

Abstract
Ketone bodies , β-hydroxybutyrate (BHB) and acetoacetate support mammalian survival during states of energy deficit by serving as alternative source of ATP1. BHB levels are elevated during starvation, high-intensity exercise or by the low carbohydrate ketogenic diet2. Prolonged caloric restriction or fasting reduces inflammation as immune system adapts to low glucose supply and energy metabolism switches towards mitochondrial fatty acid oxidation, ketogenesis and ketolysis2-6. However, role of ketones bodies in regulation of innate immune response is unknown. We report that BHB, but neither acetoacetate nor structurally-related short chain fatty acids, butyrate and acetate, suppresses activation of the NLRP3 inflammasome in response to several structurally unrelated NLRP3 activators, without impacting NLRC4, AIM2 or non-canonical caspase-11 inflammasome activation. Mechanistically, BHB inhibits NLRP3 inflammasome by preventing K+ efflux and reducing ASC oligomerization and speck formation. The inhibitory effects of BHB on NLRP3 were not dependent on chirality or classical starvation regulated mechanisms like AMPK, reactive oxygen species (ROS), autophagy or glycolytic inhibition. BHB blocked NLRP3 inflammasome without undergoing oxidation in TCA cycle, independently of uncoupling protein-2 (UCP2), Sirt2, receptor Gpr109a and inhibition of NLRP3 did not correlate with magnitude of histone acetylation in macrophages. BHB reduced the NLRP3 inflammasome mediated IL-1β and IL-18 production in human monocytes. In vivo, BHB attenuates caspase-1 activation and IL-1β secretion in mouse models of NLRP3-mediated diseases like Muckle-Wells Syndrome (MWS), Familial Cold Autoinflammatory syndrome (FCAS) and urate crystal induce body cavity inflammation. Taken together, these findings suggest that the anti-inflammatory effects of caloric restriction or ketogenic diets may be mechanistically linked to BHB-mediated inhibition of the NLRP3 inflammasome, and point to the potential use of interventions that elevate circulating BHB against NLRP3-mediated proinflammatory diseases.
The NLRP3 inflammasome controls the activation of caspase-1 and the release of the pro-inflammatory cytokines IL-1β and IL-18 in macrophages7-11. It is an important innate immune sensor that can get activated in response to structurally diverse damage-associated molecular patterns (DAMPs) such as toxins8, ATP8, excess glucose7, ceramides12, amyloids13,14 , urate15 and cholesterol crystals16. Ablation of NLRP3 attenuates type 2 diabetes12, 14,17, atherosclerosis16, multiple sclerosis18, Alzheimer's disease14, age-related functional decline19, bone loss19 and gout15. Thus, identification of endogenous mechanisms that control NLRP3 inflammasome deactivation may provide insights into the control of several chronic diseases. Although it is known that immune-metabolic interactions via inhibition of glycolysis dampen pro-inflammatory responses6, it is not known whether alternate metabolic fuels such as ketones that are produced during energy deficits impact the innate immune response.
The ketone bodies β-hydroxybutyrate (BHB) and acetoacetate (AcAc) are produced in the liver and serve as alternative energy sources for the brain, heart, and skeletal muscle in mammals during nutrient deprivation and adherence to low carbohydrate diets1,2. Circulating levels of BHB can increase up to 6-8 mM upon prolonged fasting as liver glycogen stores get utilized1,2. While these nutritional states are associated with altered immune cell function, it is unknown whether ketone bodies serve as immune effectors. To test whether BHB impacts inflammasome activation, we treated LPS-primed mouse bone marrow derived macrophages (BMDMs) with the NLRP3 activator ATP along with BHB for 60 minutes and measured caspase-1 activation using a Western blot that detects the enzymatically-active p20 subunit. BHB dose-dependently inhibited ATP-induced caspase-1 cleavage into p20 and processing of biologically active p17 form of IL-1β at concentrations similar to elevations in BHB induced by strenuous exercise or 2 days of fasting1,2 (Fig.1a and Supplementary Fig. 1a). Ketone body acetoacetate (AcAc) and the microbiota-derived short chain fatty acids (SCFAs), butyrate and acetate, that are structurally related to BHB did not affect ATP-induced NLRP3 activation (Fig. 1b). We sought to determine whether BHB specifically targets ATP-induced inflammasome activation or common signaling mechanisms in response to structurally diverse NLRP3 activators. BHB, but not butyrate, inhibited monosodium urate (MSU) crystal or particulate matter-induced caspase-1 activation (Fig. 1c and Supplementary Fig. 1b). Furthermore, BHB blocked inflammasome activation by five additional NLRP3 activators nigericin, silica particles (Supplementary Fig. 1b), lipotoxic fatty acids palmitate, ceramides and sphingosine (Fig. 1d). BHB also inhibited processing of IL-1β in response to the TLR4 pathogen associated molecular pattern (PAMP) agonist lipid A, the TLR1/2 ligand Pam3-CSK4 and the TLR2 agonist lipoteichoic acid (LTA) (Fig. 1e).
Figure 1. BHB specifically inhibits the NLRP3 inflammasome.
