

Abstract
Although oncolytic viruses have shown great promise as cancer therapeutics, results from a recent phase III clinical trial indicate that their potency may need further improvement for a clear clinical benefit. Here, we report a novel strategy to increase the bystander effect of virotherapy by arming an oncolytic virus with a secreted form of a Her2 single chain antibody linked to a self-multimerizing Fas ligand extracellular domain (Her2-COL-sFasL). The rationale is that, due to its much smaller size, this apoptosis activator can overcome obstacles such as the dense collagen in the tumor tissues to spread more freely than the viral particles. When measured in vitro, Her2-COL-sFasL was found to efficiently induce caspase cleavage, resulting in an 80% reduction in cell viability. Once incorporated into the genome of an oncolytic type 2 herpes simplex virus, FusOn-H3, Her2-COL-sFasL potentiates the therapeutic efficacy of the virus in an aggressive syngeneic mammary tumor model. Our data suggest that arming an oncolytic virus with a secretable and self-multimerizing apoptosis inducer is a feasible strategy to improve the potency of virotherapy.
INTRODUCTION
Despite the steady progress made in recent years to improve standard of care, many patients with malignant diseases nevertheless receive poor prognoses. Consequently, the urgent development of new therapies is critical. Cancer virotherapy specifically represents one of the new attempts. Unlike other biotherapeutics currently under research and development, virotherapy has a simple yet pragmatic antitumor mechanism – harnessing the intrinsic cytolytic capability of a virus for targeted killing of cancer cells. Many viruses have been modified for oncolytic purpose, including a type 2 herpes simplex virus (HSV-2) that we have used for the construction of FusOn-H2 that can selectively replicate in tumor cells with an activated Ras signaling pathway.1 In recent years several oncolytic viruses have been evaluated in clinical trials for treatment of a variety of malignant diseases. However, despite these exciting developments, the clinical outcome from a recent phase III clinical trial indicates that the therapeutic efficacy of current versions of oncolytic viruses need to be further improved before their full clinical benefit may be realized.
Arming an oncolytic virus with a molecule that can incite an additional killing effect against tumor cells is an appealing strategy to potentiate virotherapy. Indeed, it has been reported that incorporation of molecules, such as the prodrug converting enzyme thymidine kinase and fusogenic glycoproteins that can cause syncytia formation, into an oncolytic virus can enhance the therapeutic effect when tested in preclinical animal models.2–7 We reasoned that, if the incorporated molecule could be secreted and acted extrinsically on the bystander tumor cells, the additional therapeutic benefit would be more profound. First, as compared with viral particles, the incorporated molecules are likely to have a much smaller size. As such, they would be able to diffuse more freely within tumor tissues even in the presence of dense extracellular matrixes, which have been shown to severely compromise the ability of oncolytic viruses to spread.8 Second, the therapeutic effect of the secretable molecules will not be limited to act strictly on infected cells. As such, the oncolytic virus and secretable molecules can kill tumor cells separately and by different mechanisms. This is desirable as, in addition to the extra killing of tumor cells, the incited bystander effect on the heterogeneous population of malignant cells can potentially prevent tumor regrowth following virotherapy.
The extrinsic pathway of apoptosis is a well characterized pathway to activate programmed cell death via a cell surface interaction. This form of programmed cell death is activated following the binding of a transmembrane ligand located on the cell membrane with its respective transmembrane death receptor located on the surface of another cell. Two commonly studied transmembrane ligands that activate the extrinsic pathway of apoptosis are the TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL). TRAIL binds to death receptor 4 (DR4) and DR5, while FasL (also known as CD95 ligand) binds to the Fas receptor (CD95). Binding of TRAIL or FasL to their respective receptors leads to signaling inside the cell through formation of an intracellular death-inducing signaling complex (DISC) at the cytoplasmic domain of the receptors.9 DISC formation results in proteolytic processing of initiator caspase-8. Caspase-8 then activates the executioner caspases-3, and −7 that then process various proteins responsible for apoptosis.10
TRAIL has been shown to induce apoptosis in a variety of cancer cells without harming normal cells and accordingly active forms of recombinant TRAIL can be safely systemically administered.11, 12 Therefore, multiple TRAIL-based cancer therapeutics have been developed and moved into phase I clinical trials. Although, the clinical outcomes suggest translation of the tumor killing properties of TRAIL as a single agent has not proven successful.13–15 Due to the safety profile of TRAIL-based therapies, multiple investigators have effectively established oncolytic viruses that safely deliver transmembrane or soluble TRAIL transgenes for the treatment of multiple cancer subtypes in vitro and in vivo.16–18 The more important ligand for our study is FasL in which its proteolytic cleavage produces a soluble form consisting of the FasL extracellular domain that has 1,000-fold less apoptosis inducing capacity.19 Nevertheless the apoptosis signaling activity of this soluble form of FasL can be restored through forced multimerization or secondary crosslinking.19–21 Unlike TRAIL, however, FasL lacks tumor selective properties and therefore IV injection of active molecules targeting the Fas receptor results in rapid systemic toxicity.12, 19, 22 As such, safe viral delivery of FasL requires strict control of gene expression but has identified FasL as a potent anticancer agent.23–27 Yet, due to safety concerns, the use of active sFasL molecules as cancer therapeutics has not been widely explored.
We explored the possibility of potentiating FusOn-H3 with a soluble form of either TRIAL or FasL. When we compared their potency, we found that FasL was significantly more effective than TRAIL at inducing apoptotic death of several tumor cells that we tested. We thus focused on a self-multimerizing form of sFasL and we used two strategies to limit its effect to tumor cells. First, we linked this multimerized sFasL to a single chain antibody specific for the tumor antigen Her2. We then inserted it into the backbone of FusOn-H3, so that it is expressed in the context of a conditionally replicating oncolytic virus. This will safely establish a high relative concentration of this secreted multimerized sFasL specifically at the tumor site. As a result, secreted molecules will induce apoptotic death of bystander uninfected tumor cells, inciting maximal therapeutic effect of both the oncolytic virus and the chimeric sFasL molecule.
RESULTS
Construction of secreted sFasL chimeric molecules that can self multimerize as active apoptosis activators
We first designed and constructed a panel of chimeric molecules with differing combinations of functional domains. A schematic of the constructs is shown in Figure 1a. First, an optimal secretion signal and hemagglutinin tag (HA) was fused to the amino-terminus of sFasL (amino acids 139–281 of the extracellular domain) forming Secr-sFasL. Second, for the purpose of multimerization, a previously optimized collagen-based multimerization domain (COL) was inserted between the HA tag and sFasL domain, forming COL-sFasL. The COL domain consists of the hinge region of human IgG at the N terminus, a collagen-like scaffold (glycine-proline-proline)10 repeat flanked at the C terminus by the NC1 domain of type XXI collagen (see Supplementary Figure S1 for sequence details).28 Furthermore, Her2-COL-sFasL was constructed by inserting the Her2 scFv between the secretion signal and HA tag of COL-sFasL. For comparison, Her2-sFasL was constructed by inserting the Her2 scFv between the secretion signal and HA tag of Secr-sFasL. In addition two control constructs, HA-COL and Her2-COL, were made that contain differing functional domains without sFasL.
Figure 1. Design and Western blot analysis of secreted multimerized sFasL chimeric molecules.
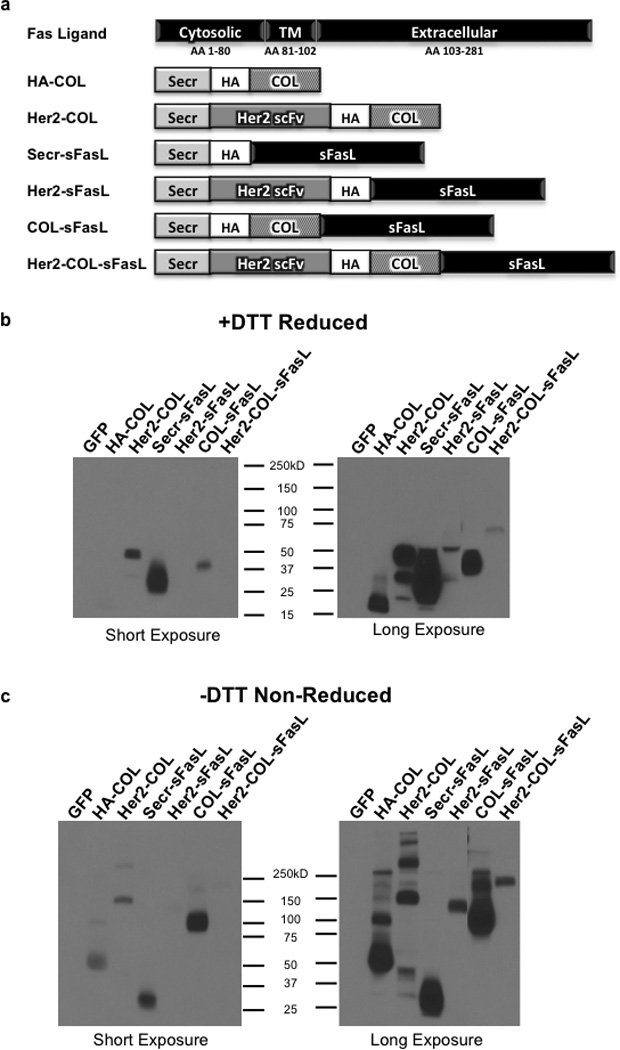
(a) Schematic representation of the sFasL chimeric molecules used in this study. A portion of the FasL extracellular domain (amino acids 139–281) was used to make constructs. Secr, optimal secretion signal; HA, hemagglutinin tag; COL, collagen-like trimerization domain; Her2 scFv, FRP5 Her2 single chain variable fragment; TM, transmembrane domain; AA, amino acids. (b and c) Western blot detection of chimeric sFasL molecules and control constructs under reducing and non-reducing conditions. HCT116 cells were transfected with mammalian expression constructs encoding the respective chimeric molecules. After 48 hours cell debris was removed from media and equal amounts of media subjected to Western blot analysis (b) with 25mM DTT and (c) without DTT. HA tag antibody was used for detection and data shown at two different exposure times for clarity. Western blot results are representative of three independent experiments.
