

Abstract
Background
The treatment of traumatic brain injury (TBI) represents an unmet medical need, as no effective pharmacological treatment currently exists. The development of such a treatment requires a fundamental understanding of the pathophysiological mechanisms that underpin the sequelae resulting from TBI, particularly the ensuing neuronal cell death and cognitive impairments. Tumor necrosis factor-alpha (TNF-α) is a cytokine that is a master regulator of systemic and neuroinflammatory processes. TNF-α levels are reported to become rapidly elevated post TBI and, potentially, can lead to secondary neuronal damage.
Methods
To elucidate the role of TNF-α in TBI, particularly as a drug target, the present study evaluated (i) time-dependent TNF-α levels and (ii) markers of apoptosis and gliosis within the brain and related these to behavioral measures of ‘well being’ and cognition in a mouse closed head 50 g weight drop mild TBI (mTBI) model in the presence and absence of post-treatment with an experimental TNF-α synthesis inhibitor, 3,6′-dithiothalidomide.
Results
mTBI elevated brain TNF-α levels, which peaked at 12 h post injury and returned to baseline by 18 h. This was accompanied by a neuronal loss and an increase in astrocyte number (evaluated by neuronal nuclei (NeuN) and glial fibrillary acidic protein (GFAP) immunostaining), as well as an elevation in the apoptotic death marker BH3-interacting domain death agonist (BID) at 72 h. Selective impairments in measures of cognition, evaluated by novel object recognition and passive avoidance paradigms - without changes in well being, were evident at 7 days after injury. A single systemic treatment with the TNF-α synthesis inhibitor 3,6′-dithiothalidomide 1 h post injury prevented the mTBI-induced TNF-α elevation and fully ameliorated the neuronal loss (NeuN), elevations in astrocyte number (GFAP) and BID, and cognitive impairments. Cognitive impairments evident at 7 days after injury were prevented by treatment as late as 12 h post mTBI but were not reversed when treatment was delayed until 18 h.
Conclusions
These results implicate that TNF-α in mTBI induced secondary brain damage and indicate that pharmacologically limiting the generation of TNF-α post mTBI may mitigate such damage, defining a time-dependent window of up to 12 h to achieve this reversal.
Introduction
Traumatic brain injury (TBI) is a common cause of morbidity and mortality across both the civilian and military populations, with a reported worldwide annual incidence of some ten million cases [1]. Indeed, within the US alone, TBI accounts for some 1.7 million emergency department visits - a number that likely underestimates its true incidence [2] - and is credited with some 30% of all injury-related deaths [3]. In essence, TBI is elicited following the unexpected application of an external force to the head. Patients who survive such injury often present with persistent long-term disabilities that require rehabilitation - a costly 52 billion dollars annual expense in the US alone [4-6]. The severity of ensuing disabilities varies and often may be associated with the severity of the injury itself [7]. Mild TBI (mTBI) accounts for some 80% to 90% of cases, and arising common disabilities include sensory-motor problems, learning and memory deficits, anxiety, and depression [8,9]. Of significant additional concern, mTBI may predispose long-term survivors to age-related neurodegenerative disorders by providing a risk factor for the development of Alzheimer’s disease, Parkinson’s disease, and post-traumatic dementia [10-14], with the older people being most vulnerable [15,16]. Despite significant ongoing research and advancements in our understanding of the molecular and cellular changes that occur after TBI, no effective pharmacological treatment is currently available [17,18].
mTBI-associated brain damage can be subdivided into two phases: an initial primary phase that is immediate and results from the mechanical force(s) applied to the skull and brain at the time of impact, potentially inducing shearing and compression of neuronal and vascular tissue that results in brain contusion, axonal injury, blood vessel rupture, and hemorrhage. This is followed by an extended second phase that involves cascades of biological processes initiated at the time of injury that may persist over subsequent days, weeks, and possibly months, consequent to ischemia, neuroinflammation, glutamate toxicity, altered blood-brain barrier permeability, oxidative stress, astrocyte reactivity, cellular dysfunction, and apoptosis [19-22]. As secondary brain injury may be reversible, in order to develop an effective treatment, it is imperative to understand the biological cascades that drive the delayed secondary phase that occurs following TBI [23-25].
