

Abstract
Objective:
To minimize pathologic heterogeneity in genetic studies of Parkinson disease (PD), the Autopsy-Confirmed Parkinson Disease Genetics Consortium conducted a genome-wide association study using both patients with neuropathologically confirmed PD and controls.
Methods:
Four hundred eighty-four cases and 1,145 controls met neuropathologic diagnostic criteria, were genotyped, and then imputed to 3,922,209 variants for genome-wide association study analysis.
Results:
A small region on chromosome 1 was strongly associated with PD (rs10788972; p = 6.2 × 10−8). The association peak lies within and very close to the maximum linkage peaks of 2 prior positive linkage studies defining the PARK10 locus. We demonstrate that rs10788972 is in strong linkage disequilibrium with rs914722, the single nucleotide polymorphism defining the PARK10 haplotype previously shown to be significantly associated with age at onset in PD. The region containing the PARK10 locus was significantly reduced from 10.6 megabases to 100 kilobases and contains 4 known genes: TCEANC2, TMEM59, miR-4781, and LDLRAD1.
Conclusions:
We confirm the association of a PARK10 haplotype with the risk of developing idiopathic PD. Furthermore, we significantly reduce the size of the PARK10 region. None of the candidate genes in the new PARK10 region have been previously implicated in the biology of PD, suggesting new areas of potential research. This study strongly suggests that reducing pathologic heterogeneity may enhance the application of genetic association studies to PD.
Family studies have identified multiple Parkinson Disease (PD) genes: α-synuclein (SNCA), parkin (PARK2), DJ1 (PARK7), PTEN induced putative kinase 1 (PINK1), and leucine-rich repeat kinase 2 (LRRK2); association studies have identified up to 28 loci meeting genome-wide significance.1–7 However, much of the heritability of PD remains unexplained.
One reason may be the amount of neuropathologic heterogeneity. PD is defined using clinical criteria that can include heterogeneous neuropathologic features, each of which may have a distinct genetic architecture.8–11 Earlier PD autopsy studies10,12 reported that approximately 75% of clinically diagnosed PD cases had neuropathic evidence of Lewy body disease. However, both of these predate modern imaging studies.13 Indeed, more recent analyses11 suggest that concordance of the neuropathologic diagnosis with the clinical diagnosis of PD can approach 90%. These layers of causal, pathologic heterogeneity in PD reduce the statistical power of genetic association tests and, as a result, limit the ability to find disease genes. This is particularly true as the samples in large PD genome-wide association studies (GWAS) have been collected over many years, often predating routine use of modern clinical diagnostic methods such as the advanced imaging of today.
To reduce potential pathologic heterogeneity, the Autopsy-Confirmed Parkinson Disease Genetics Consortium (APDGC) collected a set of individuals with PD that have neuropathologic confirmation of Lewy body PD pathology, as well as a set of “autopsy-confirmed” control individuals with no evidence of PD neuropathology. These neuropathologically confirmed cases and controls provided a level of pathologic homogeneity unavailable in previous GWAS.
METHODS
Sample selection.
To be included in the study, all autopsy cases were required to have a diagnosis of PD documented by a neurologist. All patient samples and a subset of the controls used in the analysis were contributed by 12 APDGC centers, which are listed in text e-1 on the Neurology® Web site at Neurology.org.
Ascertainment of cases into the APDGC was primarily through existing autopsy cases of patients diagnosed with PD available at the participating center, referred to neuropathologists for autopsy by neurologists, or recruited from families whose affected relative were participants in existing PD studies. Participants from the Miami Udall were enrolled prospectively for autopsy studies. APDGC controls were volunteers, often spouses of patients with PD, without symptoms of parkinsonism. The Alzheimer Disease Genetics Consortium (ADGC) provided neuropathologically examined control samples for this study as well; ADGC contributing centers and affiliate members are described in text e-1 and the contributors supplemental material, respectively.
Before statistical analysis, all neuropathologic reports were reviewed by a single neuropathologist (D.W.D.). Inclusion criteria included clinical diagnosis of PD by a neurologist before death, moderate/severe neuronal loss in the substantia nigra, and the presence of Lewy bodies. Cases were excluded if they had prominent dementia within 1 year of diagnosis14 (to minimize the inclusion of Lewy body dementia), had competing pathologic features (e.g., progressive supranuclear palsy rather than PD), or had a Braak neurofibrillary tangle stage >IV. Because of the autopsy-based ascertainment scheme, additional information on PD cases (e.g., family history data, age at onset) was limited. Controls were restricted to samples with no antemortem diagnosis of PD, no more than minimal neuronal loss in the substantia nigra, an absence of Lewy bodies, and a Braak neurofibrillary tangle stage ≤IV. The ADGC controls were previously genotyped (see below). All ADGC controls used for our study met the same neuropathologic criteria as the APDGC.
While 977 APDGC samples were genotyped by the Center for Inherited Disease Research (CIDR), Johns Hopkins University, only 791 samples remained after neuropathologic review (484 cases, 307 controls). Exclusion based on neuropathology included the following: 66 cases without Lewy bodies; 6 with excessive Alzheimer pathology; 7 had parkinsonism with other pathologies; 16 had dementia with Lewy bodies; and 9 cases were withdrawn at the request of the contributing sites. As some sites specifically sent samples that met our entrance criteria, whereas other sites sent samples from a broader set of PD (with or without Lewy bodies), these numbers should not be interpreted as population-level rates of pathologic misclassification/diagnosis. For the ADGC controls set, we requested only genotype data for samples that met our inclusion criteria.15 However, after examination of this dataset, we excluded 6 samples with pathology consistent with PD, 15 that had other diagnoses that excluded them from the analysis, and 2 controls were withdrawn at the request of the contributing sites. Because this was not a random selection of the ADGC dataset, we do not know the breakdown of the control sample exclusions in the entire ADGC control set.