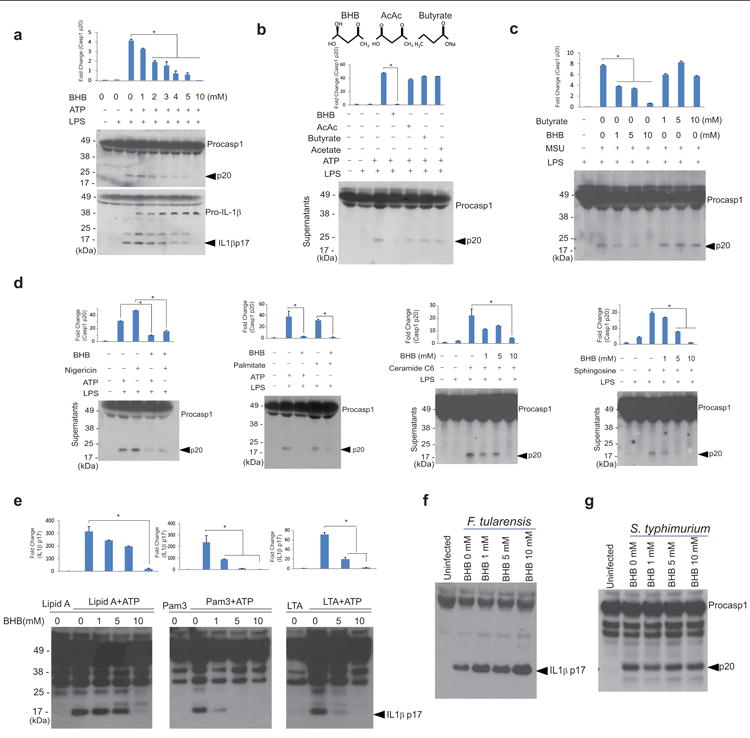
(a) Representative Western blot analysis of caspase-1 (active subunit p20) and IL-1β (active p17) in the supernatant of BMDMs primed with LPS for 4 hours and stimulated with ATP for 1 hour in the presence of various concentrations of D-BHB. (b) Western blot analysis of caspase-1 activation in BMDMs stimulated with LPS and ATP and treated with BHB (10mM), butyrate (10mM), acetoacetate (10mM) and acetate (10mM). (c) Western blot analysis of caspase-1 activation in LPS- primed BMDMs stimulated with MSU and treated with butyrate and D-BHB (d) nigericin (10 μM) for 1h, palmitate (200μM) for 24h, C6 ceramide for 6h (80μg/ml), and sphingosine (50 μM) for 1hour. (e) Western blot analysis of IL-1β activation (active subunit p17) in BMDMs primed with TLR ligands lipid A, Pam3-CSK and LTA for 4h and stimulated with ATP and increasing doses of D-BHB for 1h. Active IL1β (p17) was analysed in supernatants by western blot. BMDMs were infected with (f) F. tularensis and (g) S. typhimurium and treated with different doses of BHB and IL1β activation (p17 active form) was analyzed. Data are expressed as mean ± S.E.M (*P < 0.05) from cells derived from twelve (a-d) or six (e), three (f, g), mice with each independent experiment each carried out in triplicate (a-d, e) and duplicate (f g). All bar graphs in (a-e) represent quantitation of p20 caspase-1 band intensity as fold-change by normalizing to inactive p48 procaspase-1, or p17 IL-1β band intensity as fold change by normalizing to inactive p37 pro-IL-1β. The differences between means and the effects of treatments were determined by one-way ANOVA using Tukey's test.
We next investigated the specificity of BHB to NLRP3 as compared to other inflammasomes. The BMDMs were infected with either Francisella tularensis to activate the AIM2 or Salmonella typhimurium to activate the NLRC4 inflammasome. BHB did not inhibit either AIM2 inflammasome induced IL-1β activation (Fig. 1f) or NLRC4-mediated caspase-1 cleavage (Fig. 1g). Given inflammasome can also be activated by LPS through caspase-11 activation independently of TLR420,21, we also evaluated the non-canonical inflammasome pathway. Our data indicate that neither butyrate nor BHB blocks caspase-11 activation (Supplementary Fig. 1c).These results suggest that BHB acts on a central common signalling pathway specific to the NLRP3 inflammasome in response to PAMPs and a wide array of pro-inflammatory DAMPs.
Prolonged fasting and subsequent increases in circulating BHB are linked to a reduction in oxidative stress22, increased AMPK activity 23 and autophagy24. Furthermore, all these mechanisms have also been implicated in regulating the NLRP3 inflammasome8. Consistent with recent data25, ROS damage via rotenone or hydrogen peroxide (Fig. 2a, Supplementary Fig. 2a, b) was not sufficient to induce caspase-1 cleavage and did not abrogate the suppressive effects of BHB on ATP-induced NLRP3 inflammasome activation. Caspase-1 activation was induced by LPS priming alone in macrophages deficient in the autophagy regulator Atg5 (Fig. 2b, Supplementary Fig. 2a). However, absence of Atg5 did not alter the inhibitory effects of BHB on the inflammasome (Fig. 2b, Supplementary Fig. 2a). Consistent with these findings, the autophagy inhibitor 3-methyladenine (3-MA) and the proteasome blocker epoxomicin did not abrogate BHB's suppressive effects on ATP-induced NLRP3 inflammasome activation (Fig. 2c, Supplementary Fig. 2a). The activation of AMPK using AICAR (Fig. 2d) and inhibition of glycolysis with 2-deoxy glucose did not mimic the effects of BHB on inhibition of NLRP3 inflammasome (Supplementary Fig. 3a). Furthermore, inhibition of AMPK via compound C did not abrogate BHB's inhibitory effects on NLRP3-mediated caspase-1 activation. (Fig. 2d and Supplementary Figure 2a, c, Supplementary Figure 3a). BHB also did not impair the viability of BMDMs and at a concentration of 10 mM increased cellular proliferation (Fig. 2e).
Figure 2. BHB inhibits the NLRP3 inflammasome independently of Gpr109a and starvation-regulated mechanisms.
