

Abstract
Aberrant DNA methylation pattern is a well-known epigenetic marker of cancer cells. Recently, aberrant methylation was also reported in the peripheral blood of cancer patients and it could potentially serve as a biomarker for cancer risk. We investigated the methylation pattern of LINE-1 and other repetitive DNA elements in peripheral blood of cutaneous melanoma patients in order to search for an association with clinical characteristics. The patient cohort was composed by 69 unrelated melanoma patients, 28 of whom were hereditary cases (with or without CDKN2A mutations) and 41 were isolated (sporadic) melanoma cases. Methylation of LINE-1 was evaluated by pyrosequencing, whereas additional repetitive DNA sequences were assessed using Illumina 450K methylation microarray. Melanoma patients exhibited a higher, albeit heterogeneous, LINE-1 methylation level compared with controls. Hereditary melanoma patients carrying CDKN2A mutations showed a hypermethylated pattern of both LINE-1 and repetitive DNA elements compared with other patients. In particular, the methylation level at one specific CpG of LINE-1 was found to be correlated with the occurrence of metastasis. Our data suggest that LINE-1 hypermethylation in peripheral blood of melanoma patients is a potential epigenetic biomarker for metastasis occurrence.
Keywords: biomarkers, DNA methylation, leukocytes, LINE-1 elements, melanoma, metastasis, repetitive sequences
Introduction
Methylation of cytosines in CpG dinucleotides is a covalent modification associated with transcriptional repression 1. This epigenetic mark regulates several normal biological processes, such as cellular differentiation 2, and is also implicated in diseases, including cancer 3.
The transposable element LINE-1 has been used as a surrogate marker for global methylation in several cancer studies 4,5. The pattern of LINE-1 methylation in peripheral blood has been associated with cancer risk and differs among different cancer types; whereas hypomethylation has been reported for hepatocellular carcinoma 6 and gastric 7 and bladder cancer 8, hypermethylation has been described for renal 9 and colorectal cancer 10. Recently, a study was performed in leukocytes of melanoma patients, which did not detect differences in LINE-1 methylation between patients and controls 11.
We analyzed the methylation patterns of LINE-1 and other repetitive DNA elements as potential biomarkers for cutaneous melanoma in a cohort of Brazilian patients classified according to clinical characteristics.
Patients and methods
Patients and control groups
This retrospective study was performed at the A. C. Camargo Cancer Center (São Paulo, Brazil) after obtaining approval from the Internal Ethics Committee Board. Melanoma patients were referred to the Skin Cancer Department of the A. C. Camargo Cancer Center, which surveyed sex, age at blood draw, number of melanomas, Breslow thickness of melanoma high stage, and metastasis occurrence. Family history of cancer was assessed in the Oncogenetics Department, and genetic counseling was provided to all participants.
The study was based on blood samples from 69 unrelated cutaneous melanoma patients who had undergone no previous chemotherapy, who were classified in three groups: familial melanoma patients who were carriers of pathogenic CDKN2A mutations (n=8), familial melanoma patients with CDKN2A-wild type (n=20), and sporadic melanoma patients (n=41). The familial melanoma patients who were negative for CDKN2A mutations fulfilled at least one of the following criteria: they had at least three primary melanomas (n=8); they had at least two primary melanomas, one of which was before 35 years of age (n=3); and they had a family history in three or more relatives of two consecutive generations, one of whom was under 50 years of age (n=9). The control group was composed of 51 individuals without cancer history, who were matched for age (range for each 10 years) and sex with the melanoma patients.
DNA samples extracted from peripheral blood were retrieved from the A. C. Camargo Cancer Center Biobank.