Standard protocol approvals, registrations, and patient consents.
All samples were obtained with appropriate institutional review board approval and informed consent.
Genotyping/quality control.
Genotyping for all APDGC samples was performed through CIDR using the Illumina HumanOmni1-Quad beadchip (Illumina, Inc., San Diego, CA). ADGC samples were previously genotyped on a variety of platforms and centers, and are detailed elsewhere.15 For quality control (QC) purposes, there were 8 HapMap trios and 41 within-study duplicates included in the genotyping. Preliminary QC included checks for sample missingness, sample relatedness, sex inconsistencies, and a principal components analysis (PCA) to infer ancestry.16 For the PCA, we used only good quality (missingness <5%) and common (minor allele frequency [MAF] >5%) variants in low linkage disequilibrium (LD) with each other (r2 < 0.1) that were not in the major histocompatibility complex regions such as the HLA (human leukocyte antigen), LTR (long terminal repeat), chromosome 7q21-31, and chromosome 8p23.1. Samples that did not cluster with the known European ancestry group were excluded. Initial QC also included review of probe intensity data. The sample-level QC was performed on each sample and was largely independent of the dataset to which the sample belonged. Single nucleotide polymorphism (SNP)-level QC was performed on APDGC and ADGC datasets separately for all steps up to and including imputation. The initial postimputation filtering (removing monomorphic SNPs and those with low IMPUTE info scores, <0.40) was also performed on each set separately. Starting with the comparison of the control groups, all analyses were performed jointly.
The Omni1-Quad beadchip assays more than 1.1 million probes. From these, we removed 123,000 intensity-only probes, 10,000 SNPs failed CIDR's technical filter, 55,000 were monomorphic, and 22,000 showed high SNP missingness rates (>2%). Copy number analysis revealed 2 samples with trisomy-21 that were subsequently excluded. A small number of SNPs (<300) were removed since they had 2 or more discordant calls among the HapMap trios, study replicates, or had low p values for the Hardy-Weinberg equilibrium test among controls (p < 1 × 10−5). SNPs with MAF <2% were not used in the genotype imputation (141,000). Dropout from genotyping QC was <3%. After all of these QC and filtering steps, 782,456 SNPs remained before imputation.
Genotype imputation and final QC.
The IMPUTE version 2 software was used to infer genotypes at additional loci for the cases and both control sets. Data from the 1000 Genomes Project were used as a reference (March 2012 release).17 Imputation and imputation QC were independently performed within the APDGC samples and within each of the 9 ADGC control cohorts.18 After imputation, the variants were considered for further analysis if they were not monomorphic, had an IMPUTE info score greater than 0.45, and showed no significant difference in frequencies between the ADGC controls and the APDGC controls (genotypic χ2 test p value >0.05). Thus, control subgroup discordance or pseudoassociation did not contribute to overall significance levels. In addition, SNPs with missingness >5%, MAF <1%, and positions with PLINK's info score <0.8 were removed from further analysis; for imputed loci, MAF was estimated based on allelic dosage.19 Initial imputation started with 23,284,435 loci, but after filters, using conservative thresholds to avoid biasing the association results by adding unmatched controls typed with different platforms, a final 3,922,209 variants were used in the association analysis. After all genotype and phenotype-based QC, the primary analysis dataset consisted of 484 neuropathologically confirmed PD cases and 1,145 neuropathologically confirmed control samples (307 controls from the APDGC and 838 controls from the ADGC).
Statistical association.