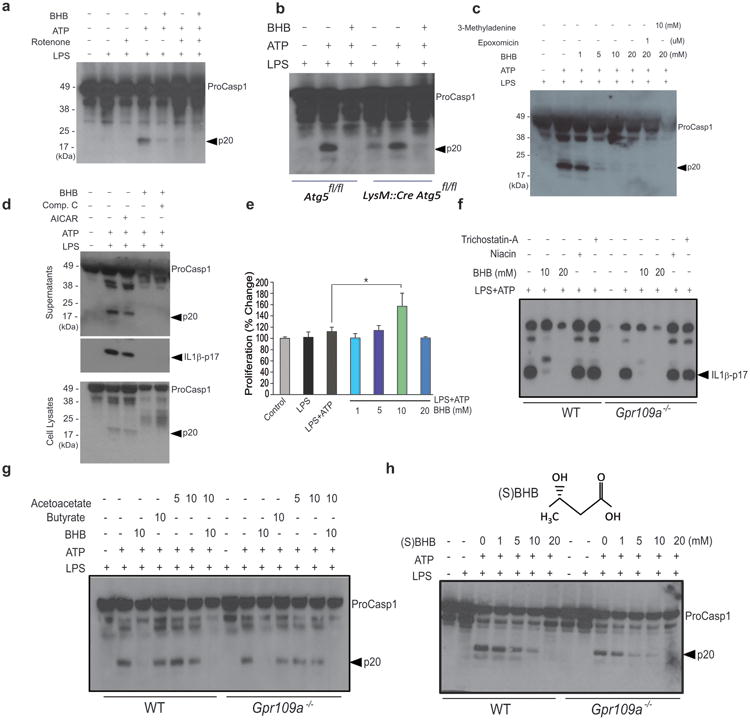
(a) Western blot analysis of caspase-1 activation in LPS- primed BMDMs treated with rotenone (10μM), ATP (5μM) together with BHB (10mM). (b) Western blot analysis of caspase-1 activation in BMDMs derived from control Atg5fl/fl and LysM:Cre Atg5fl/fl mice primed with LPS and stimulated in presence of ATP and BHB (10mM) (c) The BMDMs were primed with LPS and pretreated with 3MA and epoxomicin for 30min and stimulated in presence of ATP and BHB. The caspase-1 activation was measured by immunoblot analysis. (d) Western blots of caspase-1 and IL-1β activation in LPS-primed BMDM stimulated with ATP and BHB (10mM) in presence of AMPK activator (AICAR, 2mM) and AMPK antagonist Compound C (25 μM). (e) Proliferation of BMDMs in response to increasing concentrations of BHB. (f ,g) Western blot analysis of caspase-1 and IL-1β activation in BMDMs from control and Gpr109a deficient mice activated with LPS and ATP and co-incubated with TSA (50nM), niacin (1mM), butyrate (10mM), acetoacetate (10mM) and BHB (10, 20mM) (h) Western blot analysis of caspase-1 activation in BMDMs of WT and Gpr109a-/- mice treated with LPS for 4h and stimulated with ATP in presence of BHB chiral enantiomer (S) BHB for 1h. Data are expressed as mean ± S.E.M (*P < 0.05) from cells derived from six (a) 4 (b) ten (c, d, e) and four (f-h) mice with each independent experiment each carried out in triplicate. Due to space limitations the quantitation of p20 caspase1 and p17 IL1β band intensity from each experiment are presented in the Supplementary Fig. 2A. The differences between means and the effects of treatments were determined by one-way ANOVA using Tukey's test.
It has been suggested that BHB can act as signaling molecule by binding the G protein coupled receptor GPR109a26 or by serving as a histone deacetylase (HDAC) inhibitor22. Inhibition of HDACs using trichostatin A (TSA) did not did not impact inflammasome activation in LPS primed and NLRP3 agonist treated macrophages (Fig. 2f, Supplementary Fig. 2a despite induction of H3 acetylation by BHB in macrophages (Supplementary Fig. 3b). To understand the role of GPR109a in BHB's effects on macrophages, we used niacin, a GPR109a ligand that has been reported to inhibit colonic inflammation 27. We found that, unlike BHB, niacin did not block the NLRP3 inflammasome activation (Fig. 2f, Supplementary Fig. 2a).The effects of BHB on the inflammasome were not altered in Gpr109a-deficient BMDM (Fig. 2f, g, Supplementary Fig. 2a), and neither butyrate nor acetoacetate altered NLRP3 inflammasome activity in Gpr109a-sufficient or –deficient cells (Fig. 2g, Supplementary Fig. 2a). BHB is a chiral compound and its enantiomer, (S)-BHB, does not enter TCA cycle but binds Gpr109a with high affinity26. The enantiomer (S)-BHB exhibits similar effects on the inflammasome as D-(BHB) and does not require Gpr109a to block NLRP3 (Fig. 2h).
Compared to fatty acids, oxidation of BHB is energetically more efficient as all reducing equivalents generated by ketone oxidation are delivered through NADH to complex-I within the mitochondrial electron transport chain2. Furthermore, ketone oxidation increases the redox span between complex-I and complex-III by keeping mitochondrial ubiquinone oxidized28. We next asked whether BHB oxidation, entry into the TCA cycle or reduced mitochondrial stress mediate its effects on the inflammasome. Macrophages express ketogenic and ketolytic enzymes Acat1, Bdh1, Bdh2 and Hmgcl (Supplementary Figure. 4a, b, and c). In terms of macrophage polarization, classically activated M1 macrophages show reduction expression of Acat1, Bdh1, Bdh2 and Hmgcl as compared to M2 macrophages, suggesting ketones may affect macrophage polarization (Supplementary Figure. 4b). In addition, LPS induced protein expression of the ketolytic enzyme, Scot (succinyl-CoA:3-oxoacid CoA transferase , encoded by Oxct) and the ketogenic enzyme Hmgcl in BMDMs (Supplementary Figure. 4c). However, the TCA cycle entry inhibitor aminoxyacetate (AOA) did inhibit the effects of BHBon the inflammasome (Supplementary Figure. 4d), and the enantiomer (S)- BHB that does not enter the TCA cycle efficiently blocked NLRP3 inflammasome activation (Supplementary Figure. 4e). To directly assess the role of BHB oxidation, we specifically deleted the ketolytic mitochondrial enzyme Scot27 in macrophages (Fig. 3a, Supplementary Figure 4f). These experiments confirmed that TCA intermediates generated through ketone body oxidation in macrophage mitochondria do not mediate the suppressive effects of BHB on the NLRP3 inflammasome (Fig. 3a, Supplementary Figure 4f).
Figure 3. BHB inhibits ASC oligomerization and speck formation without undergoing mitochondrial oxidation.
