

Abstract
Purpose
Severe combined immunodeficiency (SCID) encompasses a group of disorders characterized by reduced or absent T-cell number and function and identified by newborn screening utilizing T-cell receptor excision circles (TRECs). This screening has also identified infants with T lymphopenia who lack mutations in typical SCID genes. We report an infant with low TRECs and non-SCID T lymphopenia, who proved upon whole exome sequencing to have Nijmegen breakage syndrome (NBS).
Methods
Exome sequencing of DNA from the infant and his parents was performed. Genomic analysis revealed deleterious variants in the NBN gene. Confirmatory testing included Sanger sequencing and immunoblotting and radiosensitivity testing of patient lymphocytes.
Results
Two novel nonsense mutations in NBN were identified in genomic DNA from the family. Immunoblotting showed absence of nibrin protein. A colony survival assay demonstrated radiosensitivity comparable to patients with ataxia telangiectasia.
Conclusions
Although TREC screening was developed to identify newborns with SCID, it has also identified T lymphopenic disorders that may not otherwise be diagnosed until later in life. Timely identification of an infant with T lymphopenia allowed for prompt pursuit of underlying etiology, making possible a diagnosis of NBS, genetic counseling, and early intervention to minimize complications.
Keywords: Nijmegen breakage syndrome, TREC, SCID, exome sequencing, nibrin, T lymphopenia
Introduction
Severe combined immunodeficiency (SCID) refers to a group of life-threatening fatal, congenital disorders characterized by absent or reduced T-cell number and reduced or non-functional B-cells. An early diagnosis of SCID allows for avoidance of exposure to infections, live virus vaccines and non-irradiated blood products, and also makes possible early hematopoietic stem cell transplantation (HSCT) or other immune system restoring treatments, adenosine deaminase enzyme replacement, or gene therapy [1].
Since beginning in 2008 in Wisconsin, newborn screening for SCID has been implemented in 23 states in the U.S., as well as in the Navajo Nation [2]. The assay employs PCR quantification of circular DNA byproducts of T-cell receptor gene rearrangement, T-cell receptor excision circles (TRECs). Although TRECs have no identifiable function, their presence serves as a marker for the maturation of T-cells [3]. Insufficiency of TRECs characterizes SCID and other T-lymphopenic disorders including complete DiGeorge Syndrome [4] and some cases of ataxia telangiectasia (AT) [5].
Like AT, Nijmegen breakage syndrome (NBS) [6–9] (formerly known as an AT Variant 1) is a disorder of DNA repair associated with chromosomal instability and immune compromise that may be identified through TREC screening. Herein, we describe a baby with a complex perinatal history and low TRECs near birth. Flow cytometry confirmed modest T-lymphopenia. Whole exome sequencing identified heterozygous mutations in NBN, the gene that encodes nibrin, a component of a molecular complex involved in the early recognition and subsequent repair of DNA damage [10]. Nibrin was absent from nuclear lysates on immunoblots, and the patient’s cells were hypersensitive to ionizing radiation by colony survival assay [11, 12].
Case Report
A 41-week gestational age male (NBS20LA), weighing 2860 grams, was born via normal vaginal delivery to a 23-year-old, gravida 1, paragravida 0 mother following prenatal identification of increased cerebrospinal fluid on ultrasound. Postnatally, the infant was hospitalized for 1 week for meconium aspiration. By 4 weeks of age progressive hydrocephalus required a ventriculo-peritoneal shunt. At 17 weeks, he was hospitalized for a presumed urinary tract infection; renal ultrasound showed left hydronephrosis. During the first year of life he also exhibited developmental delay and failure to thrive with weight and length for age persistently below the 3rd percentiles and head circumference at 8 months of age below the 2nd percentile.
At 3 weeks of age, SCID newborn screening results were interpreted as “incomplete”: TREC, 13 copies/mcL (normal >25) and actin control, 9,510 copies/mcL (normal >10,000). Repeat screening was “positive”: TREC, 12 copies/mcL and actin, 19,800 copies/mcL. Subsequent flow cytometry at 6 weeks of life demonstrated persistent T-lymphopenia and elevated NK cell percentages and numbers (Table 1).