All cohorts were jointly analyzed for association between the SNPs and PD status. Statistical association was performed using logistic regression with age at death, sex, and the first eigenvector for population substructure included as covariates in the model. No additional vectors were used in the analysis because they accounted for only a small proportion of the variation (principal component 1 [PC1] accounted for only 0.4% of variation). Association testing was performed using PLINK.19
To test for independent genetic effects in regions with multiple associated variants, a forward-backward selection procedure was used: each SNP was tested for association independently and then the most associated variant was added to the model. Each of the remaining variants was separately added to the model, with the most associated being added to the multiple variable regression models. These “forward” selection steps are interrupted by “backward” selection steps to determine whether variants included in the current model should be removed. In the backward step, any variables in the model that are no longer associated with a p value threshold of 0.05 are removed from the model; this sometimes occurs when a signal of strong effect is better described by 2 moderate effects. When no new SNPs are being added to the model, and no current SNPs are being removed, the model represents the independent effects at the locus. LD calculations were performed using the Haploview software.20
RESULTS
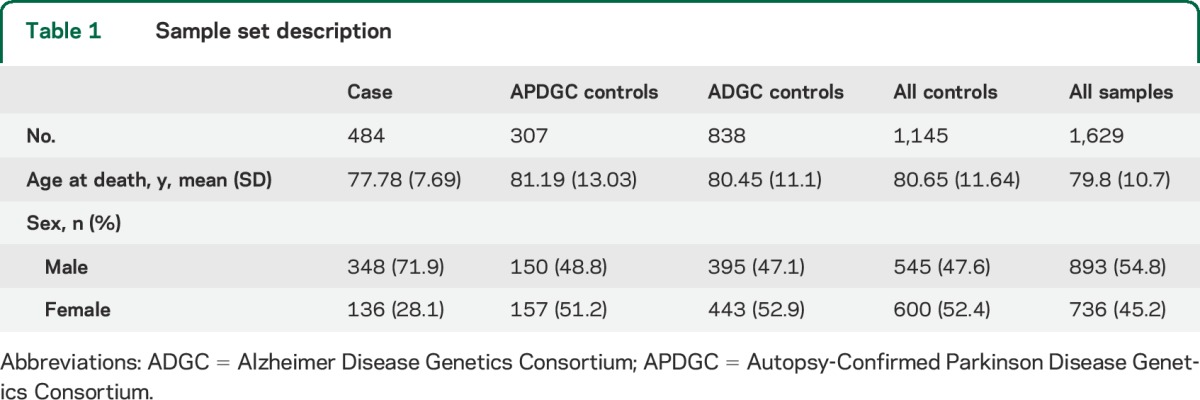
A description of the characteristics of patients and controls is shown in table 1. Population substructure analysis revealed only minor substructure within this European-descended sample (PC1 accounted for less than 0.4% of the genetic variation) that was not associated with disease (p value between PC1 and disease = 0.53). A plot of the first 2 principal components is shown in figure e-1. The genomic inflation factor (λ value) was 1.12 (see Q-Q plot, figure e-2). This is comparable with typical GWAS (1.05–1.10) and much less than those expected in studies with issues in population substructure (>1.2).
Table 1.
Sample set description
A 100-kilobase (kb) LD block, lying within the 10.6-megabase linkage region of the known PD locus PARK10 (50,700,000–61,300,000 base pairs [bp]; OMIM: %606852), was found to be strongly associated with the risk of PD (figure 1). The most significant SNP (rs10788972) in this LD block is located within the TCEANC2 gene (table 2), achieving near genome-wide significance (chr1: 54,572,243, MAF = 0.43; odds ratio [OR] = 0.64, p value = 6.2 × 10−8). As we have merged control datasets, we tested this SNP using the APDGC dataset alone, and also found strong association (p value = 5 × 10−5). The association peak lies well within the 2 previous PARK10 linkage regions, very near the maximum logarithm of odds (LOD) score results (figure 2). Rs10788972 and a second strongly associated marker (rs6588502) are in strong LD (e.g., r2 = 0.59 for rs10788972; r2 = 0.77 for rs6588502) with the marker rs914722, previously reported to be associated with age at onset in PD.21 This is shown in figure e-3 using data from the 1000 Genomes Project.17 To confirm the LD in our dataset, we genotyped rs914722 in a subset of our samples (n = 357), and confirmed the LD with rs914722 seen in the 1000 Genomes Project (figures e-4 and e-5). There was no evidence of multiple, independent association signals in the region.
Figure 1. Regional association plot around rs10788972 on chromosome 1.

A regional association plot surrounding the rs10788972 single nucleotide polymorphism (SNP) is shown. The x-axis represents the position in base pair on chromosome 1. The diamond points represent genotyped or imputed variants, with the y-axis denoting −log p value, base 10. The shades of red in the SNP markers show linkage disequilibrium between variants and the top SNP (rs10788972). The blue line shows the recombination rate at different positions across the region.
Table 2.
Top 20 PARK10 SNPs
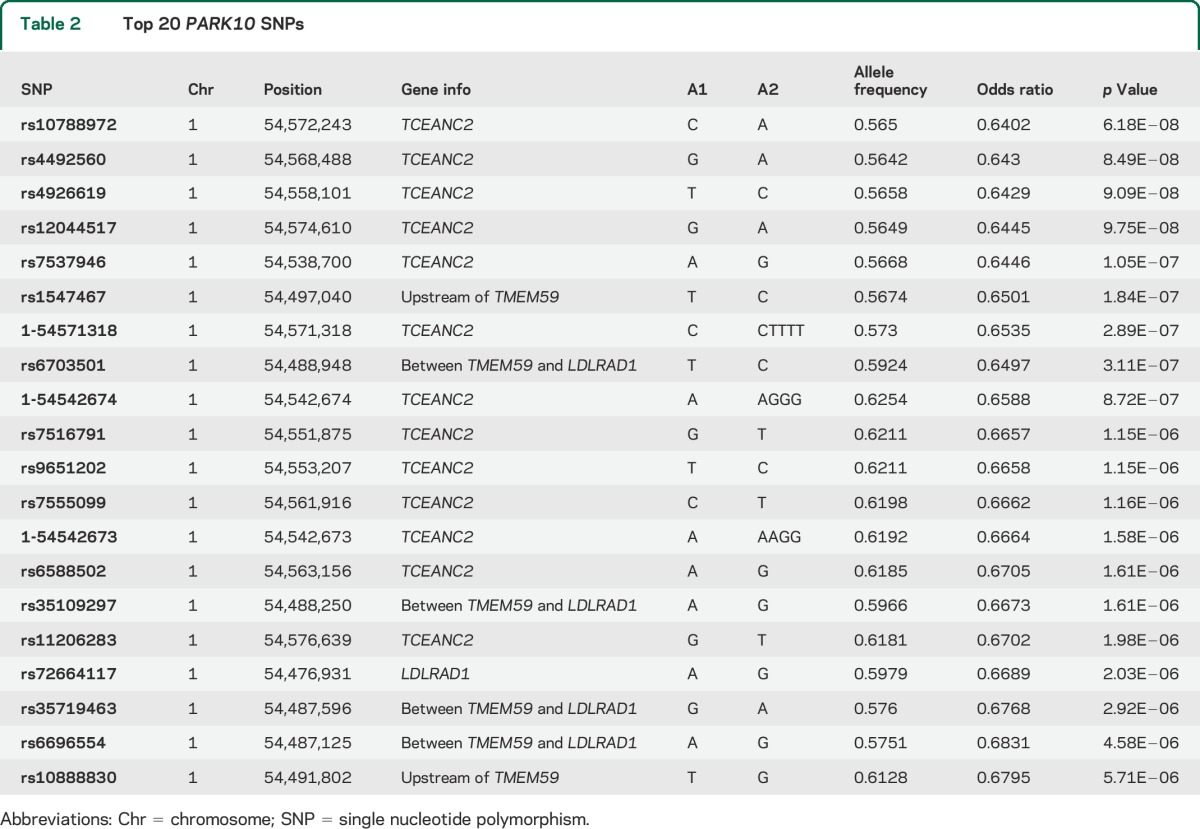
Figure 2. Plot of association results and linkage results.