Pyrosequencing and HM450K genome-wide methylation analysis
Bisulfite modification was performed on 500 ng of DNA using the EZ DNA methylation kit (Zymo Research, Irvine, California, USA). Quantitative bisulfite pyrosequencing for LINE-1 was performed in DNA blood samples from 69 melanoma patients and 51 controls using PyroMark Q96-CpG LINE-1 (Qiagen, Hilden, Germany), which analyzed four CpGs sites (position 305 to 331 – GenBank accession X58075). The methylation level was obtained using PyroMark Q96-CpG Software (Qiagen), which provides the percentage of methylated cytosines relative to the sum of methylated and unmethylated cytosines. All experiments were performed twice and samples with a variation coefficient greater than 10% were excluded.
The methylation profile of several repetitive DNA elements was performed in a subset of 39 melanoma patients (eight familial melanoma with CDKN2A mutations, 19 familial melanoma CDKN2A-wild type, and 12 sporadic cases) and 12 controls using Infinium HumanMethylation 450K BeadChips (Illumina, San Diego, California, USA), according to the manufacturer’s instructions. The Bioconductor IMA package was applied for quality control of array methylation data. We removed from further analyses probes that (a) lacked β-values, (b) contained single-nucleotide polymorphisms, (c) mapped at sexual chromosomes, or (d) had detection P-values greater than 1e−05. Normalization and probe type bias adjustment were achieved by beta mixture quantile dilation using the ChAMP package (http://www.bioconductor.org/packages/release/bioc/html/ChAMP.html). Information regarding HM450K probes mapped within repetitive sequences was obtained from the data set GSE42409 (available at the Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/), and only probes with total overlap (50 mers) were retrieved.
We evaluated the influence of both sex and age on the methylation levels of LINE-1 using Fisher’s test. DNA methylation differences between groups were tested for significance using either the Mann–Whitney test or the Kruskal–Wallis with Dunn’s multiple comparison post-test. After considering LINE-1 as a continuous variable, we also categorized the LINE-1 methylation level based on the median of the methylation values of the control group: hypomethylated when patients exhibited LINE-1 level lower than that of controls and hypermethylated when patients had LINE-1 level at least the same level as controls; differences were tested for significance using Fisher’s test. All statistical analyses were performed using the GraphPad PRISM (GraphPad Software Inc.,La Jolla, California, USA) statistics software package.
Results
Quantitative bisulfite pyrosequencing for evaluation of the methylation pattern of four LINE-1 CpGs was performed for 69 patients and 51 control individuals. After exclusion of samples with variation coefficient above 10% between replicates, data from 48 melanoma patients and 42 controls were recovered. Considering all CpGs, neither controls nor melanoma patients exhibited significant differences in LINE-1 methylation level according to sex (controls P=0.06; patients P=0.17) or age at blood draw (<40 or≥40 years; controls P=0.99; patients P=0.17). These two characteristics were not associated with LINE-1 methylation level in either controls or melanoma patients (sex P=1; age at blood draw P=0.38). Therefore, statistical adjustments for these factors were not performed.
Table 1 presents the methylation data for the entire LINE-1 sequence (average of the four investigated CpGs) and each CpG position, comparing melanoma patients with controls, and groups of patients with different clinical characteristics with each other. The P-values in Table 1 refer to the statistical analysis of the means of the methylation levels of compared categories. Methylation levels were slightly heterogeneous between controls and melanoma patients across the four analyzed CpGs (Fig. 1a), and the CpG position 1 showed a statistically significantly higher methylation level compared with the other CpG positions in both groups (controls P<0.01, patients P<0.01). However, melanoma patients exhibited a significantly higher average LINE-1 methylation level compared with controls, as well as at the CpG positions 2 and 4 (Fig. 1a; Table 1).
Table 1.
LINE-1 methylation levels of melanoma patients and association with clinical characteristics
Fig. 1.