To verify secretion and to characterize multimerization of the chimeric molecules, HCT116 cells were transfected with the indicated constructs and 48 hours later the transfected cell media was collected for Western blot analysis. HCT116 cells were primarily chosen for this and subsequent experiments for their high sensitivity to FasL, Her2 expression on their cell surface, and permissiveness to FusOn-H3 replication (data not shown). All the chimeric molecules are produced and secreted by the transfected cells at their respective predicted molecular weights (Supplementary Table S1), with the relative secretion levels varying between chimeric proteins (Figure 1b). Although Her2-sFaL and Her2-COL-sFaL are the least produced molecules, we can’t completely rule out the possibility that this may be due to the HA tag placement (in the middle of these two chimeric molecules) that may affect the binding affinity of the anti-HA antibody. As predicted, under non-reducing conditions the chimeric molecules that contain the COL domain (HA-COL, Her2-COL, COL-sFasL, and Her2-COL-sFasL) are multimerized into trimeric structures whereas Secr-sFasL and Her2-sFasL are not (Figure 1c). It is also noticed that, although most of the COL domain containing molecules multimerize into trimers, disulfide bond formation at the NC1 domain between two trimers allows hexamer formation as seen by the higher molecular weight band in COL-sFasL (Figure 1c). Thus, the chimeric molecules are expressed, multimerized, and secreted at a readily detectable level into the transfected cell media.
The multimerized forms of sFasL-containing chimeric molecules can induce caspase activation and cell death
Caspase-8 is an initiator caspase specific to the extrinsic pathway, whereas caspase-3 is an executioner caspase that activates the downstream events resulting in cell death through apoptosis.29 Therefore, we tested the ability of the secreted molecules to induce caspase cleavage to activate apoptosis by measuring the cleavage of caspase-8 and caspase-3 into their respective active cleaved forms (p18 and p19 respectively). An equal volume of the 48 hour HCT116 cell media used for Figures 1b and c was added to freshly seeded HCT116 cells, which were then incubated for 24 hours. Afterwards, cells were collected and samples with equal amounts of protein were loaded for Western blot analysis. HCT116 cells treated with media containing control sFasL molecules that lack a multimerization domain or control molecules lacking the sFasL domain show negligible caspase-8 or caspase-3 cleavage (Figure 2a). Whereas, the transfer of media containing Her2-sFasL, COL-sFasL, or Her2-COL-sFasL to HCT116 cells led to significant cleavage of caspase-8 and caspase-3 into their respective active products. These results thus indicate that the ability of these sFasL containing chimeric molecules to self multimerize into an active form is crucial for them to activate caspase cleavage.
Figure 2. Chimeric sFasL molecules induce caspase activation and cell death in vitro.
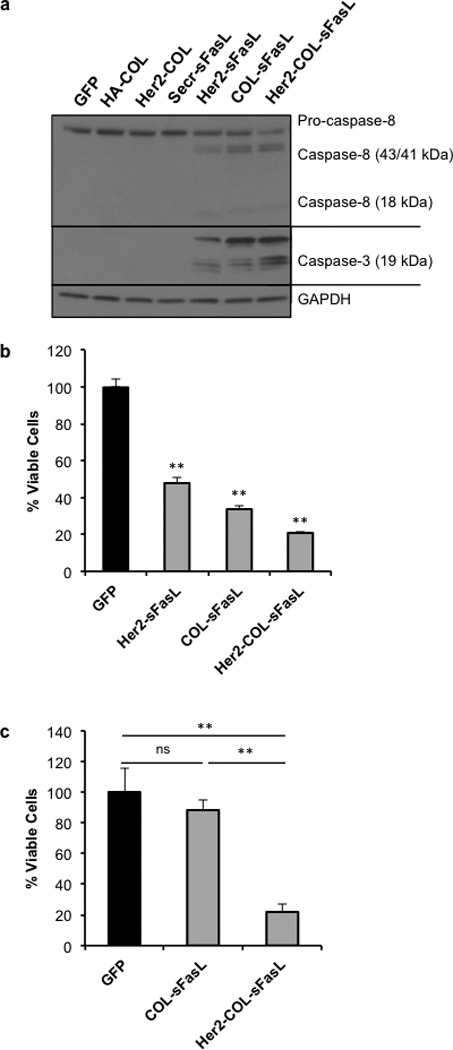
HCT116 cells were transfected with respective molecules. Media was collected after 48 hours and cellular debris removed. (a) Caspase-8 and −3 cleavage following treatment with chimeric sFasL molecules. Western blot analysis of equal amounts of HCT116 cellular protein after 24 hours of specified media treatment. Caspase-8 and caspase-3 antibodies were used to detect active cleavage products specified (p18 and p19, respectively). GAPDH was used as loading control. In (b and c) viable HCT116 cells counted using trypan blue exclusion assay 48 hours after specified media treatment. Then triplicate values were averaged and percent viable cells calculated by normalization to GFP control media treated cells. (b) Reduced HCT116 cell viability following supernatant transfer. Data shown is the average of three independent experiments. ** p<0.001 as compared with GFP transfected cell media treatment controls using student’s T test. (c) HCT116 cells treated in triplicate with equal amounts of COL-sFasL and Her2-COL-sFasL. Data are representative of two independent experiments. ns, not significant; ** p<0.001 using student’s T test as compared to cells treated with GFP media. Graphs show mean ±SEM.
Next, we examined if the induced caspase cleavage by these three molecules affected cell viability. Supernatants were collected the same way as in Figure 2a and subsequently added to freshly seeded HCT116 cells in triplicate, which were then incubated for 48 hours. Afterwards, cell viability was determined by trypan blue exclusion assay and the obtained results were normalized to the GFP transfected cell media treated cell control. The result shows that all three molecules shown to activate caspase cleavage also can significantly reduce the amount of viable HCT116 cells (Figure 2b). However, the most potent one among the three active molecules is Her2-COL-sFasL. It reduces the amount of viable cells to a very low level, at 21% of the control. These results, together with those shown in Figure 2a, indicate that, in addition to the ability to multimerize, the addition of the Her2 scFv to the chimeric molecule also potentiates its ability to induce apoptosis.
Considering the chimeric molecules were secreted at different quantities in the supernatants (Figure 1b), we then conducted a supernatant transfer experiment in which two of the most potent molecules, COL-sFasL and Her2-COL-sFasL, were adjusted to an equal amount (Supplementary Figure S2). With the adjusted quantity, Her2-COL-sFasL still showed a similar potency in decreasing the cell viability (to 22%), while a minimal decrease in percent viable cells (to 88%) is seen with an equivalent amount of COL-sFasL (Figure 2c). This result indicates that Her2-COL-sFasL is intrinsically more potent than the other chimeric molecules in inducing tumor cell death. As such, it was chosen as the prototype for insertion into the oncolytic virus for further examination. Three other molecules, Her2-COL, Her2-sFasL, and COL-sFasL were also inserted into the virus as controls for the purpose of comparison.
Construction and verification of recombinant oncolytic viruses containing apoptosis-inducing chimeric molecules
The parental virus used throughout these experiments as the backbone for all recombinant viruses is FusOn-H3. FusOn-H3 is derived from a previously reported HSV-2 based oncolytic virus, FusOn-H2 (ref. 1), by removing the GFP gene contained in the parental virus (Supplementary Figure S3). Initially the gene encoding red fluorescent protein (RFP) flanked by two Cre recombinase recognition sequences that cannot cross-interact (loxp and lox2272), was inserted into a locus adjacent to the modified ICP10 gene cassette in the genome of FusOn-H3 through homologous recombination (Supplementary Figure S3). As depicted in Figure 3a, the new virus is designated as FusOn-H3-RFP. For further virus construction, recombinase-mediated cassette exchange (RMCE) was used to rapidly produce recombinant viruses.30 In the presence of Cre recombinase and a molar excess of the donor plasmid containing the desired insert flanked by the two sites as described in Figure 3b, the insert is rapidly and specifically exchanged into the viral backbone at the location of the corresponding recognition sites. Therefore co-transfection of linear donor plasmid (inserts shown in Figure 3b) with purified FusOn-H3-RFP DNA into a Cre-overexpressing Vero cell line results in rapid production of recombinant viruses.