A regional association plot of the PARK10 locus. The x-axis represents the position in base pair on chromosome 1. The points represent genotyped or imputed variants, with the left y-axis denoting the corresponding −log p value, base 10. The solid blue line represents the Parkinson disease age-at-onset linkage peak23 and the dashed blue line represents a linkage peak from the deCODE Icelandic study.22 The right y-axis denotes the corresponding logarithm of odds (LOD) score. MB = megabase.
No association test met a stringent genome-wide significance threshold (p value < 5 × 10−8). We did note strong association with several known PD loci, including the SNCA locus (rs13140923, OR = 1.45, p value = 0.0005513), the microtubule-associated protein tau (MAPT) locus (rs1052553, OR = 0.65, p value = 0.000776), and the GAK locus (rs5572858, OR = 0.64, p = 0.000171). Because of poor imputation in the ADGC set at the MAPT locus, only data from the APDGC dataset were available for the MAPT-specific analysis. In addition, across the genome, there were 39 variants that showed association with PD with a p value < 2.5 × 10−6 (see table e-1). These loci await validation in a second autopsy-confirmed PD dataset. None of these loci were in a previously identified “PARK” locus.
DISCUSSION
This study uses both autopsy-confirmed cases and controls to reduce pathologic heterogeneity and improve the chances of making a novel discovery. We confirm the PARK10 locus as a contributing risk locus for PD cases with Lewy body pathology. The SNP rs10788972 is close to genome-wide significance, and the association peak lies close to both of the reported maximum linkage LOD score markers,22,23 with an OR of 0.64. This effect size is comparable to effect estimates for SNCA and MAPT (SNCA: rs356165, OR = 0.74; MAPT: rs242559, OR = 0.78; in terms of the protective allele). Furthermore, we demonstrate that rs10788972 and other top-associated alleles are in strong LD with rs914722 in the PARK10 haplotype, which previously demonstrated significant association with age at onset in PD.21 Thus, multiple independent lines of evidence support the importance of the PARK10 locus in Lewy body PD.
The first report of linkage for a PD risk locus in this chromosome 1 region was in a large Icelandic family.22 The authors named the locus PARK10. This was followed by a second report23 of linkage for AAO genes in PD to the same region. The substantial overlap between these 2 linkage peaks is notable (figure 2), suggesting that the PARK10 locus confers both risk and age at onset effects in PD.
Subsequently, using a dataset independent of the present study, we published an association analysis with age at onset in PD in the PARK10 region.21 Two C1orf8 (TMEM59) haplotypes were found to be significantly associated (p = 0.004 and 0.009) with age at onset in PD (rs3766466 [SNP192]-rs914722 [SNP193]-rs2236512 [SNP194], table 10 in original report). However, examination of these 2 haplotypes reveals that only rs914722 varies between the 2 haplotypes, and is the marker driving the association. Thus, we have confirmed the association with this haplotype in PARK10 and PD.
The associated region from this study reduces the size of the PARK10 locus by 100-fold. No obvious candidate genes are observed in the overall region, and clearly further work is needed to identify any actual PARK10 risk variant. Four genes are known to lie in this smaller candidate region. None have been previously implicated in the biology of PD: the transcription elongation factor A (SII) N-terminal and central domain containing 2 (TCEANC2), transmembrane 59 (TMEM59 or C1orf8), low density lipoprotein receptor class A domain containing protein 1 (LDLRAD1) genes and the microRNA miR-4781 (figure 1). Little is known about TCEANC2, but it appears to be involved in RNA processing. LDLRAD1 is a member of the low-density lipoprotein receptor family involved in the binding site for low-density lipoprotein and calcium. In silico analysis of miR-4781 targets does not implicate any predicted or known PD target genes.24,25 TMEM59 (C1orf8) was shown to be overexpressed (3.6-fold) in the substantia nigra of patients with PD compared to controls.26 Localized in the Golgi body, it is involved in ectodomain shedding of amyloid protein precursor.27
To assure that the Alzheimer controls were not solely responsible for the observed association, we repeated the association analysis for TMEM59 with just the APDGC dataset and detected association (p = 5 × 10−5) in this dataset alone. To look for independent genetic effects in the region, a multiple linear regression was performed using a forward-backward SNP selection scheme. The forward-backward selection did not indicate additional independently associated SNPs in the region (data not shown).