It is widely recognized that inflammatory cytokines, chemokines, and growth factors play significant roles in the pathophysiology of TBI. Albeit that initiation of an inflammatory response can be essential to promote neuroreparative mechanisms in response to a physiological insult [26-28], if this is excessive or unregulated, it can augment neuronal dysfunction and degeneration by inducing a self-propagating pathological cascade of neuroinflammation [29-31]. Shortly following TBI, substantial synthesis and release of proinflammatory cytokines occur from astrocytes and microglia, particularly tumor necrosis factor-α (TNF-α) with mRNA and protein levels becoming acutely elevated within as little as 17 min after injury seen in post-mortem brains from patients who died shortly after TBI [32]. A parallel rapid sequence has been described in rodent TBI animal models in which a TNF-α rise precedes the appearance of ensuing cytokines [33-35]. Depending on its signaling pathway, TNF-α can exacerbate trauma and oxidative stress within the brain and contribute to glutamate release and blood-brain barrier dysfunction that can lead to further influx of inflammatory factors from blood to brain [36].
Inhibiting the generation of TNF-α may thus reinforce its role in mTBI and define its value as a potential treatment target, as it is considered a master regulator of the inflammatory response. Sudden and substantial rises in TNF-α can induce a diverse array of cell death processes, including NF-kB activation, apoptosis, and necrosis [37]. In addition, an increase in TNF-α levels trigger glutamate release from astrocytes, which can lead to glutamate excitotoxicity [38]. Although the elevation of TNF-α levels in the early hours post TBI can be harmful [39-41], cytokine balance has been reported as essential for long-term recovery from injury [40-42]. In this current study, rather than utilizing a TNF-α antibody approach to capture and clear it before it can potentially reach its target, as is effectively achieved in the treatment of rheumatoid arthritis, the experimental drug 3,6′-dithiothalidomide was employed to reduce TNF-α synthesis [43] and thereby maintain but dramatically lower its physiological release pattern. In our previous studies, we effectively used 3,6′-dithiothalidomide to ameliorate cognitive deficit following mTBI [44]. However, our previous work did not define the therapeutic window for 3,6′-dithiothalidomide, the extended time course of TNF-α overproduction, and the histochemical changes in neurons and glia correlated with injury. We extend our previous finding in the present study, correlating the potential role of mTBI-induced TNF-α release with neuronal loss, apoptosis, and astrocyte elevation, and defining a window of opportunity for potential treatment.
Materials and methods
Animals
Male ICR mice (30 to 40 g of weight and 6 to 8 weeks of age) were bred and raised within the vivarium of Tel Aviv University, Israel, originally derived from breeding pairs purchased from HSD Jerusalem, Israel. They were housed four to six per cage, maintained at a constant 22 ± 1°C, had ad libitum access to food and water, and kept on a 12:12 h light/dark cycle. Lighting during the light phase remained constant, and all experimental manipulations were undertaken during this light phase of the cycle. A minimum number of animals were included into studies, and all efforts were made to minimize potential suffering. Each animal was used for only a single experiment, and all experimental procedures and housing conditions were approved by the Institutional Animal Care and Use Committee of Tel Aviv University (M-10-006).
Mild traumatic brain injury
Mice were subjected to mTBI using a weight drop device that has previously been described [44-46]. Mice were anesthetized with isoflurane (Merck & Co., Inc., Whitehouse Station, NJ, USA) and then placed under the device. The weight drop apparatus comprised of a cylindrical-shaped 50-g piece of metal with a rounded spherical tip, which was dropped through a vertical metal guide tube (diameter 13 mm × length 80 cm). Anesthetized mice were carefully positioned with their head supported and immobilized by a sponge so that the right temporal side of the head, between the corner of the eye and the ear, was directly below the guide tube opening. The sponge allowed anterior/posterior motion of the head without rotational movement at the moment of impact following weight drop [44-46]. Sham mice were submitted to the same procedure as described for mTBI, but without release of the weight.