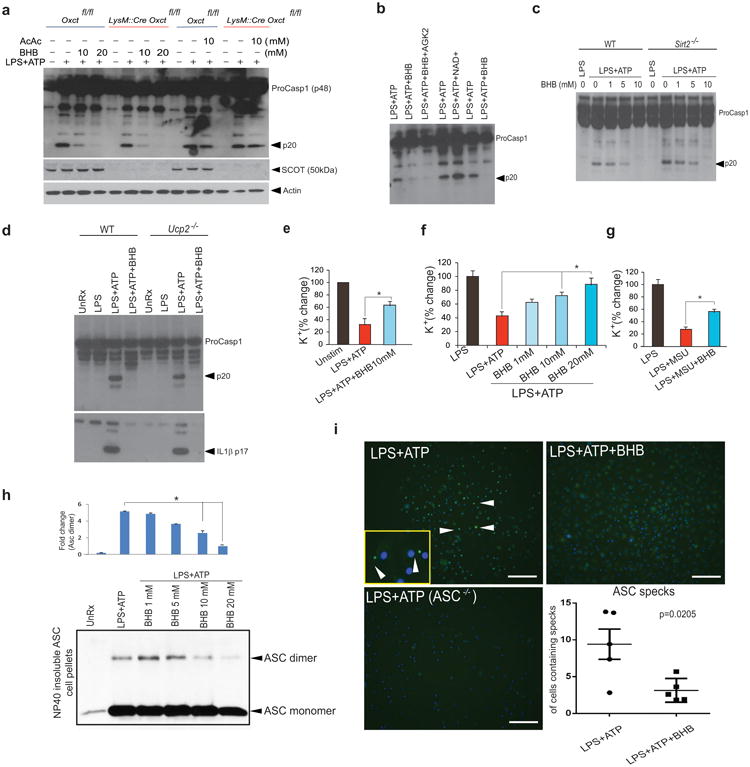
(a) Western blot analysis of caspase-1 activation, SCOT and actin in BMDMs of Oxctfl/fl and LysM:Cre Oxct5fl/fl mice treated with LPS for 4h and stimulated with ATP in presence of BHB and acetoacetate. (b) Western blot analysis of caspase-1 activation in LPS-primed BMDM treated with ATP in presence of BHB (10mM), Sirt2 antagonist AGK2 (10μM) and NAD+ (10 μM).(c, d) Western blot analysis of caspase-1 activation in BMDMs of WT, Sirt2-/- and Ucp2-/- mice treated with LPS for 4h and stimulated with ATP in presence of BHB (10mM) (e) Intracellular Potassium levels in BMDMs stimulated with LPS and ATP in the presence of BHB (10 mM), as measured by Inductively Coupled Mass Spectrometry (ICP-MS). Intracellular potassium levels in LPS-primed BMDMs treated with (f) ATP, (g) MSU and BHB for 1 h. as assessed using a APG-1 dye that selectively binds potassium with an excitation emission spectra of 488-540nm. (h) Representative immunoblot analysis of disuccinimidyl suberate (DSS) cross-linked ASC in the Nonidet P-40-insoluble pellet of BMDM that were primed with LPS (4 h) and stimulated with ATP and BHB for 1 h. The bar graphs represent the quantification of band intensity of the ASC dimer compared to LPS+ATP stimulation (i) Representative immunofluroescent images of ASC speck formation in BMDMs in the presence of BHB (10mM). Data are expressed as mean ± S.E.M (*P < 0.05) from cells derived from five (a) six (b,d), eight (e-g) and four (h) mice with each independent experiment carried out in triplicate. The differences between means and the effects of treatments were determined by one-way ANOVA using Tukey's test.(i) Data are shown as mean ± SEM and are representative of two independent experiments. Statistical differences were calculated by student's t-test.
The NAD dependent deacetylase Sirt2 regulates acetylation of α-tubulin, which controls microtubule-driven apposition of Nlrp3 and Asc29. Inhibition of Sirt2 by the small molecule AGK2 activates the Nlrp3 inflammasome and supplementation with NAD+ lowers IL-1β secretion from macrophages29. Inhibition of Sirt2 by AGK2 or ablation of Sirt2 or uncoupling protein 2 (UCP2) did not abrogate the effects of BHB on the inflammasome, and addition of NAD+ did not block caspase-1 activation in response to LPS and ATP (Fig. 3b-d, Supplementary Figure 4f). These findings rule out a major role for mitochondrial ROS in the effects of ketone bodies on the inflammasome.
BHB is a strongly anionic endogenous molecule2 and exerts anti-epileptic effects by reducing neuronal excitability via regulation of intracellular potassium cations30. Consistent with recent studies that demonstrate K+ efflux triggers NLRP3 inflammasome activation8, 25, BHB prevented the decline in intracellular K+ in response to incubation with the NLRP3 activators ATP, MSU and ceramides (Fig. 3e, f, g and Supplementary Figure 5a). Furthermore, the NLRP3-dependent ASC nucleation-induced polymerization or oligomerization is considered a common mechanism of NLRP3 inflammasome activation31,32. BHB prevents ATP-induced ASC oligomerization (Fig. 3h) and speck formation (Fig. 3i). Our data suggest that BHB blocks NLRP3 inflammasome activation by controlling an unknown upstream event that reduces K+ efflux from macrophages and by inhibiting ASC polymerization, speck formation and assembly of the inflammasome.
Next we investigated whether delivery of BHB can inhibit the NLRP3 inflammasome in human monocytes and mouse models of NLRP3-driven inflammation in vivo. BHB dose-dependently inhibited IL-1β and IL-18 (Fig. 4a) secretion in LPS-stimulated human monocytes without significantly affecting the TNFα production (Supplementary Figure 5b). In vivo administration of BHB is insufficient to achieve sustained high serum concentration due to rapid clearance1,2,30. To reduce clearance BHB was complexed with nanolipogels (nLGs) to improve its bioavailability.33 BHB nLGs inhibited NLRP3 inflammasome activation in macrophages (Fig. 4b). In mice the NLRP3 inflammasome was activated following intraperitoneal (i.p.) injection of MSU crystal, resulting in the influx of neutrophils into the peritoneum and increased secretion of IL-1β 4 h after injection34. Compared to mice given nLGs alone, BHB nLGs reduced neutrophil infiltration into the peritoneum (Fig. 4c and Supplementary Figure 5c, 5d and 5e) without directly impairing neutrophil migration (Supplementary Figure 5f) suggesting direct effects in vivo on NLRP3-driven neutrophil influx. Peritoneal cells from MSU-injected mice treated with BHB-nLGs produced less IL-1β (Fig. 4d) and the concentration of IL-1β in the serum was reduced following BHB nLG treatment (Fig. 4e).