Table 1.
Laboratory values of patient NBS20LA from 1.5 months to 1 year of age
| Patient Age | 1.5 month | 3 months | 5.5 months | 10 months | Reference Ranges | ||
|---|---|---|---|---|---|---|---|
| Birth to 2 months | 2 months to 4 months | 4 months to 1 year | |||||
| Total WBC cells/uL | 6800 | 5120 | 5500 | 7880 | 5000–17,000 | 5000–17,000 | 5000–17,000 |
| %Neutrophils | 47 | 44 | 58 | 56 | 25–45 | 25–45 | 25–45 |
| %Lymphocytes | 33 | 27 | 23 | 22 | 46–66 | 46–66 | 46–66 |
| %Monocytes | 8 | 19 | 12 | 14 | 2–8 % | 2–8 % | 2–8 % |
| %Eosinophils | 12 | 10 | 7 | 8 | 0–3 | 0–3 | 0–3 |
| CD 3+ T cells/mL (%) | 924 (42) | 259 (24) | 289 (18) | 381 (22) | 1375–7129 (69–93) | 2533–6778 (55–79) | 2889–7995 (59–80) |
| CD 4+ T cells/mL (%) | 660 (30) | 65 (5) | 224 (15.1) | 320 (19) | 1169–5623 (48–75) | 1392–5210 (35–62) | 1786–5141 (38–61) |
| CD 8+ T cells/mL (%) | 242 (11) | 99 (8) | 63 (4) | 72 (4) | 267–1860 (13–33) | 652–2449 (14–30) | 946–2791 (14–34) |
| CD19+ B-cells/mL (%) | 88 (4) | 26 (2) | 72 (5) | 174 (10) | 104–1448 (4–26) | 745–3499 (14–39) | 858–3774 (16–36) |
| CD3-CD16/56+ NK cells /mm3 (%) | 638 (29) | 680 (66) | 1123 (73) | 1169 (68) | 60–434 (2–14) | 194–994 (3–14) | 182–1581 (3–13) |
| IgG (mg/dL) [Reference Range] | 220 [198–577] | 202 [165–781] | 316 [282–1026] | ||||
| IgM (mg/dL) [Reference Range] | 24 [16–100] | 34 [31–103] | 70 [39–142] | ||||
| IgA (mg/dL) [Reference Range] | <7 [1–52] | 7 [8–83] | 16 [16–83] | ||||
| Ab Responses: | |||||||
| Tetanus (IU/mL) (RR: 0.1–7) | 2.19 | 2.71 | |||||
| H. influenzae b (mcg/mL) (RR: 1.0–10.0) | 0.15 | 1.16 | |||||
| LPA responses: | |||||||
| Phytohemaglutinin (SI) [1:125] | 173 | 44 | 42 | ||||
| Pokeweed mitogen (SI) | 23 | 75 | 23 | ||||
| Tetanus antigen (SI) | 2 | 1 | |||||
| Candida antigen (SI) | 4 | 6 | |||||
Targeted sequencing of SCID associated genes ADA, AK2, CD3D, CD3zeta, DCLRE1C, ILRG, IL7R, JAK3, LIG4, NHEJ1, PNP, PTPRC, RAC2, RAG1, RAG2, RMRP, and ZAP70 (GeneDx, Gaithersburg, MD) and enzyme assays for adenosine deaminase and nucleoside phosphorylase revealed no abnormalities.
Methods
Subjects and Samples
Informed consent was obtained for research, including cellular immune studies and whole exome sequencing (WES), for the infant and both parents under approved protocols at Children’s Hospital Los Angeles (CHLA) and the University of California San Francisco (UCSF). Genomic DNA from EDTA-anticoagulated whole blood was prepared using a Gentra Puregene Blood kit (Qiagen USA: Germantown, MD).
