW, Langenfeld J, Dmitrovsky E. Overexpression of the epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression. Clin Cancer Res. 1997;3:515–22. [PubMed] [Google Scholar]
- 2.Klijn JG, Berns PM, Schmitz PI, Foekens JA. The clinical significance of epidermal growth factor receptor (EGF-R) in human breast cancer: a review on 5232 patients. Endocr Rev. 1992;13:3–17. doi: 10.1210/edrv-13-1-3. [DOI] [PubMed] [Google Scholar]
- 3.Yao M, Shuin T, Misaki H, Kubota Y. Enhanced expression of c-myc and epidermal growth factor receptor (C-erbB-1) genes in primary human renal cancer. Cancer Res. 1988;48:6753–7. [PubMed] [Google Scholar]
- 4.Shin DM, Ro JY, Hong WK, Hittelman WN. Dysregulation of epidermal growth factor receptor expression in premalignant lesions during head and neck tumorigenesis. Cancer Res. 1994;54:3153–9. [PubMed] [Google Scholar]
- 5.Rubin Grandis J, Melhem MF, Barnes EL, Tweardy DJ. Quantitative immunohistochemical analysis of transforming growth factor-alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer. 1996;78:1284–92. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1284::AID-CNCR17>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 6.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359:1143–54. doi: 10.1056/NEJMra0707975. [DOI] [PubMed] [Google Scholar]
- 7.Noonberg SB, Benz CC. Tyrosine kinase inhibitors targeted to the epidermal growth factor receptor subfamily: role as anticancer agents. Drugs. 2000;59:753–67. doi: 10.2165/00003495-200059040-00003. [DOI] [PubMed] [Google Scholar]
- 8.Saba NF, Khuri FR, Shin DM. Targeting the epidermal growth factor receptor. Trials in head and neck and lung cancer. Oncology (Williston Park) 2006;20:153–61. discussion 62, 66, 69 passim. [PubMed] [Google Scholar]
- 9.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–22. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4:431–6. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 11.Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J Clin Oncol. 2005;23:254–66. doi: 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- 12.Cohen EG, Almahmeed T, Du B, Golijanin D, Boyle JO, Soslow RA, et al. Microsomal prostaglandin E synthase-1 is overexpressed in head and neck squamous cell carcinoma. Clin Cancer Res. 2003;9:3425–30. [PubMed] [Google Scholar]
- 13.Chan G, Boyle JO, Yang EK, Zhang F, Sacks PG, Shah JP, et al. Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res. 1999;59:991–4. [PubMed] [Google Scholar]
- 14.Chen Z, Zhang X, Li M, Wang Z, Wieand HS, Grandis JR, et al. Simultaneously targeting epidermal growth factor receptor tyrosine kinase and cyclooxygenase-2, an efficient approach to inhibition of squamous cell carcinoma of the head and neck. Clin Cancer Res. 2004;10:5930–9. doi: 10.1158/1078-0432.CCR-03-0677. [DOI] [PubMed] [Google Scholar]
- 15.Saba NF, Choi M, Muller S, Shin HJ, Tighiouart M, Papadimitrakopoulou VA, et al. Role of cyclooxygenase-2 in tumor progression and survival of head and neck squamous cell carcinoma. Cancer Prev Res (Phila) 2009;2:823–9. doi: 10.1158/1940-6207.CAPR-09-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papadimitrakopoulou VA, William WN, Jr, Dannenberg AJ, Lippman SM, Lee JJ, Ondrey FG, et al. Pilot randomized phase II study of celecoxib in oral premalignant lesions. Clin Cancer Res. 2008;14:2095–101. doi: 10.1158/1078-0432.CCR-07-4024. [DOI] [PubMed] [Google Scholar]
- 17.Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69(Suppl 1):28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- 18.Williams CS, Watson AJ, Sheng H, Helou R, Shao J, DuBois RN. Celecoxib prevents tumor growth in vivo without toxicity to normal gut: lack of correlation between in vitro and in vivo models. Cancer Res. 2000;60:6045–51. [PubMed] [Google Scholar]
- 19.Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 20.Nathan CO, Leskov IL, Lin M, Abreo FW, Shi R, Hartman GH, et al. COX-2 expression in dysplasia of the head and neck: correlation with elF4E. Cancer. 2001;92:1888–95. doi: 10.1002/1097-0142(20011001)92:7<1888::aid-cncr1706>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 21.Ondrey FG, Juhn SK, Adams GL. Inhibition of head and neck tumor cell growth with arachidonic acid metabolism inhibition. Laryngoscope. 1996;106:129–34. doi: 10.1097/00005537-199602000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Scioscia KA, Snyderman CH, Rueger R, Reddy J, D'Amico F, Comsa S, et al. Role of arachidonic acid metabolites in tumor growth inhibition by nonsteroidal antiinflammatory drugs. Am J Otolaryngol. 1997;18:1–8. doi: 10.1016/s0196-0709(97)90041-7. [DOI] [PubMed] [Google Scholar]