Figure 3. Construction of FusOn-H3 recombinant viruses and secretion of chimeric sFasL molecules following infection.
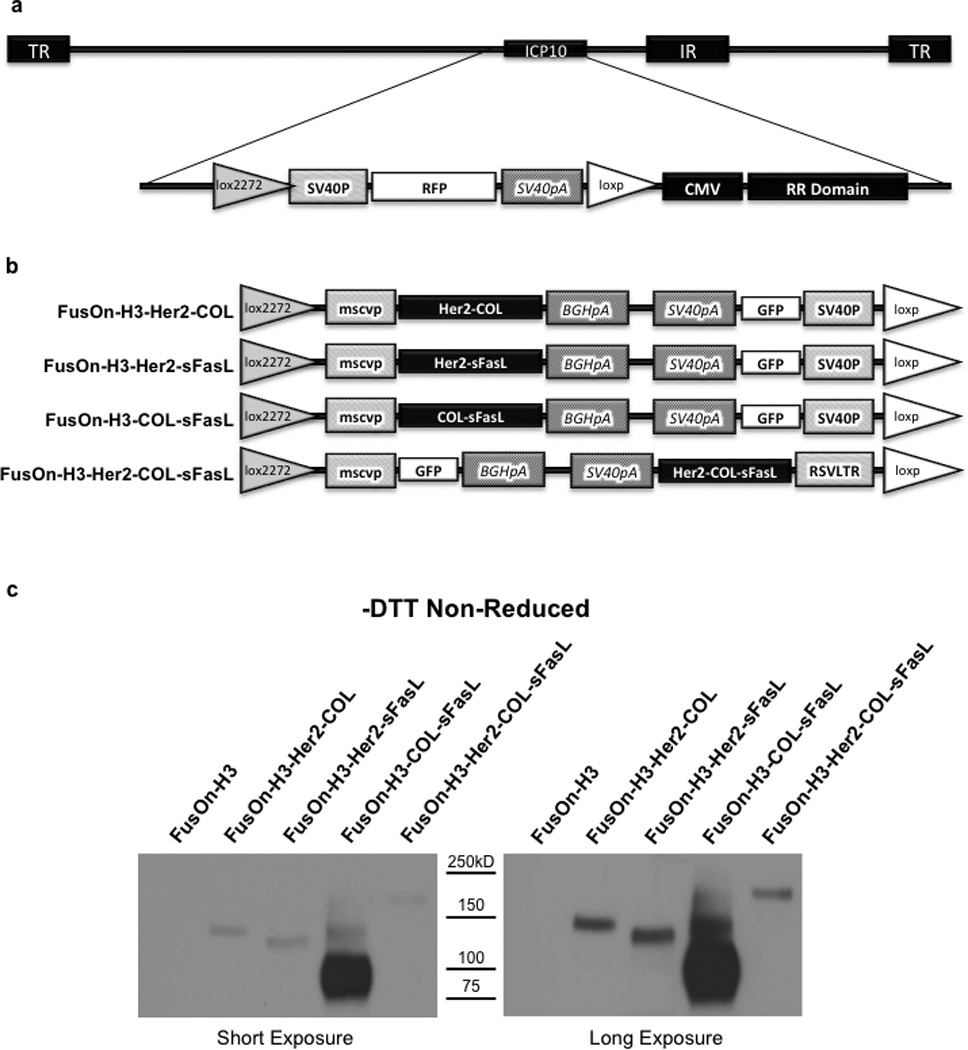
(a) Schematic representation of FusOn-H3-RFP virus genome. The modified region adjacent to the ICP10 gene locus is enlarged showing the lox2272/loxp flanked RFP cassette. TR, terminal repeat; IR, internal repeat; ICP10, infected cell protein 10 gene; loxp and lox2272, cre recombinase binding sites; SV40P, SV40 promoter; RFP, red fluorescent protein gene; SV40pA, SV40 polyadenylation sequence; CMV, cytomegalovirus promoter; RR1, ribonucleotide reductase carboxyl terminus. (b) PCR2.1 donor plasmid inserts for recombinant FusOn-H3 construction. Mscvp, murine stem cell virus promoter; BGHpA, bovine growth hormone polyadenylation signal; GFP, green fluorescent protein; RSVLTR, rous sarcoma virus long terminal repeat. (c) Western blot confirmation of sFasL chimeric molecule secretion by recombinant FusOn-H3 virus infected cells. HCT116 cells were infected at an MOI of 1. Media was collected 24 hours post infection and equal volumes subjected to Western blot under non-reducing conditions. Proteins were detected using an HA tag antibody and are shown at two different exposure times for clarity. Data are representative of two independent experiments.
Using this strategy, viruses expressing Her2-COL, Her2-sFasL, COL-sFasL, and Her2-COL-sFasL were constructed using the donor plasmids shown in Figure 3b, creating FusOn-H3-Her2-COL, FusOn-H3-Her2-sFasL, FusOn-H3-COL-sFasL, and FusOn-H3-Her2-COL-sFasL recombinant oncolytic viruses, respectively. Upon plaque formation, recombinants were easily identified through a change in fluorescence marker from RFP to GFP. The recombinant viruses were subsequently purified to homogeneity by multiple rounds of plaque purification in Vero cells. PCR amplification of viral DNA purified from the recombinants was then used to further verify gene insertion at the correct locus (Supplementary Figures S4 and 5). Secretion of the molecules was verified by infecting HCT116 cells at a multiplicity of infection (MOI) of 1 for 24 hours. An equal volume of media was used for Western blot analysis under non-reducing conditions using the HA tag for detection (Figure 3c). Under non-reducing conditions the molecules secreted following viral infection are also multimerized as seen in Figure 1c and described in Supplementary Table S1. Similar to what was seen in the transfected HCT116 cell media shown in Figure 1, the relative amount of molecules secreted following infection with the recombinant viruses is also not equivalent. These data demonstrate that these apoptosis inducing chimeric molecules can be efficiently expressed from the oncolytic FusOn-H3 once the gene cassettes were inserted into the virus backbone.
Arming of FusOn-H3 with apoptosis activators increases caspase activation in infected cells while not severely inhibiting virus replication
In order to determine if the sFasL-containing chimeric molecules expressed by the recombinant viruses could induce apoptosis activation, HCT116 cells were infected with the viruses at an MOI of 0.1 for 30 hours. Several viruses, including the HSV-1 based oncolytic virus Baco-1 (ref. 31), wild type HSV-2 (wt186),32 and the parental FusOn-H3 were included as controls. Cells were then harvested and caspase-8 and −3 cleavage was examined. The results in Figure 4a show that, unlike Baco-1 or wt186, FusOn-H3 induced a measurable level of caspase-8 cleavage. This result supports our previous finding that the deletion introduced into the N-terminal region of the ICP10 gene in FusOn-H2 increased the ability of the virus to induce apoptotic death of the infected cells.1 However, both FusOn-H3-COL-sFasL and FusOn-H3-Her2-COL-sFasL induced a higher level of caspase-8 cleavage than FusOn-H3. Moreover, both FusOn-H3-COL-sFasL and FusOn-H3-Her2-COL-sFasL induced noticeable caspase-3 cleavage while no caspase-3 cleavage was visible in samples harvested from cells infected with FusOn-H3 (Figure 4a). Similar to the findings from supernatant transfer, FusOn-H3-Her2-COL-sFasL is particularly potent at inducing cleavage of both caspase-8 and −3. In contrast, FusOn-H3-Her2-sFasL, which contains the non-trimerized Her2-sFasL molecule, did not significantly increase the cleavage of either caspase. This further reinforces the importance of multimerization in the ability of these sFasL-containing molecules to induce apoptosis.
Figure 4. Arming of FusOn-H3 with apoptosis activators increases caspase activation without severely effecting replication.