Exome Sequencing and Analysis
WES was performed as previously described [5]. Briefly, libraries prepared by ligating TruSeq adaptors (Illumina: San Diego, CA) to genomic DNA fragments of 200–300 bp were enriched with 10 cycles of PCR, pooled and submitted to exon capture using a Roche Nimblegen version 3.0 capture array. After 10 additional amplification cycles, 100 bp paired end sequence reads were generated (HiSeq2000, llumina), yielding 3.8 % duplicates and a mean of >100 reads covering the targeted regions, with >95 % of target regions having ≥10 reads.
Reads were aligned against GRCh37 (Aug 2009 release) using BWA (v0.6.2) [13]. The results were converted to BAM, sorted by coordinate, indexed, and marked for PCR duplicate reads using the Picard toolkit (v 1.81) (http://picard.sourceforge.net). Local realignment was performed around known indel locations, and base quality scores were re-calibrated using GATK (v 2.6.5) [14, 15].
Variants were called using GATK UnifiedGenotyper and freebayes (vesion 0.9.10) [16]. GATK variant quality scores were re-calibrated by VQSR (Variant Quality Score Recalibration) using the trio of exomes in this report, as well as 65 others sequenced at our site. HapMap v3.3 [17], 1000 genomes high confidence SNPs (phase1 v3 2010-11-23) and Omni chip array sets were used as training data, and to provided truth sites for SNPs, while the Mills dataset [18] from the 1000 genomes was used for indels, using a truth sensitivity cutoff of 99 %. Variant annotation (including region, effect, allele frequency, disease phenotype annotation, and conservation) was performed using our custom tool Varant (http://compbio.berkeley.edu/proj/varant/).
Particular attention was given to high-confidence, rare, likely-damaging, protein-altering variants in genes associated with primary cellular immunodeficiency [19] and to those conforming to models of autosomal or X-linked recessive inheritance in the family, or de novo mutations that may be dominant. Candidate variants in NBN were given priority and confirmed by Sanger sequencing.
Immunoblotting
Peripheral blood lymphoblastoid cell lines (LCL) from the patient NBS20LA and controls were made with Epstein-Barr virus as described [11], and 50 μg of nuclear protein lysate from each was electrophoresed on a 6 % SDS-polyacrylamide gel (PAGE), blotted onto PVDF membrane (BioRad, Hercules, CA) and incubated with antibodies to nibrin (Novus, NB100-143 at 1:5000, Littleton, CO) overnight at 4 °C. The immunoblots were subsequently incubated with an HRP-conjugated anti-rabbit secondary antibody at room temperature for 40 min for detection by enhanced chemiluminescence (ECL) (Amersham Pharmacia, Piscataway, NJ). Under the conditions of the assay, as little as 1 μg of nibrin protein could be detected. p84 was used as a loading control (Genetex GTX70220 at 1:3000). On the same blot, MRE11 and Rad50 were also measured (Novus 100–142 at 1:15,000 and Novus 100–154 at 1:500, respectively).
Peripheral blood lymphocytes from the patient NBS20LA and controls were immortalized with Epstein-Barr virus as previously described [11], and 50 μg of nuclear lysate protein from the each lymphoblastoid cell line (LCL) was electrophoresed on a 6 % SDS-polyacrylamide gel (PAGE), blotted onto PVDF membrane (BioRad, Hercules, CA) and incubated with antibodies to nibrin and DNA repair proteins MRE11, RAD50 and ATM (Novus, Littleton, CO) overnight at 4 °C. The immunoblots were subsequently incubated with an HRP-conjugated anti-rabbit secondary antibody at room temperature for 40 min for detection by enhanced chemiluminescence (ECL) (Amersham Pharmacia, Piscataway, NJ). Under the conditions of the assay, as little as 1 μg of nibrin protein can be detected.