Drug administration
Synthesis of 3,6′-dithiothalidomide (Merck & Co., Inc., Whitehouse Station, NJ, USA) was achieved by a published synthetic route [43], and chemical characterization confirmed the structure of the final product with a chemical purity of 99.8%. The agent was prepared as a suspension in 1% carboxymethyl cellulose (formulated in isotonic saline; Merck & Co., Inc., Whitehouse Station, NJ, USA) immediately prior to daily use in each study to provide a final dose of 28 mg/kg (0.1 ml/10 g) body weight. Either 3,6′-dithiothalidomide or similarly prepared vehicle was administered by the intraperitoneal (i.p.) route from 1 to 18 h post injury or sham procedure, depending on the measures evaluated (whether for ELISA, immunohistochemistry, or behavioral studies).
TNF-α analysis by ELISA
To verify the occurrence of TNF-α elevation in our mTBI model and define its time dependence, mice were subjected to mTBI and brains were removed at specific times thereafter (1 to 18 h; n = 4 to 5 per time). The right cortex was immediately frozen in liquid nitrogen and homogenized with appropriate protease inhibitors (Halt Protease Inhibitor Cocktail; Sigma-Aldrich, St. Louis, MO, USA). The samples were then quantified for TNF-α levels by ELISA assay (BioLegend, San Diego, CA, USA).
Physiological parameters of well-being
Rectal temperature was recorded with a mouse thermometer. Baseline values (°C) were evaluated 30 min before 3,6′-dithiothalidomide administration and at 1 and 4 h following mTBI or sham procedure.
Anxiety-like behavior and motor activity were evaluated by elevated plus maze. The maze was elevated 60 cm above the floor level and comprised of 4 arms (30 × 5 × 15 cm) along which mice could walk that formed a ‘+’ shape [47]. Two conjoined arms were open (without walls) and the other two were closed (with walls but no ceiling). On evaluation days, mice were placed at the center of the plus-maze, facing one of the open arms and their time spent within the open arms was recorded over a 5-min period. The maze was cleansed with 70% ethanol (ETOH; v/v) between animals.
Cognitive behavioral tests
Two behavioral paradigms were evaluated: Y-maze and novel object recognition (NOR).
Y-maze test
Spatial memory was evaluated by Y-maze, as initially described by Dellu and colleagues [48], and is a task that takes advantage of a preference of rodents to explore novel rather than familiar places. The Y-maze was erected from black Plexiglas and comprised of three alike arms (30 × 8 × 15 cm length, set at an angle of 120° from one another). Evaluation comprised of two trials separated by a 2-min interval (during which the mouse was returned to its home cage). The initial ‘familiarization’ trial was of 5-min duration with only two arms open (one termed the ‘start’ arm and the other the ‘old’ arm), with the third (‘novel’) arm blocked by a door. The second trial was of 2-min duration, and all three arms were open. The time spent in each of the arms was recorded, and discrimination of spatial novelty was determined as a preference index [49] calculated as (time in the novel − time in the old arm)/(time in the novel + time in the old arm). The apparatus was cleansed between trials with 70% ETOH (v/v).
NOR test
An object recognition test to evaluate short-term recognition memory [50] was undertaken within an open field that comprised a black Plexiglas arena (59 × 59 cm size) surrounded by 20-cm black walls. The task takes advantage of a predisposition for rodents to explore new objects and included three trials of 5-min duration separated by a 24-h interval. On the initial day of evaluation, mice were individually placed within the empty arena for habituation. The following day, mice were placed into the same arena that had two identical objects, A and B, positioned 40 cm from one another and 10 cm from the walls. On the third day, mice were again placed into the arena; however, object A remained the same as the preceding day and new object C replaced prior object B. The arena and objects were thoroughly cleansed (70% ETOH v/v) between each trial. Object exploration (defined as rearing on the object or sniffing it at a distance of less than 2 cm and/or nose touching it) was recorded and discrimination of recognition novelty was determined as a preference index [49]: (time exploring the new object − time exploring the old object)/(total time exploring an object). Mice that explored objects for less than 10% of the total available time were excluded from analyses.