The Web site PDGene (pdgene.org) presents a meta-analysis for risk loci. Examination of these data reveals that the meta-analysis results for these studies generated no significant associations in TCEANC2, TMEM59, miR-4787, and LDLRAD1. However, there are nominally significant associations (p < 0.05) in the adjacent gene, CDCP2, and several others in the PARK10 interval (50,700,000–61,300,000 bp). These results indicate that there is at least nominal evidence for association at PARK10 in meta-analyzed GWAS data from PD case-control samples based on clinical diagnosis only.
Why did this autopsy-confirmed GWAS confirm the PARK10 association while GWAS in clinically ascertained samples have not? The most likely reason is that we reduced the inherent genetic heterogeneity of the PD phenotype by using a more specific phenotype, i.e., autopsy-confirmed Lewy body PD. It is important to realize that this finding does not suggest any clinical misdiagnosis of these patients; all met the standard PD clinical criteria at the time of their collection. Rather, the results reflect that we limited the pathologic heterogeneity present in the phenotype.
An obvious limitation of our approach is the additional cost to phenotyping and the reduced sample size. While we did see association at SNCA, MAPT, and GAK, the moderate sample size is likely why we did not observe genome-wide p values at these loci. We would expect that if these loci are relatively less susceptible to genetic and phenotypic heterogeneity, then an increase in sample size, to increase power, should overcome any additional heterogeneity added by the larger dataset; this principle is at work in large GWAS with unselected control sets for rarer diseases. Finally, while a reduced sample size could increase the impact of population stratification, our PCA and association analyses showed no evidence of differential population substructure between cases and controls.
Unfortunately, because of the autopsy-based ascertainment scheme, age-at-onset data were not available for the majority of our sample, so we could not test the age-at-onset effect. This emphasizes the importance that all centers performing autopsies in collecting a uniform set of historical and clinical data to maximize the usefulness of these efforts for future research.
It is interesting that the 2 previous PARK10 linkage studies21,22 and the 2 association studies (including the present study) followed similar patterns. The Icelandic family data identified a risk effect for PD, while the initial association study used multiplex families from primarily North America and found association for age at onset, but not risk. It seems likely that the Icelandic study, like the current study, had reduced genetic heterogeneity by studying a more stringently defined sample (PD in a single, large Icelandic family) than the multiplex and initial association21 studies. Thus, the studies with less genetic heterogeneity saw a significant risk effect, while those containing more population complexity found an age-at-onset effect. It may be that PARK10 has a stronger age-at-onset effect relative to other genes, and thus is detected in those studies and not in the more genetically heterogeneous risk effect. However, whatever the reason for this finding, the evidence is strong that PARK10, similar to the APO ε4 allele in AD, affects both age at onset and risk of PD. The reduction in size of the PARK10 region makes it an excellent candidate for next-generation sequencing, which should provide insight into the actual variant for PARK10 and likely new directions for PD research.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all the members of the research teams, as well as the patients and their families for making this research possible. In particular: Laura Marsh, MD, Zoltan Mari, MD, and Melissa Gerstenhaber, RN-C, BSN, MAS, MSN, CCRC, played an integral role in sample collection and patient characterization from The Johns Hopkins University cohort. T.M.D. acknowledges the joint participation by the Adrienne Helis Malvin Medical Research Foundation through their direct engagement in the continuous active conduct of medical research in conjunction with the Johns Hopkins Hospital and The Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Program. In addition, the authors thank eMERGE study and Paul Crane. Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. The authors thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible.
GLOSSARY
- ADGC
Alzheimer Disease Genetics Consortium
- APDGC
Autopsy-Confirmed Parkinson Disease Genetics Consortium
- CIDR
Center for Inherited Disease Research
- GWAS
genome-wide association study
- LD
linkage disequilibrium
- MAF
minor allele frequency
- OR
odds ratio
- PCA
principal component analysis
- PC1
principal component 1
- PD
Parkinson disease
- QC
quality control
- SNP
single nucleotide polymorphism
Footnotes
Editorial, page 966
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Study design: G.W.B., W.K.S., E.R.M., J.Q.T., K.M., T.M.D., J.M.V. Sample/data collection: W.K.S., L.W., G.S., E.B.L., J.D.B., D.W.D., J.Q.T., V.M.V.D., H.I.H., D.C.M., T.G.B., J.C.T., O.P., M.P.F., G.W.B., T.M.F., L.S.H., K.M., J.P.V., S.M.G., H.V.V., Z.K.W., T.J.M., J.B.L., T.J.M., J.M.V. Data analysis: G.W.B., W.K.S., E.R.M., K.N., D.M.D., J.Q.T., M.P.F., J.P.V., B.G., G.S., J.D.B., Z.K.W., T.J.M., J.M.V. Data interpretation: G.W.B., W.K.S., E.R.M., L.W., K.N., D.M.D., J.M.V. Writing: G.W.B., D.D.W., W.K.S., E.R.M., G.S., K.N., E.B.L., J.D.B., J.Q.T., V.M.V., H.I.H., D.C.M., T.G.B., J.C.T., O.P., M.P.F., B.G., T.M.F., L.S.H., K.M., J.P.V., S.M.G., H.V.V., O.A.R., Z.K.W., L.W., D.M.D., M.A.P.-V., T.J.M., J.B.L., T.M.D., J.M.V.