Colony Survival Assay
LCLs from wild type (healthy controls), individuals with AT (radiosensitive controls), and NBS20LA were maintained in RMPI 1640 medium with 10 % fetal bovine serum and 1 % penicillin-streptomycin-L-glutamine [12]. After plating into paired 96-well trays, one of each pair was irradiated with 1 Gray. After incubation at 37 °C for 2 weeks, the trays were stained with 0.1 % 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. Individual wells with at least one colony composed of >32 cells (i.e., >5 divisions) were scored as positive. The number of positive wells in irradiated- versus non-irradiated plates was compared to determine the survival fraction. Results were compared to measurements performed by Sun et al. using 104 AT patients and 29 healthy controls [12].
Results
WES analysis by our Varant tool identified two nonsense mutations in NBN on chromosome 8q21.3 in the patient, suggesting a diagnosis of NBS. The patient and his father shared a thymine to guanine transversion cDNA 842 T > G (NM_002485.4(NBN):c.842 T > G) in exon 6, changing leucine 281 to a stop codon (protein L281X) (ClinVar Provisional Accession: SCV000196640), while the infant and mother shared a cytosine to thymine transition cDNA 1030C > T (NM_002485.4(NBN):c.1030C > T) in exon 9, changing glutamine 344 to a stop codon (Q344X) (ClinVar Provisional Accession: SCV000196641) [20]. A rare X-linked CD40LG gene variant noted in our patient and mother NM_000074 (CD40LG):c.542G > C (chrX: 135741330, G > C; R181P in CD40LG) was predicted damaging by PolyPhen-2 software [21], but was inconsistent with the patient’s phenotoype. No other high quality DNA variants in the targeted exome regions were as consistent with the phenotype. Neither variant had been reported in other patients with NBS nor in the 1000 genomes [22], ESPDB [23] or dbSNP (v137) [24] databases. The patient’s paternally inherited allele carried L281X, while his maternally inherited allele carried Q344X. Confirmatory Sanger sequencing was performed by the CLIA-approved Molecular Genetics Laboratory at the University of California Los Angeles School of Medicine.
An immunoblot showed no detectable nibrin protein in NBS20LA (Fig. 1, penultimate lane). Interestingly, MRE11 and Rad50 protein levels were also reduced; this reflects the instability of the MRE11-Rad50-nibrin (MRN) complex whenever nibrin is absent. Lane 2 contains LCL protein from an AT patient whose cell lines were radiosensitive in Fig. 2. ATM protein was absent in the ATM LCL, but was present in NBS20LA (not shown). All other LCLs in Fig. 1 were from healthy individuals and showed normal amounts of MRN-complex proteins and the loading control, p84. These findings provided functional confirmation that the patient’s NBN variants were both null mutations that abrogated protein expression, and that his cells scored as radiosensitive.
Fig. 1.
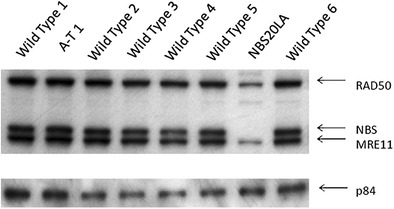
Immunoblot allows comparison of nibrin (NBS), MRE11 and Rad50 protein levels in an NBS cell line (NBS20LA) to wild type and AT cells. Note absence of nibrin in NBS LCL. Protein loading control was p84
Fig. 2.