- 23.Panje WR. Regression of head and neck carcinoma with a prostaglandin-synthesis inhibitor. Arch Otolaryngol. 1981;107:658–63. doi: 10.1001/archotol.1981.00790470006003. [DOI] [PubMed] [Google Scholar]
- 24.Wirth LJ, K J, Li Y, Othus M, Moran AE, Dorfman DM, Norris CM, Jr, Goguen L, Posner MR, Haddad RI, Bertagnolli MM. A pilot surrogate endpoint biomarker study of celecoxib in oral premalignant lesions. Cancer Prev Res (Phila) 2008;1:339–48. doi: 10.1158/1940-6207.CAPR-07-0003. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Chen ZG, Choe MS, Lin Y, Sun SY, Wieand HS, et al. Tumor growth inhibition by simultaneously blocking epidermal growth factor receptor and cyclooxygenase-2 in a xenograft model. Clin Cancer Res. 2005;11:6261–9. doi: 10.1158/1078-0432.CCR-04-2102. [DOI] [PubMed] [Google Scholar]
- 26.Choe MS, Zhang X, Shin HJ, Shin DM, Chen ZG. Interaction between epidermal growth factor receptor- and cyclooxygenase 2-mediated pathways and its implications for the chemoprevention of head and neck cancer. Mol Cancer Ther. 2005;4:1448–55. doi: 10.1158/1535-7163.MCT-04-0251. [DOI] [PubMed] [Google Scholar]
- 27.Shin DM, Zhang H, Saba NF, Chen AY, Nannapaneni S, Amin AR, et al. Chemoprevention of head and neck cancer by simultaneous blocking of epidermal growth factor receptor and cyclooxygenase-2 signaling pathways: preclinical and clinical studies. Clin Cancer Res. 2013;19:1244–56. doi: 10.1158/1078-0432.CCR-12-3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetic parameters. III. Monoexponential model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm. 1983;11:303–19. doi: 10.1007/BF01061870. [DOI] [PubMed] [Google Scholar]
- 29.White-Koning M, Civade E, Geoerger B, Thomas F, Le Deley MC, Hennebelle I, et al. Population analysis of erlotinib in adults and children reveals pharmacokinetic characteristics as the main factor explaining tolerance particularities in children. Clin Cancer Res. 2011;17:4862–71. doi: 10.1158/1078-0432.CCR-10-3278. [DOI] [PubMed] [Google Scholar]
- 30.Lu JF, Eppler SM, Wolf J, Hamilton M, Rakhit A, Bruno R, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80:136–45. doi: 10.1016/j.clpt.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Thomas F, Rochaix P, White-Koning M, Hennebelle I, Sarini J, Benlyazid A, et al. Population pharmacokinetics of erlotinib and its pharmacokinetic/pharmacodynamic relationships in head and neck squamous cell carcinoma. Eur J Cancer. 2009;45:2316–23. doi: 10.1016/j.ejca.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 32.Beal S SL, Bookman A, Bauer RJ. NONMEM User's Guides. ICON Development Solutions; Ellicott City, MD, USA: 2009. 1989-2009. [Google Scholar]
- 33.Kao J, Genden EM, Chen CT, Rivera M, Tong CC, Misiukiewicz K, et al. Phase 1 trial of concurrent erlotinib, celecoxib, and reirradiation for recurrent head and neck cancer. Cancer. 2011;117:3173–81. doi: 10.1002/cncr.25786. [DOI] [PubMed] [Google Scholar]
- 34.Dogne JM, Hanson J, Supuran C, Pratico D. Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des. 2006;12:971–5. doi: 10.2174/138161206776055949. [DOI] [PubMed] [Google Scholar]
- 35.Lin DT, Subbaramaiah K, Shah JP, Dannenberg AJ, Boyle JO. Cyclooxygenase-2: a novel molecular target for the prevention and treatment of head and neck cancer. Head Neck. 2002;24:792–9. doi: 10.1002/hed.10108. [DOI] [PubMed] [Google Scholar]
- 36.Amin AR, Kucuk O, Khuri FR, Shin DM. Perspectives for cancer prevention with natural compounds. J Clin Oncol. 2009;27:2712–25. doi: 10.1200/JCO.2008.20.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JW, Amin AR, Shin DM. Chemoprevention of head and neck cancer with green tea polyphenols. Cancer Prev Res (Phila) 2010;3:900–9. doi: 10.1158/1940-6207.CAPR-09-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amin AR, Wang D, Zhang H, Peng S, Shin HJ, Brandes JC, et al. Enhanced anti-tumor activity by the combination of the natural compounds (-)-epigallocatechin-3-gallate and luteolin: potential role of p53. J Biol Chem. 2010;285:34557–65. doi: 10.1074/jbc.M110.141135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24:2187–97. doi: 10.1007/s11095-007-9361-x. [DOI] [PubMed] [Google Scholar]
- 40.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13:143–51. doi: 10.1208/s12248-011-9255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.