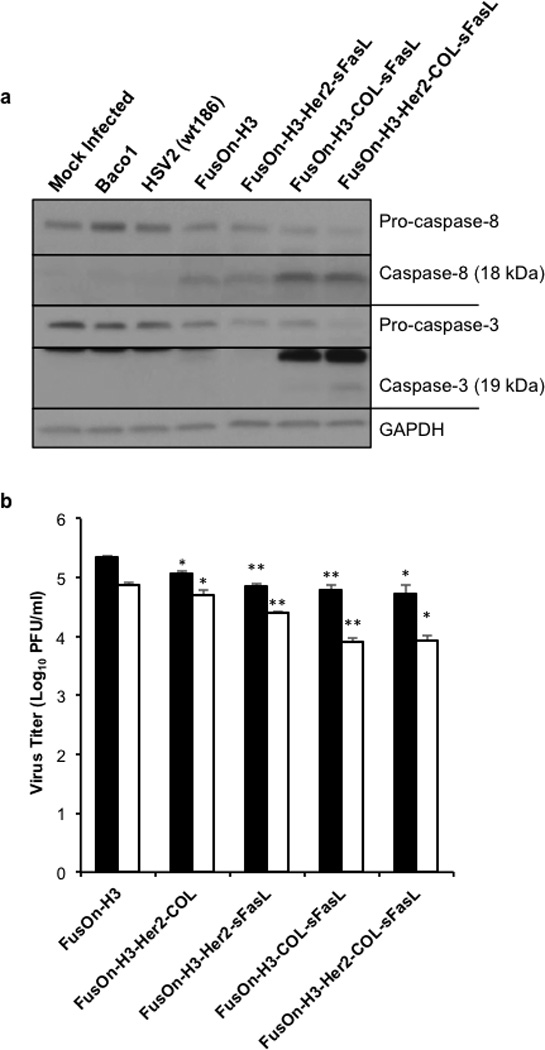
(a) Western blot of caspase-8 and caspase-3 cleavage following recombinant virus infection. HCT116 cells were infected at an MOI of 0.1, cells collected 30 hours post infection, and lysates subjected to Western blot analysis. Caspase-8 and capsase-3 antibodies were used to detect specified cleavage products. GAPDH was used as a loading control. Data shown is representative of two independent experiments (b) Viral titer of FusOn-H3 and recombinant viruses expressing chimeric sFasL molecules. HCT116 cells were infected in triplicate with designated virus at an MOI of 1. Cells were collected 24 (black bars) and 48 hours post infection (white bars) and total infectious virus quantified through titration in Vero cells. *p< 0.01; **p<0.001 compared to respective 24 or 48 hour FusOn-H3 titer according to student’s T test. Error bars represent mean ±SEM.
Next we examined if the addition of these molecules to the oncolytic virus affects viral replication. HCT116 cells were infected with the recombinant viruses at an MOI of 1 for 24 or 48 hours followed by subsequent virus titration in Vero cells. As shown in Figure 4b, all the recombinant viruses had lower virus yield as compared with the parental FusOn-H3 at both time points, indicating that insertion of the gene cassettes containing these chimeric molecules affects virus replication to a certain degree. The results also showed a tendency of inverse correlation between the ability of the molecules to induce caspase cleavage and the virus yield in the tumor cells, indicating that the impact of the molecules on the tumor cells also affects virus replication.
Arming of FusOn-H3 with Her2-COL-sFasL can enhance and extend the therapeutic effect of the oncolytic virus in vivo
For initial in vivo studies, we chose the FusOn-H3-Her2-COL-sFasL recombinant virus that contains the most active apoptosis-inducing activator. We initially compared FusOn-H3-Her2-COL-sFasL with the parental FusOn-H3 for their therapeutic effect against HCT116 xenograft tumors established subcutaneously in NSG mice. When HCT116 tumors reached the approximate diameter of 5mm, mice were randomly separated into three treatment groups as follows: PBS control group, FusOn-H3, and FusOn-H3-Her2-COL-sFasL. The viruses were intratumorally injected at a relatively low dose of 1×105 pfu to allow the additional antitumor effect from the transgene to be fully displayed. Tumors were measured twice a week following treatment and the results are shown in Figure 5. At this relatively low dose, FusOn-H3-Her2-COL-sFasL almost completely eradicated the tumor. Although the tumors treated with the parental FusOn-H3 virus are much smaller than those in the PBS control group, at the end of the experiment they still had a sizable mass remaining (Figure 5). Consequently, these results demonstrate that incorporation of Her2-COL-sFasL can potentiate the therapeutic effect of the backbone oncolytic virus.
Figure 5. Arming of FusOn-H3 with Her2-COL-sFasL can enhance and extend the therapeutic effect of the oncolytic virus in vivo.
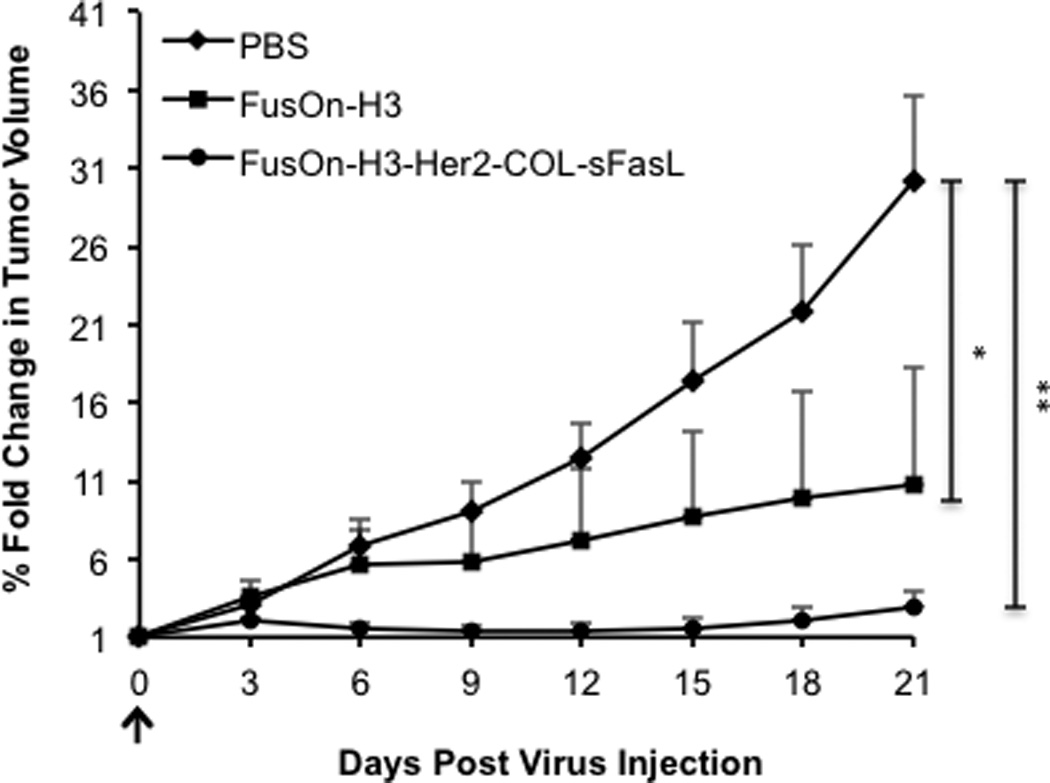
HCT116 subcutaneous tumors were established in the right flank of NSG mice by subcutaneous injection of 2.5×106 cells. Once tumors reached the diameter of 5mm they were intratumorally injected (arrow) with PBS as a control, 1×105 pfu FusOn-H3, or 1×105 pfu FusOn-H3-Her2-COL-sFasL. Tumors were measured every 3 days with a caliper. Percent change in tumor volume was calculated by dividing the daily tumor volume by the tumor volume measurement at day 0. These measurements were then averaged (n=5 mice per group). *p<0.05; **p<0.001 on day 21 according to student’s T test. Error bars represent ±SEM.
FusOn-H3-Her2-COL-sFasL retrieved from in vivo passage in tumor-bearing mice maintains high titer and shows enhanced efficacy against a syngeneic murine tumor
The results in Figure 4b showed that incorporation of Her2-COL-sFasL into the FusOn-H3 backbone affected the virus replication by almost a log. Previous studies by Shah et al have shown that in vivo passage of transgene encoding oncolytic HSVs can improve virus replication.33 We thus passaged FusOn-H3-Her2-COL-sFasL in vivo by injecting the virus into HCT116 tumors and retrieving it 30 days after virus injection. The retrieved virus was then compared with FusOn-H3 and the unpassaged FusOn-H3-Her2-COL-sFasL virus for replication in 4T1 mouse mammary gland tumor cells, which were previously found to be semi-permissive to FusOn-H2 (ref. 34). Figure 6a shows that the passaged virus (herein referred to as FusOn-H3-Her2-COL-sFasL*) replicates closer to the level of FusOn-H3 parental virus in vitro in 4T1 cells, indicating the strategy to improve virus replication through in vivo passage also applies to this sFasL-containing oncolytic HSV.
Figure 6. In vivo passaging of FusOn-H3-Her2-COL-sFasL results in a virus adapted for enhanced in vitro replication and significantly extends the therapeutic effect of the oncolytic virus in a 4T1 immunocompetent in vivo model.