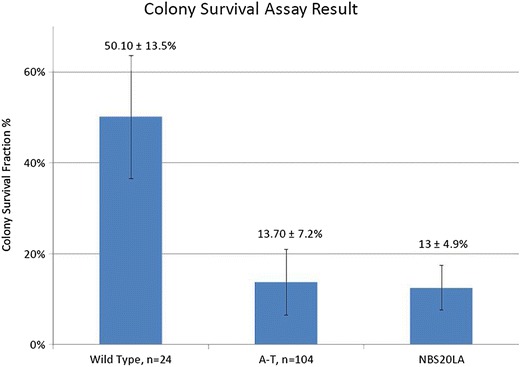
Colony Survival Assay for AT and NBS B lymphoblastoid cell lines showing that both the AT control cells and those of patient NBS20LA are in the radiosensitive range, as defined by Sun et al. 2002 [12]
Discussion
Although the primary goal of newborn screening with TRECs has been the pre-symptomatic diagnosis of SCID, thereby allowing early intervention, this screening has also identified individuals with non-SCID T-lymphopenia. In California, the first 2 years of newborn screening for SCID identified 50 infants with T-lymphopenia, defined as <1500 T cells/mcL: 11 had typical SCID, five had idiopathic T-lymphopenia or variant SCID (defined as significant T cell defects without mutation in known SCID genes), eight had partial and one had complete DiGeorge syndrome, four had trisomy 21, and three had AT [5]. Twenty-three patients had preterm birth or other congenital anomalies [4].
Recently, exome sequencing was utilized to identify AT as the cause of T-lymphopenia in two patients with positive SCID screens [5]. In this same study retrospective analysis of older AT patients suggested that approximately half of patients with AT will have a positive newborn SCID screen. NBS is a distinct DNA breakage/chromosomal instability disorder caused by defects in nibrin, part of the same DNA repair pathway as AT mutated protein. The immune defects in AT and NBS are similar and include impaired VDJ recombination, which affects T-cell receptor and immunoglobulin rearrangement. T-lymphopenia and decreased TRECs as seen in patients with AT would thus also be expected in patients with NBS.
In our patient, the TREC newborn screening for SCID was positive, but an extensive search for SCID gene mutations was unrevealing. In contrast, exome sequencing revealed novel, compound heterozygous NBN mutations that were associated with absence of nibrin and radiosensitivity. A R181P missense variant in CD40LG, the gene associated with X-linked hyper-IgM syndrome, was also predicted to be damaging but was inconsistent with the patient’s clinical and laboratory data including absent nibrin and a total serum IgG concentration in the normal range and positive IgG antibody responses to tetanus and H. influenzae b. Although it is not impossible for a patient to have two primary immune disorders, NBN mutations fully explain the low T cells, absent nibrin and cellular radiosensitivity that establish the diagnosis of NBS in this patient, while the features of CD40L deficient hyper-IgM syndrome are absent.
The clinical phenotype of NBS is variable. Most patients are full term [7], but may have microcephaly at birth with subsequent growth failure. Most patients meet early developmental milestones but have varying degrees of intellectual disability later in life [7, 25–27]. Perinatal hydrocephalus as seen in our patient has been reported in two children with NBS [8]. Hydronephrosis, renal anomalies such as horseshoe or dysplastic kidneys, skeletal anomalies such as clinodactyly and partial syndactyly, café au lait spots and vitiligo have all been seen, but are not constant features and are also found in other conditions [8, 28]. It is therefore difficult to identify NBS based on early clinical signs. Lymphoid malignancy affects up to 40 % of patients with NBS by their 21st year [28]. Onset of malignancy can vary in these patients depending on the mutation inherited and degree of nibrin dysfunction [28].
Most patients with NBS show some degree of immunodeficiency. In a review of 57 patients from the Polish Nijmegen Breakage Syndrome Registry, 19 % had normal total serum immunoglobulins, while 25 % had severe hypogammaglobulinemia [29]. At least one IgG subclass was deficient in all patients: 89 % IgG4, 73 % IgG2 and 54 % IgG1. Similarly, our patient had variably low total serum immunoglobulins that trended upward with age (Table 1), but IgG3 and IgG4 remained low (data not shown). Specific antibody response after two H. influenza type b immunizations was negative at 6 months, but positive at 10 months following an additional immunization. Tetanus toxoid titers were positive by 6 months. Our patient’s absolute CD19+ B cells were reduced during the first year of life, as seen in 72 % of Polish registry patients [29]. In the Polish cohort, 84 % of patients received immunoglobulin replacement therapy for frequent or prolonged respiratory tract infections, which have not occurred in our patient up to the present.