STUDY FUNDING
Primary funding was by the NIH. A complete description of the Alzheimer Disease Genetics Consortium funding is below. This project was supported by the following: P50 NS072187, P50 AG016574 (D.W.D., O.A.R., Z.K.W.); P50 NS053488 (J.Q.T., V.M.V., H.I.H.); U24 NS072026 (T.G.B.); NS38377 and the JPB Foundation (T.M.D., J.C.T., O.P.); AG06781, AG005136, and NS062684 (T.J.M.); P50 AG16570 (H.V.V.); P01 AG007232 (L.S.H.); AG005136 and NS062684 (J.B.L.); R01NS37167, P30AG10133, and U24AG021886 (T.F., B.G.); P50 AG005134 and P50 NS038372 (M.P.F.); NS039764 and NS071674 (G.W.B., W.K.S., E.R.M., L.W., K.N., D.M.D., J.M.V.). The Alzheimer Disease Genetics Consortium was supported by the NIH, National Institute on Aging (NIH-NIA), which supported this work through the following grants: ADGC, U01 AG032984, RC2 AG036528; NACC, U01 AG016976; NCRAD, U24 AG021886; NIA LOAD, U24 AG026395, U24 AG026390; Banner Sun Health Research Institute, P30 AG019610; Boston University, P30 AG013846, U01 AG10483, R01 CA129769, R01 MH080295, R01 AG017173, R01 AG025259, R01AG33193; Columbia University, P50 AG008702, R37 AG015473, P01 AG007232; Duke University, P30 AG028377, AG05128; Emory University, AG025688; Group Health Research Institute, UO1 AG06781, UO1 HG004610; Indiana University, P30 AG10133; Johns Hopkins University, P50 AG005146, R01 AG020688; Massachusetts General Hospital, P50 AG005134; Mayo Clinic, P50 AG016574; Mount Sinai School of Medicine, P50 AG005138, P01 AG002219; New York University, P30 AG08051, MO1RR00096, UL1 RR029893, 5R01AG012101, 5R01AG022374, 5R01AG013616, 1RC2AG036502, 1R01AG035137; Northwestern University, P30 AG013854; Oregon Health & Science University, P30 AG008017, R01 AG026916; Rush University, P30 AG010161, R01 AG019085, R01 AG15819, R01 AG17917, R01 AG30146; TGen, R01 NS059873; University of Alabama at Birmingham, P50 AG016582, UL1RR02777; University of Arizona, R01 AG031581; University of California, Davis, P30 AG010129; University of California, Irvine, P50 AG016573, P50, P50 AG016575, P50 AG016576, P50 AG016577; University of California, Los Angeles, P50 AG016570; University of California, San Diego, P50 AG005131; University of California, San Francisco, P50 AG023501, P01 AG019724; University of Kentucky, P30 AG028383, AG05144; University of Michigan, P50 AG008671; University of Pennsylvania, P30 AG010124; University of Pittsburgh, P50 AG005133, AG030653; University of Southern California, P50 AG005142; University of Texas Southwestern, P30 AG012300; University of Miami, R01 AG027944, AG010491, AG027944, AG021547, AG019757; University of Washington, P50 AG005136; Vanderbilt University, R01 AG019085; and Washington University, P50 AG005681, P01 AG03991. The Kathleen Price Bryan Brain Bank at Duke University Medical Center is funded by National Institute of Neurological Disorders and Stroke grant NS39764, NIMH MH60451 and by GlaxoSmithKline. Genotyping of the TGEN2 cohort was supported by Kronos Science. The TGen series was also funded by NIA grant AG034504 to A.J.M., The Banner Alzheimer's Foundation, The Johnnie B. Byrd, Sr., Alzheimer's Institute, the Medical Research Council, and the state of Arizona and also includes samples from the following sites: Newcastle Brain Tissue Resource (funding via the Medical Research Council, local NHS trusts, and Newcastle University), MRC London Brain Bank for Neurodegenerative Diseases (funding via the Medical Research Council), South West Dementia Brain Bank (funding via numerous sources including the Higher Education Funding Council for England [HEFCE], Alzheimer's Research Trust [ART], BRACE, as well as North Bristol NHS Trust Research and Innovation Department and DeNDRoN), The Netherlands Brain Bank (funding via numerous sources including Stichting MS Research, Brain Net Europe, Hersenstichting Nederland Breinbrekend Werk, International Parkinson Fonds, Internationale Stiching Alzheimer Onderzoek), Institut de Neuropatologia, Servei Anatomia Patologica, Universitat de Barcelona. ADNI funding for ADNI is through the Northern California Institute for Research and Education by grants from Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson & Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc., F. Hoffmann-La Roche, Schering-Plough, Synarc, Inc., Alzheimer's Association, Alzheimer's Drug Discovery Foundation, the Dana Foundation, and by the National Institute of Biomedical Imaging and Bioengineering and NIA grants U01 AG024904, RC2 AG036535, and K01 AG030514. The authors thank Drs. D. Stephen Snyder and Marilyn Miller from NIA who are ex officio ADGC members. Support was also from the Alzheimer's Association (LAF, IIRG-08-89720; MP-V, IIRG-05-14147) and the US Department of Veterans Affairs Administration, Office of Research and Development, Biomedical Laboratory Research Program. P.S.G.-H. is supported by Wellcome Trust, Howard Hughes Medical Institute, and the Canadian Institute of Health Research.