Immunohistochemistry/immunofluorescence brain slice studies
A cohort of mTBI and sham mice were anesthetized at 72 h following the procedure by excess ketamine + xylazine administration and were immediately perfused transcardially with PBS followed by 4% paraformaldehyde ((PFA) in 0.1 M phosphate buffer, pH 7.4). Their brains were removed, fixed overnight (4% PFA in 0.1 M phosphate buffer, pH 7.4), and then placed in 30% sucrose for 48 h. Coronal sections (30 μm) were cut on a cryostat, placed in cryoprotectant, and stored at −20°C until use. Thereafter, 5 sections of cortex and 5 of hippocampus were blocked by incubation with 0.1% Triton X-100 in phosphate-buffered saline (PBST) and 10% normal horse serum for 1 h at 25°C. The primary antibodies, mouse anti-neuronal nuclei (NeuN; 1:50, Millipore, Danvers, MA, USA, Cat#MAB3377), mouse anti-glial fibrillary acidic protein (GFAP; 1:10,000, Millipore, Cat#MAB3402), and rabbit anti-BH3-interacting domain death agonist (BID; 1:50, Cell Signaling, Danvers, MA, USA, Cat#9942), were then dissolved in PBST and 2% normal horse serum and incubated with the sections for 48 h at 4°C. Following rinsing in PBST, sections were incubated for 1 h at 25°C with DyLight™ 594-conjugated AffinityPure Donkey Anti-rabbit IgG and DyLight™ 488-conjugated AffinityPure Donkey Anti-mouse IgG (1:300; Jackson Laboratories, Bar Harbor, ME, USA). After rinses in PBST, sections were mounted on dry gelatin-coated slides and evaluated for fluorescence with a Zeiss LSM 510 confocal microscope with × 20 and × 63 lens (Carl Zeiss, Jena, Germany). For each brain, three to five sections were taken and the average numbers of cells within the hippocampus and the temporal cortex were calculated within defined fields of either 1402 or 4402 μM. Evaluation of immunohistochemical slides for immunofluorescence was undertaken in a blinded manner, and the omission of primary antibodies was routinely undertaken in the generation of negative control sections. Analyses were performed by Imaris program for color quantification (Bitplane AG, Zurich, Switzerland).
Data analyses
Results throughout are presented as mean ± SEM values and were analyzed by SPSS 18 software (Genius Systems, Petah Tikva, Israel). One-way ANOVAs were performed to compare between all groups, followed by least significant difference (LSD) post hoc tests. ANOVA-repeated measures were performed to compare rectal temperatures.
Results
Evaluation of well-being
‘Basic well-being,’ a concept that underlies the combined health and wellness of an animal [51], was evaluated across all mice groups and combined subjective measures, such as the grooming and appearance, righting skills, ambulation, and blinking reflex, with objective ones that included the parameters of weight, body temperature, anxiety-like behavior, and motor skills.
Subjectively and in accord with prior studies [45], mice subjected to this type of mTBI were indistinguishable from those subjected to the sham procedure when evaluated at 1 or 24 h later, irrespective of 3,6′-dithiothalidomide or vehicle administration. Rectal temperature measurements were used to monitor potential core temperature changes induced by either brain injury or 3,6′-dithiothalidomide administration, and no significant difference (NS) was found either between animal groups [F(2,12) = 0.084, NS] or across measurement times (30 min before injury and 1 and 4 h post-mTBI/injection) [F(2,12) = 3.630, NS] (data not shown).
The elevated plus maze was used to examine anxiety-like behavior and motor activity. No differences were found between any groups in anxiety-like behavior at 72 h and 7 days post-injury [F(5,56) = 0.791, NS] [F(5,47) = 0.765, NS], respectively (data not shown). Likewise, no differences were evident between any groups in relation to motor skills evaluated at 72 h and 7 days post-injury [F(5,56) = 1.13 NS] [F(5,47) = 0.798, NS], respectively (data not shown). Together these result indicate that mice were healthy and that neither mTBI nor 3,6′-dithiothalidomide impacted their well-being.