Lymphocyte subset analysis in NBS has shown decreased T cells, with CD4+ T cells affected most severely [29]. This is consistent with our patient’s findings (Table 1). The elevated proportion of NK cells seen in our patient was also noted previously in 25 out of 40 patients [30].
The treatment of choice for SCID is most often HSCT. To date, however, there is limited experience with HSCT in patients with NBS [31]. Indications for attempting transplantation have included severe immunodeficiency and hematologic malignancy; pre-emptive HSCT has not been reported. Of particular concern has been the use of DNA-damaging chemotherapeutic conditioning regimens and the potential to induce de novo malignancies in patients with a pre-existing DNA repair deficit. There has been some success with reduced intensity pre-HSCT conditioning regimens, but studies to date are limited by their retrospective nature, small sample size and relatively short term follow-up.
Conclusions
With widespread adoption of TREC screening for SCID, NBS may be identified prospectively, enabling the population incidence and natural history to be better understood. NBS has been reported primarily in Eastern Europeans, where founder mutations occur [28]. Our case expands our appreciation of the association of NBS with hydrocephalus and hydronephrosis and illustrates the utility of WES as applied to diagnose the underlying cause of idiopathic T lymphopenia found by TREC screening. There are more than 200 known genetic defects for primary immunodeficiency [19, 32], and testing of individual genes can be costly and time intensive. WES resulted in prompt detection of two nonsense mutations in NBN and definitive diagnosis in our patient, who presented with atypical features of NBS. Establishing the diagnosis allowed avoidance of live virus vaccines and exposure to ionizing radiation, as well as anticipatory monitoring for serious infections and malignancy.
Trimethoprim/sulfamethoxazole was started to prevent Pneumocystis jiroveci pneumonia; however he has not received immunoglobulin infusions or HSCT. Exome sequencing of the parents also established the NBN mutations as inherited versus de novo and allowed for accurate and informative counseling regarding the risk of future offspring inheriting both affected genes and having NBS. The parents were informed that other family members could be carriers of these same mutations.
Acknowledgments
The authors thank Dr. Robert Currier and the members of the California SCID Newborn Screening Program, Genetic Disease Laboratory, California Department of Public Health as well as Karly Kondratowicz and Antonia Kwan, who provided excellent assistance. Dr. Puck and Dr. Brenner received support from the National Institute of Allergy and Infectious Diseases R01 AI105776. Dr. Puck received support from the National Institute of Allergy and Infectious Diseases R01 AI078248. Dr. Puck and Dr. Church received support from the Jeffrey Modell Foundation, and Dr. Puck received support from the Lisa and Douglas Goldman Fund. Dr. Gatti’s testing was partially supported by APRAT (Clermont-Ferrand, France). Dr. Brenner received support from Tata Consultancy Services.
References
- 1.Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129(3):597–604. doi: 10.1016/j.jaci.2011.12.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwan A, Abraham RS, Currier R, Brower, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–38. doi: 10.1001/jama.2014.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J Allergy Clin Immunol. 2012;129(3):607–16. doi: 10.1016/j.jaci.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. 2013;132(1):140–50. doi: 10.1016/j.jaci.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mallott J, Kwan A, Church J, Gonzalez-Espinosa D, Lorey F, Tang LF, et al. Newborn screening for SCID identifies patients with ataxia telangiectasia. J Clin Immunol. 2013;33(3):540–9. doi: 10.1007/s10875-012-9846-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weemaes C, Hustinx T, Scheres J, Munster P, Bakkeren J, Taalman R. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr. 1981;70(4):557–64. doi: 10.1111/j.1651-2227.1981.tb05740.x. [DOI] [PubMed] [Google Scholar]