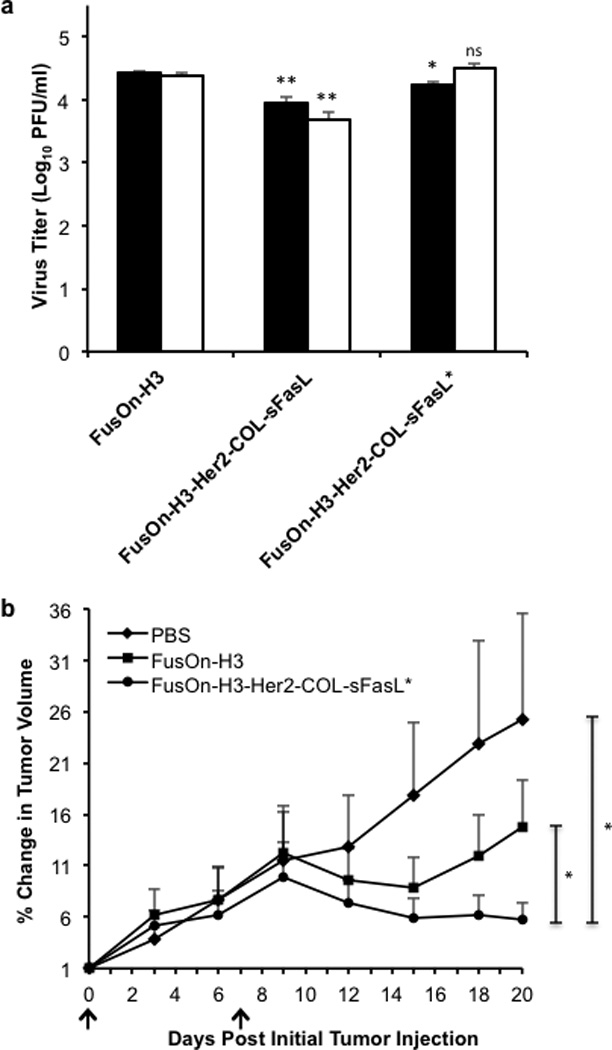
(a) Comparative analysis of in vitro replication efficiencies of control and in vivo passaged viruses in 4T1 cells. 4T1 cells were infected in triplicate with designated virus at an MOI of 10. Cells were collected at 24 (black bars) and 48 hours post infection (white bars) and total infectious virus quantified through titration in Vero cells. ns, not significant; *p< 0.01; **p<0.001 as compared to respective 24 or 48 hour FusOn-H3 titer according to student’s T test. (b) Enhanced therapeutic efficacy of FusOn-H3-Her2-COL-sFasL* in 4T1 syngeneic in vivo model. 4T1 subcutaneous tumors were established in right flanks of BALB/c mice by injection of 1×105 cells per mouse. Once tumors reached the average diameter of 4mm, tumors were intratumorally injected with PBS as a control, 1×107 pfu FusOn-H3, or 1×107 pfu FusOn-H3-Her2-COL-sFasL* on day 0 and day 7 (black arrows). Tumors were measured every 3 days with a caliper. Percent change in tumor volume was calculated by dividing the daily tumor volume by the tumor volume measurement at day 0. These measurements were then averaged (n=5 mice per group). No statistical difference between FusOn-H3 and FusOn-H3-Her2-COL-sFasL virotherapy was detected prior to day 20. *p<0.05 on day 20 according to student’s T test. Error bars represent ±SEM.
Next, we evaluated the therapeutic effect of FusOn-H3-Her2-COL-sFasL* and compared it with that of FusOn-H3 in the syngeneic 4T1 tumor model. Following subcutaneous 4T1 tumor implantation in BALB/c mice, tumors grew to approximately 4mm diameter then were randomly separated into three treatment groups as follows: PBS control group, FusOn-H3, and FusOn-H3-Her2-COL-sFasL*. The tumors were intratumorally injected twice, on day 0 and day 7, using a relatively high dose, 1×107 pfu, as these murine tumor cells are only semi-permissive to the viruses. Tumors were then measured twice weekly and the results are shown in Figure 6b. FusOn-H3-Her2-COL-sFasL* is able to successfully achieve 4T1 tumor regression until the end of the experiment including one tumor free mouse by day 15. In contrast, the therapeutic effect from the parental FusOn-H3 virus diminishes by day 15 in which tumors began to regrow. Taken together, these results demonstrate that secretion of Her2-COL-sFasL by an in vivo passaged oncolytic virus safely intensifies the therapeutic efficacy of the parental oncolytic virus in a syngeneic model of breast cancer.
DISCUSSION
Interest in oncolytic virotherapy has gained considerable popularity in recent years, and there is an increasing chance that it may become an invaluable cancer therapeutic. However, recent phase III clinical trial results suggest that further improvement on its potency is necessary before this may become a reality. To date, multiple strategies have been applied to enhance the potency of oncolytic viruses.35, 36 However, most of these strategies have been designed in such that they act on the same tumor cells the virus infects. As such, there is a limited gain on additional bystander effect. In this study, we designed a novel strategy to arm an HSV-2 based oncolytic virus with a multimerized secreted sFasL molecule that acts externally. Owing to the relatively small size, this molecule would be able to diffuse freely throughout the tumor tissues as an active form following production by infected tumor cells, inciting additional bystander effect. Our in vitro data showed that, sFasL molecules that could self-multimerize, but not those that do not contain the multimerization domain, can effectively induce caspase activation. And this resulted in efficient killing of tumor cells. When inserted into the genome of FusOn-H3, Her2-COL-sFasL-containing virus (FusOn-H3-Her2-COL-sFasL) is also the most effective among other viruses in inducing caspase cleavage (Figure 4a). In vivo evaluation suggests that FusOn-H3-Her2-COL-sFasL and FusOn-H3-Her2-COL-sFasL* are more effective than the parental FusOn-H3 in treating both xenograft and syngeneic tumors. In the xenograft tumor model, the armed virus almost completely eradicated the tumor when given as a relatively low dose, while the parental virus only reduced the tumor size. In the syngeneic murine mammary tumor model, the armed virus initially shrank and then held the tumor growth for an extended period of time, while tumors treated by FusOn-H3 initially shrank and then started to regrow. Together these results demonstrate that arming oncolytic viruses with secretable FasL extrinsic apoptosis activators is a promising strategy to potentiate virotherapy by producing a better and more sustained antitumor effect than the unarmed virus.
It has been described in the literature that linking a scFv to sTRAIL or sFasL can increase target antigen specific bioactivity.37–40 Consistent with these reports, incorporation of the Her2 scFv alone to sFasL enhanced its killing effect on Her2-positive colon carcinoma cells but not the Her2-negative 4T1 cells (Supplementary Figure S6). On the contrary, the potentiating effect from the combined addition of both a scFv and a multimerization domain seems to be more diverse. Trebing et al show that addition of both a multimerization domain and scFv to sTRAIL enhances its potency on cells expressing the surface target antigen, but the molecule also maintains activity on target antigen negative cells.41 Consistent with their findings, we noticed that Her2-COL-sFasL, which contains both a Her2 scFv and COL multimerization domain, has enhanced in vitro cell killing in both Her2 positive HCT116 cells (Figure 2b) and Her2 negative 4T1 cells (Supplementary Figure S6) in contrast to molecules with either single domain. We speculate that the Her2 scFv may increase the potency of COL-sFasL either by efficiently immobilizing it to the cell surface or by structural stabilization of the chimeric molecule itself. Regardless of its mechanism, our data clearly demonstrates that the Her2 scFv can further potentiate the killing effect of multimerized sFasL chimeric molecules.
Insertion of the sFasL-containing chimeric molecules into the viral genome seems to affect virus yield. Although there was no problem attaining the recombinant viruses, there is nearly a log decrease in the titer of FusOn-H3-Her2-COL-sFasL as compared with that of the parental FusOn-H3 (Figure 4b and Figure 6a). Despite this reduced virus yield, our in vivo data suggests this replication deficit seen in vitro does not prevent a visible in vivo therapeutic enhancement in the HCT116 model (Figure 5). Our data subsequently show that the reduced virus yield could be fully restored by a single round of in vivo passaging of FusOn-H3-Her2-COL-sFasL. The retrieved virus maintains the ability to produce functional transgene (data not shown), and when evaluated in a syngeneic tumor model, demonstrates a superior therapeutic efficacy than FusOn-H3 (Figure 6b). Previous studies by other researchers have shown that in vivo passage of oncolytic HSV can improve virus replication in less permissive tumor cells 33. The reason why FusOn-H3-Her2-COL-sFasL, after being passaged once through the permissive HCT116 tumor, becomes more effective at replicating in less permissive tumor cells such as 4T1 is not clear. The Her2-COL-sFasL transgene is an unlikely contributing factor for this, as FusOn-H3 obtained from HCT116 tumor from the same experiment also shows a 4-fold increase in virus titer than the un-passaged virus (data not shown). One plausible explanation is that, due to lack of selection pressure from permissiveness, “fitter” viruses have been selected during this in vivo passage.