Figure 4. BHB suppresses NLRP3-mediated inflammatory disease in vivo and the inflammasome in in human monocytes.
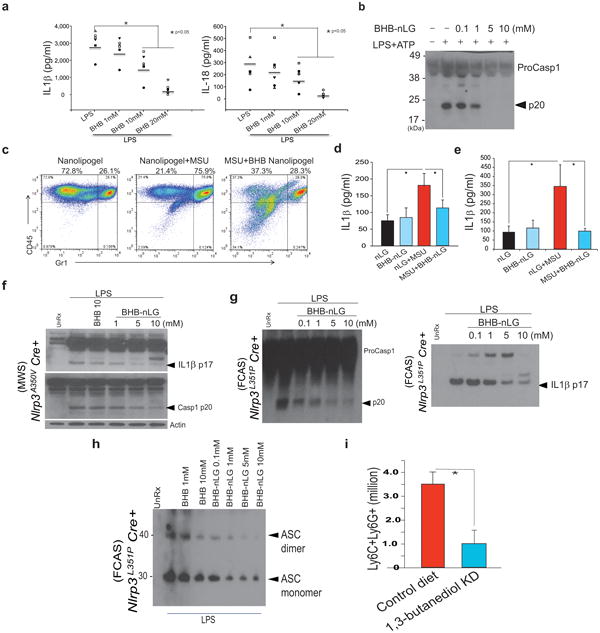
(a) Analysis of IL-1β and IL-18 secretion in culture supernatants of human monocytes stimulated with vehicle or LPS (1μg/mL) for 4h in presence of increasing concentrations of BHB (n =6/treatment). (b) BHB-complexed nanolipogels (nLGs) block the NLRP3 inflammasome activation and caspase-1 cleavage (n=3, repeated twice). (c) Frequency of CD45+ and Gr1+ immune cells in the peritoneum of mice treated with MSU (3 mg) and BHB-nLGs (125 mg/kg/bw), as assessed by FACS (N =6/group). (d) IL-1β secretion from peritoneal cells cultured overnight and (e) serum IL-1β levels from mice challenged with MSU and treated with BHB-nLGs (n =6/group) (f) Western blot analysis of caspase-1 and IL-1β activation in the BM cells stimulated in presence of LPS and BHB-nLGs from mice harbouring the MWS mutation NLRP3A350V and (g) FCAS mutation (n = 6, repeated twice) (h)Representative immunoblot analysis of disuccinimidyl suberate (DSS) cross-linked ASC in the Nonidet P-40-insoluble pellet of BMDM from FCAS mice (n = 6) that were primed with LPS (4 h) and treated with increasing concentrations of BHB-nLGs. (i) Neutrophil numbers in peritoneum of FCAS mice fed a chow or ketone diester diet (1,3-butanediol) for one week. (n =6/group).Data are expressed as mean ± S.E.M (*P < 0.05) and statistical differences between means and the effects of treatments were determined by one-way ANOVA using Tukey's test (a,d, e) and t-test (k).
Missense mutations in NLRP3 cause systemic inflammatory diseases like Muckle Wells Syndrome and Familial Cold Autoinflammatory syndrome (FCAS) that are characterized by overproduction of IL-1β and IL-1835. We tested the efficiency of BHB in knockin mice that mimic the human MWS and FCAS due to gain of function mutation A350V and L351P in the NLRP3 which renders inflammasome constitutively active without the requirement of NLRP3 ligands 35. As described before35, in FCAS mouse model, the tamoxifen-induced Cre recombinase-mediated excision of floxed neomycin cassette induces the expression of activating NLRP3L351P mutation and inflammasome activation in macrophages (Supplementary Fig. 6a).
Treatment of BMDMs of MWS (NLRPA350V) (Fig. 4f, Supplementary Fig. 5g) and FCAS (NLRPL351P) (Fig. 4g, h Supplementary Fig. 5g) mice with BHB-nLGs led dose dependently inhibited constitutive NLRP3 inflammasome activation. When complexed with nLGs, D-BHB inhibited inflammasome activation in FCAS macrophages at lower dose (Supplementary Fig. 6b, c). BHB directly prevents the ASC oligomerization in macrophages upon Cre mediated induction of NLRP3L351P by tamoxifen treatment in vitro. To model a ketogenic diet and elevate BHB levels in vivo, the FCAS mice were fed 1,3-butanediol ketone diesters (KD) for one week prior to induction of missense NLRP3 mutation. The KD in mice increases the serum BHB levels to fasting levels of 0.75-1mM. As reported previously, FCAS mice (Nlrp3L351P Cre+) develop severe neutrophila35 in the peritoneum 3 days after the induction of mutant NLRP3. Compared to chow fed FCAS mice, the KD treatment protects the these animals from neutrophilia (Fig.4i) and hyperglycemia (Supplementary Fig. 6d), without any effects on the infiltration of CD11b+F4/80+ peritoneal macrophages (Supplementary Fig. 6e). Furthermore, the KD diet did not impact the overall frequency of splenic T cells, macrophages or neutrophils (Supplementary Figure 7a,b,c,d). Given FCAS is caspase-1 dependent and IL-1β and IL-18 do not impact all of the observed pathology35, ketogenic diets that elevate BHB may improve therapeutic outcome in patients by inhibiting the inflammasome.