- 7.Chrzanowska K, Kleijer W, Krajewska‐Walasek M, Białecka M, Gutkowska A, Goryluk‐Kozakiewicz B, et al. Eleven polish patients with microcephaly, immunodeficiency, and chromosomal instability: the Nijmegen breakage syndrome. Am J Med Genet. 1995;57(3):462–71. doi: 10.1002/ajmg.1320570321. [DOI] [PubMed] [Google Scholar]
- 8.Nijmegen breakage syndrome The international Nijmegen breakage syndrome study group. Arch Dis Child. 2000;82(5):400–6. doi: 10.1136/adc.82.5.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Resnick IB, Kondratenko I, Togoev O, Vasserman N, Shagina I, Evgrafov O, et al. Nijmegen breakage syndrome: clinical characteristics and mutation analysis in eight unrelated Russian families. J Pediatr. 2002;140(3):355–61. doi: 10.1067/mpd.2002.122724. [DOI] [PubMed] [Google Scholar]
- 10.Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93(3):467–76. doi: 10.1016/S0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 11.Chun HH, Sun X, Nahas SA, Teraoka S, Lai C, Concannon P, et al. Improved diagnostic testing for ataxia–telangiectasia by immunoblotting of nuclear lysates for ATM protein expression. Mol Genet Metab. 2003;80(4):437–43. doi: 10.1016/j.ymgme.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Sun X, Becker-Catania SG, Chun HH, Hwang MJ, Huo Y, Wang Z, et al. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J Pediatr. 2002;140(6):724–31. doi: 10.1067/mpd.2002.123879. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv:1207.3907 2012.
- 17.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mills RE, Pittard WS, Mullaney JM, Farooq U, Creasy TH, Mahurkar AA, et al. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2011;21(6):830–9. doi: 10.1101/gr.115907.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;22(5):162. doi: 10.3389/fimmu.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42(Database issue):D756–63. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adzhubei IA, Schmidt S, Peshkin L, Ramensky V, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;4:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–9. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chrzanowska KH, Stumm M, Bekiesiska-Figatowska M, Varon R, Biaecka M, Gregorek H, et al. Atypical clinical picture of the Nijmegen breakage syndrome associated with developmental abnormalities of the brain. J Med Genet. 2001;38(1):E3. doi: 10.1136/jmg.38.1.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maraschio P, Danesino C, Antoccia A, Ricordy R, Tanzarella C, Varon R, et al. A novel mutation and novel features in Nijmegen breakage syndrome. J Med Genet. 2001;38(2):113–7. doi: 10.1136/jmg.38.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green AJ, Yates JR, Taylor AM, Biggs P, McGuire GM, McConville CM, et al. Severe microcephaly with normal intellectual development: the Nijmegen breakage syndrome. Arch Dis Child. 1995;73(5):431–4. doi: 10.1136/adc.73.5.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, Kalina MA, Digweed M. Nijmegen breakage syndrome (NBS) Orphanet J Rare Dis. 2012;7(1):13. doi: 10.1186/1750-1172-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregorek H, Chrzanowska KH, Dzierżanowska-Fangrat K, Wakulińska A, Pietrucha B, Zapaśnik A, et al. Nijmegen breakage syndrome: long-term monitoring of viral and immunological biomarkers in peripheral blood before development of malignancy. Clin Immunol. 2010;135(3):440–7. doi: 10.1016/j.clim.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 30.Michałkiewicz J, Barth C, Chrzanowska K, Gregorek H, Syczewska M, Weemaes C, et al. Abnormalities in the T and NK lymphocyte phenotype in patients with Nijmegen breakage syndrome. Clin Exp Immun. 2003;134(3):482–90. doi: 10.1046/j.1365-2249.2003.02285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Albert M, Gennery A, Greil J, Cale C, Kalwak K, Kondratenko I, et al. Successful SCT for Nijmegen breakage syndrome. Bone Marrow Transplant. 2009;45(4):622–6. doi: 10.1038/bmt.2009.207. [DOI] [PubMed] [Google Scholar]
- 32.Platt C, Geha RS, Chou J. Gene hunting in the genomic era: Approaches to diagnostic dilemmas in patients with primary immunodeficiencies. J Allergy Clin Immunol 2013. [DOI] [PMC free article] [PubMed]