Multiple investigators have shown that oncolytic viruses can be safely armed with other apoptosis activators such as transmembrane or soluble TRAIL for the treatment of multiple cancer types.16–18 Although to date, oncolytic virus delivery of FasL and especially sFasL molecules has not been as widely explored due to safety concerns. The few reports of arming other oncolytic virus platforms with FasL only describe the use of the transmembrane form.23, 42 Although Li et al describe a bystander effect with transmembrane FasL, we chose the secreted form of sFasL to maximize the bystander effect.23 To ensure safety, we chose to specifically deliver the secretable FasL molecule to tumor cells using our well-characterized HSV-2 oncolytic virus so that the molecule may only be expressed in the local tumor tissue. However, one concern is that the promoter that drives Her2-COL-sFasL expression, RSVLTR, is a constitutive promoter. This can potentially lead to the transgene expression in normal cells, and hence unwanted toxicity. Nevertheless, both FusOn-H3-Her2-COL-sFasL and FusOn-H3-Her2-COL-sFasL* seem to be well tolerated by the treated animals even when administered at a high titer (1×107 pfu) for the treatment of the 4T1 tumors. This is probably because the activity of a constitutive promoter in the context of an oncolytic virus will still be dictated by the ability of the virus to replicate. In normal cells where the virus can’t replicate, there is most likely only one copy of the virus genome (hence one copy of the transgene) per cell. As the virus won’t be able to spread in normal tissues either, the number of virus infected cells in a given organ tissue is very limited and hence the amount of transgene expression. But in tumor cells where the virus can replicate, the virus genome (and hence the transgene) can be replicated hundreds of times within a 24 hour period after infection. Additionally, the replicated virus can spread to the surrounding tumor cells. As such, the difference of transgene expression between tumor and normal tissues can be enormous. Taking these considerations together, we believe that delivery of Her2-COL-sFasL by the oncolytic virus in vivo is a safe and a well tolerated strategy with no apparent visible illness or treatment limiting side effects. Therefore, although Fas receptor activating therapies are typically considered unsafe for systemic administration, this study should encourage more work on establishing new treatment strategies aimed at harnessing the potent effects of soluble forms of FasL as cancer therapeutics using specific delivery methods such as the oncolytic virus described here.
MATERIALS AND METHODS
Cell culture
HCT116 (human colorectal carcinoma), Jurkat (immortalized T lymphocytes), and Vero (African green monkey kidney) cells were obtained from the American Type Culture Collection (Manassas, VA). 4T1 cells were kindly provided by Dr. Fred Miller (Wayne State University, Detroit, MI). CreGH cell line was previously established in our lab as a Cre recombinase overexpressing Vero cell line. HCT116, Vero, 4T1, and CreGH cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin unless specified during experiments. Jurkat cells were maintained in RPMI supplemented with 10% FBS and 1% penicillin-streptomycin.
Cloning of sFasL molecules
The chimeric fusion constructs were cloned using Phusion (NEB, Ipswich, MA) for PCR of the specific domains with primers containing restriction sites followed by subsequent digestion and ligation into the pcDNA3.1 mammalian expression vector (Invitrogen, Grand Island, NY). The optimal secretion signal used in all constructs encodes amino acids: MWWRLWWLLLLLLLLWPMVWA.43 The hemagglutinin (HA) tag was inserted via in frame ligation of annealed oligos encoding HA tag protein sequence (YPYDVPDYA). The Her2 scFv used is the FRP5 clone.44 The human codon optimized COL domain28 was synthesized by DNA2.0 (Menlo Park, CA). For cloning of the full-length human FasL gene (BC017502.1), first RNA was extracted from stimulated Jurkat cells (50ng/mL phorbol myristate acetate and 1ug/mL Ionomycin) using trizol reagent (Invitrogen). The full-length human FasL cDNA was then amplified and resulting PCR product sequenced. The sFasL domain fused into the constructs consists of the FasL DNA encoding amino acids 139–281 of the extracellular domain. Following cloning, all chimeric sFasL constructs were verified by sequencing.
Viruses and virus purification
Baco-1 (ref. 31) and wild type HSV-2 (wt186)32 have been previously described. FusOn-H3 virus used as the base for all recombinant viruses described was derived from FusOn-H2 (ref. 1) through homologous recombination. The resulting FusOn-H3 virus has the GFP portion of the GFP-ICP10 ribonucleotide reductase domain coding sequence replaced with a short in-frame linker. Thus, the linker-ICP10 ribonucleotide reductase domain is still driven by the CMV promoter but now lacks a fluorescent marker.
To establish the recombinase mediated cassette exchange (RMCE) system30 in the FusOn-H3 backbone, two non-interacting Cre binding sites, loxp45 and lox2272 (ref. 46), were inserted into FusOn-H3 adjacent to the modified ICP10 locus. Between the loxp and lox2272 sites an RFP expression cassette was inserted as a negative fluorescence marker for recombinants. The lox2272-RFP-loxp fragment was then inserted into a plasmid containing the respective homologous regions (∼1000 bases each) upstream and downstream from the desired insertion site. When linear plasmid is co-transfected into Vero cells with FusOn-H3 viral DNA, homologous recombination occurs at the specified location in the viral DNA to produce the FusOn-H3-RFP virus described.
Donor plasmids for RMCE were constructed in the PCR2.1 backbone (Invitrogen) by the insertion of two expression cassettes: one encoding the GFP florescent marker and the other encoding the chimeric molecule specified. Prior to transfection, plasmids were linearized using ScaI (NEB) followed by PCR purification (QIAGEN, Valencia, CA). Purified FusOn-H3-RFP viral DNA47 was co-transfected with the linear donor plasmids at a ratio of 10:1 (viral : donor plasmid) into the CreGH cells using FuGENE HD (Promega, Madison, WI). At 48 hours post transfection GFP fluorescent plaques were collected and further passaged in Vero cells until virus could be purified from a single plaque in a 96 well plate. Pure virus was amplified and recombinant viral DNA purified. PCR was then performed using hot start taq polymerase (NEB) to ensure correct insertion location and verify chimeric molecule insertion.
All viruses were amplified in Vero cells. After the cytopathic effect, cells were scraped off dishes, washed, resuspended in PBS, subjected to 3 freeze/thaw cycles, and cell debris clarified from lysate through centrifugation. The lysate was titrated in Vero cells and then stored at −80°C until use. For titrations, HCT116 or 4T1 cells were infected at an MOI of 1 or 10 respectively for 24 or 48 hours in triplicate wells. Total virus was collected and then titrated on Vero cells covered with 1% carboxymethylcellulose (CMC). After 48 hours cells were stained with crystal violet, plaques were counted, triplicate results averaged, and the results were expressed as log10 plaque forming units (pfu)/mL.
Media transfer treatment and trypan blue exclusion assay
HCT116 cells were transfected using FuGENE HD (Promega) in half of suggested media volume per well in 5% FBS DMEM (resulting in 2X concentrated secreted proteins). Media was collected 48 hours post transfection and clarified by brief centrifugation. Fresh HCT116 or 4T1 cells were seeded in a 96-well plate and triplicate wells treated with 100µl of media from cells transfected with specified constructs. 48 hours after treatment cells were washed, trypsinized, and resuspended in DMEM containing 10% FBS. An equal volume of trypan blue staining dye (Cellgro, Manassas, VA) was added to samples and then viable cells counted on a hemocytometer. Blue non-viable cells were not counted. Each treatment was performed in triplicate and viable cell counts averaged. Percent viable cells were derived from dividing the average of each treatment group by the average GFP transfected media treated cell control. Results are representative of 3 independent experiments.
For normalization of Her2-COL-sFasL and COL-sFasL molecule levels, differing dilutions of COL-sFasL cell media (from transfected HCT116 cells as described above) was diluted as follows: undiluted, 1:2, 1:5, 1:10, and 1:25 using media from GFP transfected controls as the dilution media. An equal volume of each diluted sample was subjected to Western blot analysis using HA tag antibody C29F4 primary antibody (Cell Signaling #3724, Danvers, MA) and HRP-conjugated anti-rabbit secondary antibody (Cell Signaling). ImageJ (National Institutes of Health, Bethesda, MD) was used to measure and quantify which dilution had the closest density to the undiluted Her2-COL-sFasL sample. Data shown is from 1:10 COL-sFasL dilution which is about 1.8X higher than Her2-COL-sFasL according to densitometric analysis. The treatments were performed in triplicate, percent viable cell counts quantified at 48 hours post treatment, and normalized to GFP transfected media treated cells. Representative figure is shown for two independent experiments.
SDS-PAGE and Western blot analysis
To analyze secretion of chimeric molecules from transfected HCT116 cells, HCT116 cells were transfected using FuGENE HD (Promega) in half of suggested media volume per well in 5% FBS DMEM (resulting in 2X concentrated secreted proteins) and media collected 48 hours post transfection. To analyze chimeric molecule secretion following infection, HCT116 cells were infected at an MOI of 1 in half volume of media and media collected 24 hours post infection. Medias were centrifuged at low speed to remove cell debris and 40ul of sample prepared in native Laemmli buffer +/− 25mM DTT for reducing and non-reducing conditions, respectively. Samples were then boiled at 95°C for 5 minutes then loaded on Biorad mini protean TGX precast 4–20% gradient SDS-polyacrylamide gels. Protein gels were transferred to PVDF membranes (Biorad, Hercules, CA) for analyzing by Western blot using HA tag primary antibody and HRP-conjugated anti-rabbit secondary antibody. Amersaham ECL prime Western blot detection reagent (GE Healthcare, Pittsburgh, PA) was used for detection.