These findings suggest that a fasting or exercise -induced metabolite, BHB, inhibits the NLRP3 inflammasome in macrophages independently of binding to surface Gpr109a receptors or mitochondrial oxidation, which may avoid competition for receptor occupancy and a requirement of ATP generation. Thus, in states of extreme energy deficit such as starvation, metabolic signals like BHB can dampen innate immune responses, sparing ATP for functioning of ketone-dependent organs such as the brain and heart (Supplementary Figure 8a). These findings provide insight into immunological functions of metabolic signals such as BHB and suggest that dietary or pharmacological approaches to elevate BHB, without inducing the generalized starvation response, holds promise in reducing the severity of multiple NLRP3 mediated chronic inflammatory diseases.
Online Methods
Mice and animal care
The global Nlrp3-/-, Gpr109a-/-, Ucp2-/- and Sirt2-/- knockout mice have been described before12,19,20. Oxct1fl/fl 28 and Atg5 fl/fl mice were crossed with LysM-Cre (B6.129P2-Lyz2tm1(cre)Ifo/J) animals for macrophage specific gene ablations. NLRP3L351P gain of function Familial Cold Autoinflammatory Syndrome (FCAS) and NLRP3A350V Muckle-Wells Syndrome (MWS) knockin mutations have been described before32,33. Briefly, the Nlrp3L351PneoR/+ and Nlrp3A350VPneoR/+ mutation was conditionally activated by breeding these animals with tamoxifen-inducible Cre mice (B6.Cg-Tg(CAG-cre/Esr1*)5Amc/J) or in vitro by treating cells with 4-hydroxy tamoxifen. Mice were fed 1,3-butanediol ketone diesters (KD) (B84785, Sigma )for one week after weaning and injected with tamoxifen for 3 days and analysed. The WT littermates and mutant cohorts were housed with a 12-hour light/12- hour dark cycle at 22°C. The mice were multi-housed and were either fed ad libitum normal chow diet consisting of 4.5% fat (5002; LabDiet) or ad libitum normal chow diet mixed in with 20% 1,3-butanediol ketone diesters and aged in the specific-pathogen free barrier facility in ventilated cage racks that delivers HEPA filtered air to each cage with free access to sterile water through a hydropac system. Sentinel mice in our animal rooms were negative for currently tested standard murine pathogens (Ectromelia, EDIM, LCMV, Mycoplasma pulmonis, MHV, MNV, MPV, MVM, PVM, REO3, TMEV and Sendai virus) at various times while the studies were performed (RADIL, Research Animal Diagnostic Laboratory, Columbia, MO). All experiments and animal use were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Yale and Washington University.
Human monocytes
CD14+ monocytes were sorted from cryopreserved peripheral blood monocuclear cells using the Human Monocyte Isolation Kit II from Miltenyi (130-091-153). A total of 6 healthy subjects, (67 yr female, 31 yr female, 44 yr female, 67 yr male, 31 yr male and 35 yr male) were used for monocyte isolations. Monocytes were seeded in 6 well plates at a concentration of 3 million/mL of RPMI1640 media (11875119, Life Technologies) with 10% FCS (10437028, Life Technologies) and antibiotic/antimycotic mixture (10378-016 , Life Technologies) and stimulated with LPS (62325, Sigma) for 4 h. The supernatants were used to measure IL-1β, IL-18 and TNF-α using the Human IL-1β, IL-18 and TNF-α Platinum ELISA from Ebioscience (BMS 267/2, BMS2034, BMS224). Human peripheral blood was collected by the Health Apheresis Unit and the Clinical Core Laboratory, the National Institute on Aging-Intramural Research Program, under Human Subject Protocol # 2003054 and Tissue Procurement Protocol # 2003-071, consent was obtained.
Cell culture
All steps were performed using sterile technique. Femurs were collected in RPMI (22400105, Life Technologies) + 10% FBS (R10; Omega Scientific). Both ends of the femur were then cut and the femur was flushed with R10. The bone marrow was centrifuged at 450 g for 5 min, the supernatant was decanted and red blood cells were lysed using ACK lysis buffer (118-156-101, Quality Biological). After neutralization with R10, bone marrow cells were centrifuged, resuspended in 10 ml of R10 and placed into a 6 well plate. Non-adherent cells were collected the following morning. The non-adherent cells were resuspended at 4×106 cells/ml in media consisting of 10 ml supernatant of non-adherent cells, 7.2 ml L929 conditioned media, 6.8 ml R10 and MCSF (10 ng/ml; 416-ML, R&D Systems,). An additional 2 ml of fresh media was added 4 d after isolation. Non-adherent cells were collected on day 7, separated by density gradient separation using Fico/Lite (I40650, Atlanta Biologicals) and mononuclear cells were collected. Cells were rinsed twice with Dulbecco's PBS + 2% FBS, and resuspended at 1×106 cells/ml. Cells were treated with ultrapure LPS (L6529-1mg, Sigma) alone or in combination with 5 mM ATP (1A7699-1G, Sigma) or 200 μM palmitate-BSA (P9767, Sigma). The BMDMs were also primed with ultrapure lipid A (10ug/ml; tlrl-mpla, Invivogen), lipoteichoic acid (10ug/ml; tlrl-pslta, Invivogen), or Pam3-CSK4 (10ug/ml; (tlrl-pms, Invivogen) for 4 hours and stimulated with various NLRP3 activators (ATP 5mM-1hr, MSU 250 ug/ml- 5hr; tlrl-msu, Invivogen, Silica 200 ug/ml -5hr; tlrl-sio,Invivogen, palmitate-BSA 200uM-24hr; P9767, Sigma), Ceramide C6 80ug/ml-6hr; 62525-10, Cayman, and sphingosine 40uM-1hr; 62570, Cayman) together with D-BHB or S-BHB (54920, 54925, Sigma) at indicated concentration and time. The cell supernatants and cell lysates were collected after BHB treatment and analyzed for caspase-1(1:250, Genentech) and IL-1β (1:1000, GTX74034, Genetex).