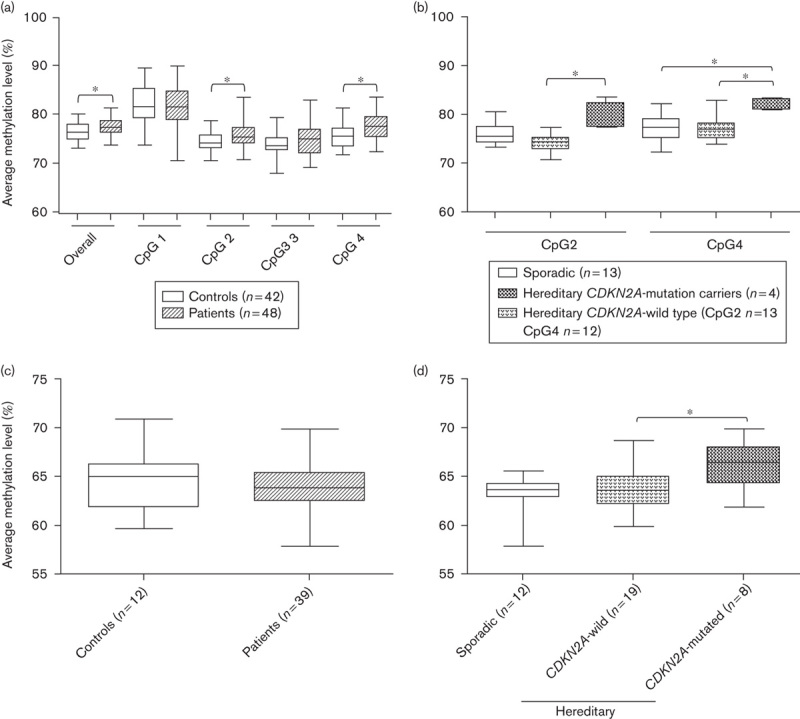
Methylation levels of LINE-1 and repetitive DNA sequences in melanoma patients. (a, b) Methylation levels of four LINE-1 CpGs (positions 331 to 305, GenBank accession X58075) obtained using quantitative bisulfite pyrosequencing. (c, d) Methylation levels of 22 436 CpGs mapped in repetitive DNA elements measured using the HM450K platform (Illumina). (a) Melanoma patients exhibited significantly higher overall and specific LINE-1 methylations levels compared with controls (the Mann–Whitney test). (b) The methylation levels at LINE-1 CpGs positions 2 and 4 were significantly different among the groups of melanoma patients (the Kruskal–Wallis with Dunn’s multiple comparison post-test). (c) Repetitive DNA elements did not show significant difference in methylation levels between melanoma patients and controls (the Mann–Whitney test). (d) Familial melanoma patients carrying CDKN2A mutations showed a hypermethylated pattern of repetitive DNA elements compared with melanoma patients without mutations (Kruskal–Wallis with Dunn’s multiple comparison post-test). *P<0.05.
Methylation levels at CpG positions 2, 3, and 4 showed statistically significant differences among melanoma groups (Table 1): CDKN2A mutation carriers showed hypermethylation compared with both CDKN2A-wild-type melanoma patients (CpG positions 2 and 4) and sporadic melanoma patients (CpG position 4) (Fig. 1b).
Subsequently, patients and controls were categorized as hypomethylated or hypermethylated on the basis of the median LINE-1 methylation levels of the control group (average LINE-1 methylation=76.26%; CpG position 1 methylation=81.50%; CpG position 2 methylation=74.20%; CpG position 3 methylation=73.49%; CpG position 4 methylation=75.59%). Considering the average LINE-1 methylation level, there was a significant association of methylation status with melanoma occurrence (P=0.03), with a higher number of hypermethylated individuals in the melanoma group (72.9%); individual CpG positions did not show significant differences in this analysis.
To investigate the relevance of LINE-1 methylation in peripheral blood as a potential biomarker for melanoma, we tested for significant differences in LINE-1 methylation according to clinical characteristics (Table 1). In melanoma patients, there was a statistically significant association of CpG1 methylation level and metastasis occurrence. The status of CDKN2A (mutated or wild type) was not significantly associated with any of the analyzed clinical characteristics (number of melanomas P=0.57; Breslow thickness P=1.00; metastasis P=0.56). However, the studied group was very small: among CDKN2A mutation carriers, only one patient showed Breslow thickness greater than 2 mm, two of them developed more than two melanomas, and metastases were not detected in this melanoma group.