For caspase cleavage analysis following supernatant transfer, HCT116 cells were treated with the transfected 48 hour HCT116 cell medias for 24 hours then were lysed in RIPA buffer, briefly sonicated at 10% amplitude for 10 seconds, and protein concentration determined using Bradford reagent (Sigma). For caspase activation following infection, HCT116 cells were infected at an MOI of 0.1 for 30 hours and cells collected in RIPA buffer as described above. The caspase-3 and caspase-8 (IC12) antibodies are both from Cell Signaling (#9662 and #9746) and detect full-length caspase and caspase cleavage products. Samples of 50ug protein were prepared in 1X Laemmli buffer and procedure for Western blot used as above. As a loading control, GAPDH ACR001PT (Acris, San Diego, CA) mouse IgG1 antibody was used.
FusOn-H3-Her2-COL-sFasL in vivo passaging and in vivo experiments
For in vivo experiments recombinant viruses were purified from the media using high speed centrifugation followed by titration for further experimentation. Animal experiments were performed following the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the University of Houston. FusOn-H3-Her2-COL-sFasL* was collected following in vivo passaging in NOD scid mice. NOD scid mice were purchased from Taconic (Hudson, NY). Mice were injected with 2.5×106 HCT116 cells in the right flank. When tumors reached 5mm they were injected with 1.0×107 pfu FusOn-H3-Her2-COL-sFasL. Tissue at the tumor site was collected 30 days post virus injection and homogenized in order to release the virus. Virus was then passaged in Vero cells, collected, and amplified. PCR was used to verify maintenance of cassette insertion and also Her2-COL-sFasL secretion was verified via Western blot.
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and BALB/c mice were purchased from Taconic. For establishment of subcutaneous tumors, HCT116 or 4T1 cells were trypsinized, washed, counted, and resuspended in PBS. NSG and BALB/c mice were injected in the right flank with 100µl containing 2.5×106 HCT116 cells and 1×105 4T1 cells, respectively. HCT116 tumors were injected with virus 8 days post tumor cell implantation at an average tumor diameter of 5mm. 4T1 tumors were injected 6 days post tumor cell implantation at an average tumor diameter of 4mm followed by a second virus injection 7 days after first virus injection with the same pfu of respective virus. For the HCT116 model in NSG mice, FusOn-H3 and FusOn-H3-Her2-COL-sFasL virus stocks were diluted to 1.0×106 pfu/mL in PBS and 100ul intratumorally injected per mouse for 1.0×105 (n=5 mice per group) treatments at day 0. For the 4T1 model in BALB/c mice, FusOn-H3 and FusOn-H3-Her2-COL-sFasL* virus stocks were diluted to 6.67×107 pfu/mL in PBS and 150ul intratumorally injected per mouse for 1.0×107 pfu treatments (n=5 mice per group) at day 0 and 7. PBS control injection volumes matched the specified volumes of virus injected.
Tumor measurements were made every three days with a caliper by measuring the tumor length and width. Tumor volume was determined using the formula (mm3) = (length (mm)) × (width (mm))2 × 0.52. The change in tumor volume was then determined by dividing each tumor volume by the tumor volume on day 0 (tumor size when virotherapy first administered). Then the change in tumor volume was averaged for each group of mice and plotted as shown.
Statistical analysis
All quantitative data are reported as means with error bars representing standard error mean (SEM). Student’s T test was used with a p<0.05 being considered statistically significant throughout.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Xinping Fu, Lihua Tao, Armando Rivera, Kim Anthony-Gonda, and Jeffrey Spencer for the sharing of their expertise for a variety of techniques. Also, we further thank Armando Rivera, Kim Anthony-Gonda, and Jeffrey Spencer for reading of the manuscript prior to submission. This work was supported by the National Cancer Institute grants R01CA106671 and R01CA132792 and also by a grant from the William and Ella Owens Medical Research Foundation (to XZ).
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
Supplementary information is available at Gene Therapy's website
REFERENCES
- 1.Fu X, Tao L, Cai R, Prigge J, Zhang X. A Mutant Type 2 Herpes Simplex Virus Deleted for the Protein Kinase Domain of the ICP10 Gene Is a Potent Oncolytic Virus. Mol Ther. 2006;13(5):882–890. doi: 10.1016/j.ymthe.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Wildner O, Blaese RM, Morris JC. Therapy of colon cancer with oncolytic adenovirus is enhanced by the addition of herpes simplex virus-thymidine kinase. Cancer Res. 1999;59(2):410–413. [PubMed] [Google Scholar]
- 3.Bauzon M, Jin F, Kretschmer P, Hermiston T. In vitro analysis of cidofovir and genetically engineered TK expression as potential approaches for the intervention of ColoAd1-based treatment of cancer. Gene Ther. 2009;16(9):1169–1174. doi: 10.1038/gt.2009.69. [DOI] [PubMed] [Google Scholar]
- 4.Fu X, Tao L, Jin A, Vile R, Brenner M, Zhang X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus provides potent synergistic anti-tumor effect. Mol. Ther. 2003;7(6):748–754. doi: 10.1016/s1525-0016(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 5.Ebert O, Shinozaki K, Kournioti C, Park MS, Garcia-Sastre A, Woo SL. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res. 2004;64(9):3265–3270. doi: 10.1158/0008-5472.can-03-3753. [DOI] [PubMed] [Google Scholar]
- 6.Simpson GR, Han Z, Liu B, Wang Y, Campbell G, Coffin RS. Combination of a fusogenic glycoprotein, prodrug activation, and oncolytic herpes simplex virus for enhanced local tumor control. Cancer Res. 2006;66(9):4835–4842. doi: 10.1158/0008-5472.CAN-05-4352. [DOI] [PubMed] [Google Scholar]
- 7.Guedan S, Grases D, Rojas JJ, Gros A, Vilardell F, Vile R, et al. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012;19(11):1048–1057. doi: 10.1038/gt.2011.184. [DOI] [PubMed] [Google Scholar]
- 8.Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and −8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007;67(22):10664–10668. doi: 10.1158/0008-5472.CAN-07-3107. [DOI] [PubMed] [Google Scholar]
- 9.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, et al. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) The EMBO journal. 1997;16(10):2794–2804. doi: 10.1093/emboj/16.10.2794. e-pub ahead of print 1997/05/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muzio M, Salvesen GS, Dixit VM. FLICE induced apoptosis in a cell-free system. Cleavage of caspase zymogens. The Journal of biological chemistry. 1997;272(5):2952–2956. doi: 10.1074/jbc.272.5.2952. e-pub ahead of print 1997/01/31. [DOI] [PubMed] [Google Scholar]
- 11.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. The Journal of clinical investigation. 1999;104(2):155–162. doi: 10.1172/JCI6926. e-pub ahead of print 1999/07/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nature medicine. 1999;5(2):157–163. doi: 10.1038/5517. e-pub ahead of print 1999/02/04. [DOI] [PubMed] [Google Scholar]
- 13.Younes A, Vose JM, Zelenetz AD, Smith MR, Burris HA, Ansell SM, et al. A Phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin's lymphoma. British journal of cancer. 2010;103(12):1783–1787. doi: 10.1038/sj.bjc.6605987. e-pub ahead of print 2010/11/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(17):2839–2846. doi: 10.1200/JCO.2009.25.1991. e-pub ahead of print 2010/05/12. [DOI] [PubMed] [Google Scholar]
- 15.Wakelee HA, Patnaik A, Sikic BI, Mita M, Fox NL, Miceli R, et al. Phase I and pharmacokinetic study of lexatumumab (HGS-ETR2) given every 2 weeks in patients with advanced solid tumors. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2010;21(2):376–381. doi: 10.1093/annonc/mdp292. e-pub ahead of print 2009/07/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamura K, Wakimoto H, Agarwal AS, Rabkin SD, Bhere D, Martuza RL, et al. Multimechanistic tumor targeted oncolytic virus overcomes resistance in brain tumors. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21(1):68–77. doi: 10.1038/mt.2012.175. e-pub ahead of print 2012/08/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou W, Zhu H, Chen W, Hu X, Pang X, Zhang J, et al. Treatment of patient tumor-derived colon cancer xenografts by a TRAIL gene-armed oncolytic adenovirus. Cancer gene therapy. 2011;18(5):336–345. doi: 10.1038/cgt.2010.83. e-pub ahead of print 2010/12/25. [DOI] [PubMed] [Google Scholar]
- 18.Cao X, Yang M, Wei RC, Zeng Y, Gu JF, Huang WD, et al. Cancer targeting Gene-Viro-Therapy of liver carcinoma by dual-regulated oncolytic adenovirus armed with TRAIL gene. Gene therapy. 2011;18(8):765–777. doi: 10.1038/gt.2011.16. e-pub ahead of print 2011/03/18. [DOI] [PubMed] [Google Scholar]