Salmonella typhimurium (SL1344, Dr. Monack-Stanford) and Francisella tularensis (U112, Dr. Teale –UTSA) was grown overnight and then subcultured to mid-Log phase. BMDMs were infected with 1 multiplicity of infection S. typhimurium and treated 1h after infection with 0, 1, 5, or 10 mM of BHB. Cell supernatants and cell lysates were collected 3h after treatment (4h after infection).
ASC oligomerization and ASC speck formation
ASC oligomerization blotting was performed as described previously32. BMDM were plated on chamber slides and allowed to attach overnight. The following day cells were primed with LPS and treated with ATP in the presence or absence of BHB (10 mM). Cells were fixed with 4% paraformaldehyde followed by ASC (ADI-905-173-100, Enzo Lifesciences) and DAPI (D9542, Sigma) staining. ASC specks were quantified using ImageJ software. At least 5 distinct fields were analyzed and a minimum of 550 cells from each treatment condition were quantified.
Neutrophil chemotaxis assay
Neutrophils were isolated from mouse bone marrow (19762, Stemcell Technologies) and 2×105 cells were plated in 3um transwell 96-well plates. Untreated cells had just RPMI (+10% FBS) in the bottom chamber. To induce chemotaxis, LPS+ATP-stimulated macrophage-conditioned media (CM) was diluted 1:1 with RPMI with or without BHB (10 mM) as indicated. Cells were incubated at 37°C for 90 minutes and cells that passed through the membrane to the lower chamber were counted with a hemocytometer.
Western Blot analysis
The BMDM cell lysates were prepared using RIPA buffer and immediately snap frozen in liquid nitrogen. Samples were vortexed every 10 mins for 1 hour. Samples were then centrifuged at 14,000g for 15 min, the supernatant was collected and the protein concentration was determined using the DC Protein Assay (Bio-RAD). The immunoblot analysis was performed as described previously12. Antibodies to Caspase 1 (1:250, 4B4.2.1 Genentech), IL-1β (GTX74034, Genetex), Caspase 11(NB120-10454, Clone 17D9, Novus Bio), Scot (12175-1-AP, Proteintech), Asc (ADI-905-173-100, Enzo Life Science), Acetylated Histone H3 ( 06-942, LYS9, EMD Millipore), HMGCL (NBP1-58026, Novus Bio), and β-Actin (4967L, Cell Signaling) were used at dilutions specified by the manufacturer. The immune complexes were visualized by incubation with horseradish peroxidase-conjugated anti-rat (PI31470, Pierce) or anti-rabbit secondary antibody (PI31460). Immunoreactive bands were visualized by enhanced chemiluminescence (PI32209, Pierce). Densitometry analysis was performed using the Image J Gel Analysis tool, where gel background was also removed individually for each band.
Gene expression analysis
Total RNA was extracted using the Trizol method and transferred to the Qiagen RNeasy mini kit and purified according to the manufactures instruction. On the columns, DNA digestion was performed to remove DNA by following the RNase-Free DNase Set's manufacturer's instructions (79254, Qiagen). Synthesis of cDNA and Q-PCR was performed as described previously (15). The primer pairs used for real-time PCR:
Bdh1 (Forward: GCTTCCTTGTATTTGCTGGC , Reverse: TTCTCCACCTCTTCACTGTTG, Probe: TGGATGGTTCTCAGTCGGTCACTCT), Bdh2 (Forward: TCTCAATGAATCTCAACGTCCG, Reverse: ATCTGTTCTCCACCCCTTTG, Probe: ATCAACATGTCGTCTGTGGCCTCC), Acat1 (forward: GGCTGCTGCAGGAAGTAAGA, Reverse: ATCCCTGCCTTCTCAATGGC), Hmgcl (Forward: CAGGTGAAGATCGTGGAAGTC, Reverse: TGGGAGAAACAAAGCTGGTG and Gapdh (Forward: TCA ACA GCA ACT CCC ACT CTT CCA, Reverse: ACC CTG TTG CTG TAG CCG TAT TCA).
BHB nanolipogel (nLG) generation and treatment
nLG is a nanoparticle that combines the advantages of both liposomes and polymer-based particles and it can provide means for delivery of two or more pharmaceutical agents at different rates, especially agents with different chemical properties and or molecular weights. nLGs were fabricated by remotely loading liposomes with BHB and cross-linkable poly(ethylene glycol) oligomers. To prepare liposomes, a molar ratio mixture of 2:1:0.1 phosphatidylcholine:cholesterol:DSPE-PEG(2000)-COOH (8400151P, 700000P, 880125P, Avanti Polar Lipids) in chloroform was evaporated under a nitrogen gas stream and then lyophilized after extrusion. Lyophilized liposomes were rehydrated with the aqueous BHB-cyclodextrin-Irgacure-PEG mixture. Vigorous mixing was applied for 30 minutes. The liposomes were then cross-linked under a 430 W UV lamp with UVA light (315–400 nm transmission filter) for 8 minutes on ice to form the nLGs, rinsed with PBS, and pelleted by ultracentrifugation. nLGs were stored at −20°C until use.
Mice were treated with BHB nLG (i.p 125mg/kg/bw). 24 hours later mice were challenged with MSU (i.p 3mg in 200ul) and given a second treatment of BHB nLG (i.p.125mg/kg/bw). 4 hours after MSU challenge, mice were sacrificed. We isolated peritoneum-infiltrating cell by performing peritoneal lavage with PBS. Lavage fluid containing cells was centrifuged at 1500rpm for 5 min. The supernatant was decanted, and the pellet was resuspended in ACK lysis buffer to lyse the red blood cells. We neutralized the reaction with RPMI (22400105, Life Technologies) + 10% FBS (R10; Omega Scientific) and filtered the cells through a 100micron filter. We pelleted the cells again by centrifuging at 1500rpm for 5min and resuspended cells in 1ml RMPI to count with a hemocytometer. Cells were stained for CD45(clone 30-F11;ebioscience), Ly6C(clone AL-21;ebioscience), and Gr1 (clone RB6-8C5;ebioscience) and analyzed using FACS Calibur. All the FACS data was analyzed by post collection compensation using FlowJO (Treestar Inc) software.