Next, we investigated whether the methylation level of other repetitive DNA elements in addition to LINE-1 could be associated with melanoma. The methylation levels of 22 436 CpG sites mapped within repetitive sequences (LINE, SINE, LTR, and others) were recovered from HM450K experiments of a subset of these patients (n=39) and controls (n=12). No significant differences were observed between melanoma patients and controls (Fig. 1c). However, there was a statistically significant difference among different melanoma subgroups: patients carrying CDKN2A mutations showed hypermethylation at repetitive DNA elements compared with patients without CDKN2A mutations (Fig. 1d).
Discussion
DNA hypomethylation in peripheral blood has been already suggested as a biomarker for cancer risk 12. Genome-wide hypomethylation might induce activation of endogenous parasitic sequences, such as transposable elements, subsequent genomic instability, and oncogene expression 13,14. Measurement of the methylation level of LINE-1, which represents 17% of the human genome, is an accurate method for evaluating the global DNA methylation pattern 15. It is worth mentioning that two independent meta-analyses have confirmed that global hypomethylation in leukocytes could be used as a biomarker for cancer risk, although LINE-1 in particular could not reflect this global methylation status 4,5. The LINE-1 methylation status associated with cancer risk appeared to vary between different cancer types, and both hypomethylation and hypermethylation were already reported 6–10. Despite LINE-1 being, or not being, a suitable indicator of global DNA methylation status, it may still be an independent biomarker for cancer.
We evaluated the possible correlation between changes in the pattern of DNA methylation in leukocytes of repetitive sequences, mainly LINE-1, and the presence of cutaneous melanoma. Our data suggest that LINE-1 hypermethylation is associated with melanoma occurrence. In addition, a higher methylation level at one single LINE-1 CpG was significantly associated with metastasis in melanoma patients. This could be partially explained by the relation between LINE-1 hypermethylation and DNA double-stranded breaks 10. Hyland et al. 11 investigated the same target LINE-1 sequence in the only other methylation study in melanoma patients. In contrast to our findings, they did not find differences between melanoma patients and controls. This divergence can be because Hyland and colleagues studied a cohort composed of melanoma-prone families, whereas our melanoma group is heterogeneous with respect to the etiology of melanoma development. In addition, Jaffe and Irizarry 16 demonstrated the importance of cell heterogeneity in the DNA methylation analysis of peripheral blood. Peripheral blood contains a mixture of lymphocyte types, and detected differences in DNA methylation levels can arise from differences in the relative proportion of lymphocyte populations. Therefore, differences in the blood cell composition could influence the contrasting findings, reinforcing the relevance of accounting for cellular heterogeneity in clinical practice. Although some previous studies have reported differences in LINE-1 methylation level according to sex 17 and age 18, they did not appear in our study.
Interestingly, melanoma patients carrying CDKN2A mutations exhibited higher methylation levels at LINE-1 and repetitive DNA elements compared with those without CDKN2A mutations. Hyland and colleagues also analyzed melanoma patients with and without CDKN2A mutations, but they did not find methylation differences. CDKN2A is the most relevant known melanoma susceptibility gene, and mutations in this gene are more commonly detected in patients with a familial melanoma history 19 or with multiple primary melanomas 20. In a preceding study, we investigated the genome-wide DNA methylation profile of CDKN2A mutation carriers, revealing in these melanoma patients few differentially methylated CpGs, which were mainly related to underlying single-nucleotide polymorphisms 21. Thus, the hypermethylated pattern found in this group of CDKN2A mutation carriers appears to be restricted to repetitive sequences. Further studies using prospective large cohorts of melanoma patients are needed to substantiate our findings and elucidate whether LINE-1 hypermethylation is a cause or a consequence of melanoma risk and metastasis occurrence.