DISCLOSURE
G. Beecham reports no disclosures relevant to the manuscript. D. Dickson is an editorial board member of Annals of Neurology, Parkinsonism and Related Disorders, Journal of Neuropathology & Experimental Neurology, and Brain Pathology. He is editor-in-chief of American Journal of Neurodegenerative Disease, and International Journal of Clinical and Experimental Pathology. Grant support: P50-AG16574, P50-NS72187, and P01-AG03949. W. Scott receives research funding through NIH (R01 AI068804, R01 EY012118, P50 NS071674), serves on the scientific review board of the Parkinson Study Group, and may accrue revenue on patents submitted by Duke University “Genetic variants increase the risk of age-related macular degeneration” in which he is an inventor. E. Martin reports no disclosures relevant to the manuscript. G. Schellenberg serves on the Medical Scientific Advisory Committee for the Alzheimer's Association, is a stockholder at Genelex (not relevant to the current publication), and receives research funding through NIH grants. K. Nuytemans reports no disclosures relevant to the manuscript. E. Larson received royalties from chapters written for UpToDate. J. Buxbaum reports no disclosures relevant to the manuscript. J. Trojanowski serves as an associate editor of Alzheimer's & Dementia. He may accrue revenue on patents submitted by the University of Pennsylvania wherein he is inventor including: Modified avidin-biotin technique, Method of stabilizing microtubules to treat Alzheimer's disease, Method of detecting abnormally phosphorylated tau, Method of screening for Alzheimer's disease or disease associated with the accumulation of paired helical filaments, Compositions and methods for producing and using homogeneous neuronal cell transplants, Rat comprising straight filaments in its brain, Compositions and methods for producing and using homogeneous neuronal cell transplants to treat neurodegenerative disorders and brain and spinal cord injuries, Diagnostic methods for Alzheimer's disease by detection of multiple MRNAs, Methods and compositions for determining lipid peroxidation levels in oxidant stress syndromes and diseases, Compositions and methods for producing and using homogenous neuronal cell transplants, Method of identifying, diagnosing and treating alpha-synuclein positive neurodegenerative disorders, Mutation-specific functional impairments in distinct tau isoforms of hereditary frontotemporal dementia and parkinsonism linked to chromosome-17: genotype predicts phenotype, Microtubule stabilizing therapies for neurodegenerative disorders, and Treatment of Alzheimer's and related diseases with an antibody. He is coinventor on patents submitted by the University of Pennsylvania wherein he is inventor that have generated income he has received from the sale of Avid to Eli Lilly including: Amyloid plaque aggregation inhibitors and diagnostic imaging agents. Finally, he receives research support from the NIH (AG 10124, AG 17586, AG-19724AG 024904, NS053488, AG029213). V. Van Deerlin received funding from AANP honoraria, NIH grant. H. Hurtig reports no disclosures relevant to the manuscript. D. Mash has stock ownership in DEMERx, Inc. T. Beach is a consultant for GE Healthcare for an unrelated topic, and has research contracts with Piramal Healthcare, Avid Radiopharmaceuticals/Eli Lilly, and GE Healthcare, all on topics unrelated to the submitted study. In 2012, received honoraria as an invited speaker to the Japanese Association of Neuropathologists and the American Academy of Neurology and as a grant reviewer for The Michael J. Fox Foundation for Parkinson's Research. Dr. Beach receives grant support from the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson's Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer's Disease Core Center), and The Michael J. Fox Foundation for Parkinson's Research. J. Troncoso and O. Pletnikova report no disclosures relevant to the manuscript. M. Frosch: member of external scientific advisory committee for Northwestern University ADC, NIA/NIH grant funding. B. Ghetti has research contracts with Piramal Healthcare, receives grant support from NIH grant. T. Foroud: consultant for University of Pennsylvania (NIA Genetics of Alzheimer's Data Storage), NIH SREA reviewer for an NHGRI CSR study section; National Advisory Council on Alcohol Abuse & Alcoholism; University of Colorado, scientific advisory board for the Center for Antisocial Drug Dependence (award DA011015), AAAS honoraria, NIH grant. L. Honig is a consultant for Johnson & Johnson; Eli Lilly; receives research support from Eli Lilly, Johnson & Johnson, Genentech, Allon, Elan, Pfizer, NIH/NIA, Alzheimer's Association, Alzheimer's Drug Discovery Foundation. K. Marder served on the editorial board of Neurology®; receives research support from the NIH (NS036630 [PI], 1UL1 RR024156-01 [Director PCIR], PO412196-G [Co-I], and PO412196-G [Co-I]). She received compensation for participating on the steering committee for U01NS052592 and from the Parkinson Disease Foundation, Huntington's Disease Society of America, the Parkinson Study Group, CHDI, and The Michael J. Fox Foundation. Dr. Marder received honoraria from Springer publications. J. Vonsattel has received research support from the NIH, and National Institute on Aging (P50AG08702, 1R01AG036040, 2P50NS038370), the Hereditary Disease Foundation, CHDI Foundation (Cure Huntington's Disease Initiative), and The Louis and Rachel Rudin Foundation. S. Goldman reports no disclosures relevant to the manuscript. H. Vinters served on the editorial board of Journal of Neuroscience Research, Korean Journal of Pathology, Neuropathology, and Neuropathology and Applied Neurobiology; receives research funding from NIH P50 AG16570 (UCLA Alzheimer Disease Research Center), NIH P01 AG12435 (recently ended), California Pediatric Neuropathology Consortium (state of California), Multi-Center AIDS Cohort Study (NIH contract); has stock ownership at 3M Corporation (medical supplies and equipment), GE (medical and imaging equipment), Teva Pharma, Pfizer, GlaxoSmithKline Beecham, and Becton Dickinson. O. Ross: NIH funding (National Institute of Neurological Disorders and Stroke P50 NS072187 and NINDS R01 NS078086) and editorial board membership for Parkinsonism and Related Disorders and the American Journal of Neurodegenerative Disease. Z. Wszolek serves as co–editor-in-chief of Parkinsonism and Related Disorders, regional editor of the European Journal of Neurology, and on the editorial boards of Neurologia i Neurochirurgia Polska, Advances in Rehabilitation, Medical Journal of the Rzeszow University, and Clinical and Experimental Medical Letters; holds and has contractual rights for receipt of future royalty payments from patents re: A novel polynucleotide involved in heritable Parkinson's disease; receives royalties from publishing Parkinsonism and Related Disorders (Elsevier, 2011, 2012, 2013) and the European Journal of Neurology (Wiley-Blackwell, 2011, 2012, 2013); and receives educational grant support from Allergan, Inc. (2011, 2012), and research support from the NIH/National Institute of Neurological Disorders and Stroke P50 NS072187, the Mayo Clinic Center for Regenerative Medicine, the Dystonia Medical Research Foundation, and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch. L. Wang reports no disclosures relevant to the manuscript. D. Dykxhoorn received funding from NIH (4R33AI088601-03). M. Pericak-Vance and T. Montine report no disclosures relevant to the manuscript. J. Leverenz is a consultant for Piramal and Navidea, received research support from Veteran Affairs, NIA (P50AG005136-27), and National Institute of Neurological Disorders and Stroke (P50NS2062684-02). T. Dawson is a consultant for CDP, chair of the SAB of the Bachmann Strauss Dystonia and Parkinson's Disease Foundation, member of the SAB of the PSP Foundation, funding from NIH/National Institute of Neurological Disorders and Stroke/NIDA/MSCRF/AHMMRF/JPB/CPT. J. Vance is a member of the scientific advisory board of the National Parkinson Foundation. He may accrue revenue on patents submitted by the Duke University wherein he is inventor including discoveries of genes causing Charcot-Marie-Tooth disease and Methods for identifying an individual at increased risk for developing coronary artery disease; research support from NIH 1P50NS071674-02, and Hope for Vision. In 2013, Dr. Vance received honoraria from AAN and royalties from Athena Diagnostics for Charcot-Marie-Tooth disease. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Pankratz N, Beecham GW, DeStefano AL, et al. Meta-analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann Neurol 2012;71:370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V, Hernandez DG, et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 2011;377:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Parkinson's Disease Genomics Consortium (IPDGC), Wellcome Trust Case Control Consortium 2 (WTCCC2). A two-stage meta-analysis identifies several new loci for Parkinson's disease. PLoS Genet 2011;7:e1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edwards TL, Scott WK, Almonte C, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 2010;74:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet 2010;42:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 2009;41:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nalls MA, Pankratz N, Lill CM, et al. Large scale meta analysis of genome-wide association data in Parkinson's disease reveals 6 novel risk loci. Nat Genet 2014;46:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol 1999;56:33–39. [DOI] [PubMed] [Google Scholar]
- 10.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson's disease. Neurology 2001;57:1497–1499. [DOI] [PubMed] [Google Scholar]
- 12.Rajput DR. Accuracy of clinical diagnosis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 1993;56:938–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holtbernd F, Eidelberg D. The utility of neuroimaging in the differential diagnosis of parkinsonian syndromes. Semin Neurol 2014;34:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. J Alzheimers Dis 2006;9:417–423. [DOI] [PubMed] [Google Scholar]
- 15.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011;43:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 17.1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007;39:906–913. [DOI] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265. [DOI] [PubMed] [Google Scholar]
- 21.Oliveira SA, Li YJ, Noureddine MA, et al. Identification of risk and age-at-onset genes on chromosome 1p in Parkinson disease. Am J Hum Genet 2005;77:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hicks AA, Petursson H, Jonsson T, et al. A susceptibility gene for late-onset idiopathic Parkinson's disease. Ann Neurol 2002;52:549–555. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Scott WK, Hedges DJ, et al. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet 2002;70:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, El Naqa IM. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics 2008;24:325–332. [DOI] [PubMed] [Google Scholar]
- 25.Maragkakis M, Alexiou P, Papadopoulos GL, et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics 2009;10:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noureddine MA, Li YJ, van der Walt JM, et al. Genomic convergence to identify candidate genes for Parkinson disease: SAGE analysis of the substantia nigra. Mov Disord 2005;20:1299–1309. [DOI] [PubMed] [Google Scholar]
- 27.Ullrich S, Munch A, Neumann S, Kremmer E, Tatzelt J, Lichtenthaler SF. The novel membrane protein TMEM59 modulates complex glycosylation, cell surface expression, and secretion of the amyloid precursor protein. J Biol Chem 2010;285:20664–20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.