BMDM cell proliferation
Proliferation of BMDMs in response to BHB treatment was analyzed using the MTT assay (CGD1-1KT, Sigma) according to manufacturer's instructions.
Intracellular K+ measurement
BMDM were incubated with Asante Potassium Green 1 (APG-1) (3604, TEFLABS) which is a fluorescent indicator with a Kd for measuring cytosolic K+ concentration. Its non-ratiometric large fluorescence dynamic range allows sensing of small changes in the concentration of K+. Optimal excitation was recorded at 517 nm (SpectraMay M5, Molecular Devices). K+ was also measured in BMDMs using an ElementXR or Agilent 7700 Inductively Coupled Mass Spectrometry (ICP-MS) as described previously25. For error analysis samples and standard solutions were run in duplicate.
Enzyme-linked immunosorbent assay (ELISA)
Peritoneal cells were incubated overnight in RPMI (22400105, Life Technologies) + 10% FBS (R10; Omega Scientific) +Antibiotic (15240062, Life Technolgies) mixture. The supernatants were analyzed for IL-1β by the Ebioscience Mouse IL-1β Platinum Elisa kit (BMS6002TWO) The sera from mice were collected and stored at -80°C and used to quantify the concentration of IL-1β in serum using the Ebioscience Mouse IL-1β Platinum Elisa kit (BMS6002TWO) ELISAs were read on the Tecan Infinite M200 i-control.
Statistical Analyses
A two-tailed Student's t test was used to examine differences between genotypes or treatments with a P < 0.05 considered statistically significant. The results are expressed as the mean ± SEM. The differences between means and the effects of treatments were determined by one-way ANOVA using Tukey's test using Sigma Stat software, which protects the significance (p < 0.05) of all pair combinations.
Supplementary Material
Acknowledgments
We thank M. Koch, Y. Kui, P. Chang, D. Albarado for technical assistance and V.M Dixit (Genentech Inc) and R. Medzhitov (Yale School of Medicine) for helpful discussions and providing knockout mice. MB and AB are supported by the National Institute on Aging -Intramural Research Program. VDD is supported in part by the grants from National Institutes of Health (AG043608, AG31797, DK090556 and AI105097).
Footnotes
Author contributions: Y.H.Y. and K.Y.N . contributed equally to the study, They designed and conducted majority of in vitro and all of in vivo experiments, analyzed and interpreted the data and participated in writing the manuscript. R.W.G. participated in design and conduct of inflammasome activation experiments. ELG performed ASC speck and neutrophil assays. MB and AB performed the human monocytes experiments. DK and TMF synthesized the BHB-nanolipogels and conducted control experiments to determine the dose response. DDA formulated the ketone diester diet. NP conducted the ICP-MS experiments to determine K+ efflux. CL and TDK conducted the F.tularensis and S. typhimurium infections experiments. TH designed the experiments and provided essential reagents for experiments involving mitochondrial ROS and UCP2. PAC generated the macrophage specific Scot deficient mice and contributed in experiment design. SK and EA designed and conducted the ASC oligomerization experiments. VDD conceived and supervised the project, interpreted the data and wrote the manuscript.
References
- 1.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. doi: 10.1016/j.tem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304:H1060–1076. doi: 10.1152/ajpheart.00646.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shido O, Nagasaka T, Watanabe T. Blunted febrile response to intravenous endotoxin in starved rats. J Appl Physiol. 1989;67:963–969. doi: 10.1152/jappl.1989.67.3.963. [DOI] [PubMed] [Google Scholar]
- 4.Johnson JB, et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med. 2007;42:665–674. doi: 10.1016/j.freeradbiomed.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mercken EM, et al. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell. 2013;12:645–51. doi: 10.1111/acel.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGettrick AF, O'Neill LA. How metabolism generates signals during innate immunity and inflammation. J Biol Chem. 2013;288:22893–22288. doi: 10.1074/jbc.R113.486464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinon F, Mayor A, Tschopp J. Annu Rev Immunol. 2009;27:229. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 8.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013;39:432–441. doi: 10.1016/j.immuni.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masters SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heneka MT, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 16.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen H, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–15. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw PJ, et al. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4610–4614. doi: 10.4049/jimmunol.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Youm YH, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18:519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 21.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013 Sep;341:1250–1253. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimazu T, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laeger T, Pöhland R, Metges CC, Kuhla B. The ketone body β-hydroxybutyric acid influences agouti-related peptide expression via AMP-activated protein kinase in hypothalamic GT1-7 cells. J Endocrinol. 2012;213:193–203. doi: 10.1530/JOE-11-0457. [DOI] [PubMed] [Google Scholar]
- 24.Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. 2005;280:25864–25870. doi: 10.1074/jbc.M502456200. [DOI] [PubMed] [Google Scholar]
- 25.Muñoz-Planillo R, et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taggart AK, et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–26652. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- 27.Singh N, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cotter DG, Ercal B, d'Avignon DA, Dietzen DJ, Crawford PA. Impact of peripheral ketolytic deficiency on hepatic ketogenesis and gluconeogenesis during the transition to birth. J Biol Chem. 2013;288:19739–19749. doi: 10.1074/jbc.M113.454868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Misawa T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013 May;14(5):454–60. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 30.Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36:32–40. doi: 10.1016/j.tins.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu A, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu JW, Wu J, Zhang Z, Datta P, Ibrahimi I, Taniguchi S, Sagara J, Fernandes-Alnemri T, Alnemri ES. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006;13:236–249. doi: 10.1038/sj.cdd.4401734. [DOI] [PubMed] [Google Scholar]
- 33.Demento SL, Siefert AL, Bandyopadhyay A, Sharp FA, Fahmy TM. Pathogen-associated molecular patterns on biomaterials: a paradigm for engineering new vaccines. Trends Biotechnol. 2011;29:294–306. doi: 10.1016/j.tibtech.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 35.Brydges SD, et al. Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity. 2009;30:875–87. doi: 10.1016/j.immuni.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.