- 19.Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, et al. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. The Journal of experimental medicine. 1998;187(8):1205–1213. doi: 10.1084/jem.187.8.1205. e-pub ahead of print 1998/05/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holler N, Tardivel A, Kovacsovics-Bankowski M, Hertig S, Gaide O, Martinon F, et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Molecular and cellular biology. 2003;23(4):1428–1440. doi: 10.1128/MCB.23.4.1428-1440.2003. e-pub ahead of print 2003/01/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berg D, Lehne M, Muller N, Siegmund D, Munkel S, Sebald W, et al. Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95L. Cell death and differentiation. 2007;14(12):2021–2034. doi: 10.1038/sj.cdd.4402213. e-pub ahead of print 2007/08/19. [DOI] [PubMed] [Google Scholar]
- 22.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, et al. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364(6440):806–809. doi: 10.1038/364806a0. e-pub ahead of print 1993/08/26. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Liu YH, Zhang YP, Zhang S, Pu X, Gardner TA, et al. Fas ligand delivery by a prostate-restricted replicative adenovirus enhances safety and antitumor efficacy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(18 Pt 1):5463–5473. doi: 10.1158/1078-0432.CCR-07-0342. e-pub ahead of print 2007/09/19. [DOI] [PubMed] [Google Scholar]
- 24.Arai H, Gordon D, Nabel EG, Nabel GJ. Gene transfer of Fas ligand induces tumor regression in vivo; Proceedings of the National Academy of Sciences of the United States of America; 1997. pp. 13862–13867. e-pub ahead of print 1998/02/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoki K, Akyurek LM, San H, Leung K, Parmacek MS, Nabel EG, et al. Restricted expression of an adenoviral vector encoding Fas ligand (CD95L) enhances safety for cancer gene therapy. Molecular therapy : the journal of the American Society of Gene Therapy. 2000;1(6):555–565. doi: 10.1006/mthe.2000.0076. e-pub ahead of print 2000/08/10. [DOI] [PubMed] [Google Scholar]
- 26.Hyer ML, Sudarshan S, Schwartz DA, Hannun Y, Dong JY, Norris JS. Quantification and characterization of the bystander effect in prostate cancer cells following adenovirus-mediated FasL expression. Cancer gene therapy. 2003;10(4):330–339. doi: 10.1038/sj.cgt.7700576. e-pub ahead of print 2003/04/08. [DOI] [PubMed] [Google Scholar]
- 27.Muruve DA, Nicolson AG, Manfro RC, Strom TB, Sukhatme VP, Libermann TA. Adenovirus-mediated expression of Fas ligand induces hepatic apoptosis after Systemic administration and apoptosis of ex vivo-infected pancreatic islet allografts and isografts. Human gene therapy. 1997;8(8):955–963. doi: 10.1089/hum.1997.8.8-955. e-pub ahead of print 1997/05/20. [DOI] [PubMed] [Google Scholar]
- 28.Fan CY, Huang CC, Chiu WC, Lai CC, Liou GG, Li HC, et al. Production of multivalent protein binders using a self-trimerizing collagen-like peptide scaffold. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22(11):3795–3804. doi: 10.1096/fj.08-111484. e-pub ahead of print 2008/07/19. [DOI] [PubMed] [Google Scholar]
- 29.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, et al. A unified model for apical caspase activation. Molecular cell. 2003;11(2):529–541. doi: 10.1016/s1097-2765(03)00051-0. e-pub ahead of print 2003/03/07. [DOI] [PubMed] [Google Scholar]
- 30.Nakano M, Odaka K, Takahashi Y, Ishimura M, Saito I, Kanegae Y. Production of viral vectors using recombinase-mediated cassette exchange. Nucleic acids research. 2005;33(8):e76. doi: 10.1093/nar/gni074. e-pub ahead of print 2005/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu X, Tao L, Jin A, Vile R, Brenner MK, Zhang X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Molecular therapy : the journal of the American Society of Gene Therapy. 2003;7(6):748–754. doi: 10.1016/s1525-0016(03)00092-3. e-pub ahead of print 2003/06/06. [DOI] [PubMed] [Google Scholar]
- 32.Milligan GN, Bernstein DI. Generation of humoral immune responses against herpes simplex virus type 2 in the murine female genital tract. Virology. 1995;206(1):234–241. doi: 10.1016/s0042-6822(95)80038-7. [DOI] [PubMed] [Google Scholar]
- 33.Shah AC, Price KH, Parker JN, Samuel SL, Meleth S, Cassady KA, et al. Serial passage through human glioma xenografts selects for a Deltagamma134.5 herpes simplex virus type 1 mutant that exhibits decreased neurotoxicity and prolongs survival of mice with experimental brain tumors. J Virol. 2006;80(15):7308–7315. doi: 10.1128/JVI.00725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Dutuor A, Fu X, Zhang X. Induction of strong antitumor immunity by an HSV-2-based oncolytic virus in a murine mammary tumor model. The journal of gene medicine. 2007;9(3):161–169. doi: 10.1002/jgm.1005. e-pub ahead of print 2007/02/03. [DOI] [PubMed] [Google Scholar]
- 35.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nature biotechnology. 2012;30(7):658–670. doi: 10.1038/nbt.2287. e-pub ahead of print 2012/07/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Molecular cancer. 2013;12(1):103. doi: 10.1186/1476-4598-12-103. e-pub ahead of print 2013/09/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider B, Munkel S, Krippner-Heidenreich A, Grunwald I, Wels WS, Wajant H, et al. Potent antitumoral activity of TRAIL through generation of tumor-targeted single-chain fusion proteins. Cell death & disease. 2010;1:e68. doi: 10.1038/cddis.2010.45. e-pub ahead of print 2011/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samel D, Muller D, Gerspach J, Assohou-Luty C, Sass G, Tiegs G, et al. Generation of a FasL-based proapoptotic fusion protein devoid of systemic toxicity due to cell-surface antigen-restricted Activation. The Journal of biological chemistry. 2003;278(34):32077–32082. doi: 10.1074/jbc.M304866200. e-pub ahead of print 2003/05/30. [DOI] [PubMed] [Google Scholar]
- 39.Bremer E, Samplonius DF, Peipp M, van Genne L, Kroesen BJ, Fey GH, et al. Target cell-restricted apoptosis induction of acute leukemic T cells by a recombinant tumor necrosis factor-related apoptosis-inducing ligand fusion protein with specificity for human CD7. Cancer research. 2005;65(8):3380–3388. doi: 10.1158/0008-5472.CAN-04-2756. e-pub ahead of print 2005/04/19. [DOI] [PubMed] [Google Scholar]
- 40.Bremer E, ten Cate B, Samplonius DF, Mueller N, Wajant H, Stel AJ, et al. Superior activity of fusion protein scFvRit:sFasL over cotreatment with rituximab and Fas agonists. Cancer research. 2008;68(2):597–604. doi: 10.1158/0008-5472.CAN-07-5171. e-pub ahead of print 2008/01/18. [DOI] [PubMed] [Google Scholar]
- 41.Trebing J, El-Mesery M, Schafer V, Weisenberger D, Siegmund D, Silence K, et al. CD70-restricted specific activation of TRAILR1 or TRAILR2 using scFv-targeted TRAIL mutants. Cell death & disease. 2014;5:e1035. doi: 10.1038/cddis.2013.555. e-pub ahead of print 2014/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sathaiah M, Thirunavukkarasu P, O'Malley ME, Kavanagh MA, Ravindranathan R, Austin F, et al. Oncolytic poxvirus armed with Fas ligand leads to induction of cellular Fas receptor and selective viral replication in FasR-negative cancer. Cancer gene therapy. 2012;19(3):192–201. doi: 10.1038/cgt.2011.77. e-pub ahead of print 2011/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barash S, Wang W, Shi Y. Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochemical and biophysical research communications. 2002;294(4):835–842. doi: 10.1016/S0006-291X(02)00566-1. e-pub ahead of print 2002/06/14. [DOI] [PubMed] [Google Scholar]
- 44.Wels W, Harwerth IM, Zwickl M, Hardman N, Groner B, Hynes NE. Construction, bacterial expression and characterization of a bifunctional single-chain antibody-phosphatase fusion protein targeted to the human erbB-2 receptor. Bio/technology (Nature Publishing Company) 1992;10(10):1128–1132. doi: 10.1038/nbt1092-1128. e-pub ahead of print 1992/10/01. [DOI] [PubMed] [Google Scholar]
- 45.Lee G, Saito I. Role of nucleotide sequences of loxP spacer region in Cre-mediated recombination. Gene. 1998;216(1):55–65. doi: 10.1016/s0378-1119(98)00325-4. e-pub ahead of print 1998/08/26. [DOI] [PubMed] [Google Scholar]
- 46.Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450(7166):56–62. doi: 10.1038/nature06293. e-pub ahead of print 2007/11/02. [DOI] [PubMed] [Google Scholar]
- 47.Goins WF, Krisky DM, Wechuck JB, Wolfe D, Huang S, Glorioso JC. Generation of replication-competent and -defective HSV vectors. Cold Spring Harbor protocols. 2011;2011(5) doi: 10.1101/pdb.prot5615. pdb.prot5615. e-pub ahead of print 2011/05/04. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.