Acknowledgements
This work was supported by grants from FAPESP (2012/13963-9; 2013/07480-8).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem 2005; 74:481–514. [DOI] [PubMed] [Google Scholar]
- 2.Kar S, Parbin S, Deb M, Shilpi A, Sengupta D, Rath SK, et al. Epigenetic choreography of stem cells: the DNA demethylation episode of development. Cell Mol Life Sci 2014; 71:1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet 2010; 70:27–56. [DOI] [PubMed] [Google Scholar]
- 4.Brennan K, Flanagan JM. Is there a link between genome-wide hypomethylation in blood and cancer risk? Cancer Prev Res (Phila) 2012; 5:1345–1357. [DOI] [PubMed] [Google Scholar]
- 5.Woo HD, Kim J. Global DNA hypomethylation in peripheral blood leukocytes as a biomarker for cancer risk: a meta-analysis. PLoS One 2012; 7:e34615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di JZ, Han XD, Gu WY, Wang Y, Zheng Q, Zhang P, et al. Association of hypomethylation of LINE-1 repetitive element in blood leukocyte DNA with an increased risk of hepatocellular carcinoma. J Zhejiang Univ Sci B 2011; 12:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hou L, Wang H, Sartori S, Gawron A, Lissowska J, Bollati V, et al. Blood leukocyte DNA hypomethylation and gastric cancer risk in a high-risk Polish population. Int J Cancer 2010; 127:1866–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cash HL, Tao L, Yuan JM, Marsit CJ, Houseman EA, Xiang YB, et al. LINE-1 hypomethylation is associated with bladder cancer risk among nonsmoking Chinese. Int J Cancer 2012; 130:1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao LM, Brennan P, van Bemmel DM, Zaridze D, Matveev V, Janout V, et al. LINE-1 methylation levels in leukocyte DNA and risk of renal cell cancer. PLoS One 2011; 6:e27361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walters RJ, Williamson EJ, English DR, Young JP, Rosty C, Clendenning M, et al. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and early-onset colorectal cancer. Epigenetics 2013; 8:748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyland PL, Burke LS, Pfeiffer RM, Mirabello L, Tucker MA, Goldstein AM, Yang XR. LINE-1 methylation in peripheral blood and the risk of melanoma in melanoma-prone families with and without CDKN2A mutations. Melanoma Res 2013; 23:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friso S, Udali S, Guarini P, Pellegrini C, Pattini P, Moruzzi S, et al. Global DNA hypomethylation in peripheral blood mononuclear cells as a biomarker of cancer risk. Cancer Epidemiol Biomarkers Prev 2013; 22:348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muñoz-Lopez M, Macia A, Garcia-Cañadas M, Badge RM, Garcia-Perez JL. An epigenetic battle: LINE-1 retrotransposons and intragenomic conflict in humans. Mob Genet Elements 2011; 1:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sigalotti L, Fratta E, Parisi G, Coral S, Maio M. Epigenetic markers of prognosis in melanoma. Methods Mol Biol 2014; 1102:481–499. [DOI] [PubMed] [Google Scholar]
- 15.Lisanti S, Omar WA, Tomaszewski B, De Prins S, Jacobs G, Koppen G, et al. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS One 2013; 8:e79044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 2014; 15:R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Maarri O, Walier M, Behne F, van Üün J, Singer H, Diaz-Lacava A, et al. Methylation at global LINE-1 repeats in human blood are affected by gender but not by age or natural hormone cycles. PLoS One 2011; 6:e16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyle KD, Guldberg P. Genetic risk factors for melanoma. Hum Genet 2009; 126:499–510. [DOI] [PubMed] [Google Scholar]
- 20.Monzon J, Liu L, Brill H, Goldstein AM, Tucker MA, From L, et al. CDKN2A mutations in multiple primary melanomas. N Engl J Med 1998; 338:879–887. [DOI] [PubMed] [Google Scholar]
- 21.De Araújo ES, Marchi FA, Rodrigues TC, Vieira HC, Kuasne H, Achatz MI, et al. Genome-wide DNA methylation profile of leukocytes from melanoma patients with and without CDKN2A mutations. Exp Mol Pathol 2014; 97:425–432. [DOI] [PubMed] [Google Scholar]