Time-dependent changes in TNF-α levels in brain tissue
As illustrated in Figure 1, mice challenged with mTBI demonstrated a time-dependent rise in brain protein levels of TNF-α that were increased by 2.5-fold, peaked at 12 h post injury, and returned to baseline by 18 h [F(3,13) = 30.529, p < 0.0001]. LSD post hoc analyses confirmed that the 12-h mTBI group was significantly different from all other groups (p < 0.0001). Levels were elevated to 132.9 pg/ml at 12 h versus a baseline value of 53.4 pg/ml. In animals subjected to mTBI and administered 3,6′-dithiothalidomide 1 h post injury, the elevated TNF-α 12 h post injury response was ameliorated. Specifically, mice treated with 3,6′-dithiothalidomide post injury had similar brain TNF-α levels as the sham group, 67.0 and 53.4 pg/ml, respectively, F(4,17) = 14.579, p < 0.0001, Figure 1B. LSD post hoc analyses confirmed that the mTBI 12-h group was significantly different from all other groups (p < 0.0001).
Figure 1.

mTBI induces a time-dependent rise in brain TNF-α levels. Right (ipsilateral to mTBI) cerebral cortex protein extracts were prepared from sham or mTBI mice at the indicated time points post injury. (A) Time-dependent brain levels of TNF-α at baseline (sham) and post injury. At 12 h post mTBI, TNF-α levels peaked (132.8 vs. 53.4 (sham) pg/ml, p < 0.0001). By 18 h post injury, TNF-α levels returned to baseline (50.5 pg/ml). (B) Treatment with 3,6′-dithiothalidomide (3,6-DT) at 1 h after mTBI prevented the TNF-α elevation evident at 12 h post mTBI (3,6′-DT + mTBI 67.1 pg/ml vs. mTBI 132.8 pg/ml, p < 0.0001). In both (A) and (B), **** was significantly different from all other groups (p < 0.0001).
mTBI- and treatment-induced changes in cognitive function
When evaluated by Y-maze at 7 days post procedure, vehicle-treated mTBI-challenged mice demonstrated a significant impairment in spatial memory, as compared to sham control animals. This mTBI-induced deficit was ameliorated by a single dose of 3,6′-dithiothalidomide administered either 1 or 12 h post injury. However, when 3,6′-dithiothalidomide administration was withheld until 18 h, mice displayed impairment and, together with the mTBI vehicle group, their preference index was significantly reduced compared to sham controls [F(4,57) = 6.462, p < 0.01] (Figure 2A). LSD post hoc analyses confirmed that the mTBI + vehicle and the mTBI + 18 h 3,6′-dithiothalidomide groups were significantly different from all other groups (p < 0.05).
Figure 2.

mTBI induces impairments in performance in both a Y-maze and novel object recognition (NOR) preference index paradigms that are ameliorated by 3,6′-dithiothalidomide when administered up to 12 but not 18 h post injury. (A) Performance of mice was quantitatively assessed in a Y-maze and (B) in a NOR paradigm at 7 days following mTBI as a preference index that was calculated as (time associated with the novel − time with the old arm or object)/(time with the novel + time with the old arm or object). Values are mean ± SEM values; a one-way ANOVA indicates that mTBI animals had a deficit in spatial (Y-maze) and visual (NOR) memory performance compared with all the other groups (*p < 0.05) with the exception of animals dosed with 3,6′-dithiothalidomide at 18 h post injury. No differences were found between any of the other groups (control (sham) 1 and 12 h 3,6′-dithiothalidomide dosing), suggesting complete amelioration by 3,6′-dithiothalidomide when administered within 12 h of injury.
As illustrated in Figure 2B, the spatial deficit evident in vehicle-treated mTBI mice in the Y-maze was also seen with the NOR paradigm. Here too, the administration of a single dose of 3,6′-dithiothalidomide to mTBI mice 1 or 12 h following injury fully mitigated the deficit, but delaying administration to 18 h post injury did not. Specifically, the mTBI vehicle and mTBI + 18 h 3,6′-dithiothalidomide groups displayed a significantly reduced index preference versus sham controls [F(4,57) = 8.975, p < 0.001]. LSD post hoc analyses established that the mTBI + vehicle and the mTBI + 18 h 3,6′-dithiothalidomide groups were significantly different from all other groups (p < 0.05).
Together these results extend the work of Baratz and colleagues [44] and define a therapeutic window of up to 12 h post mTBI to mitigate cognitive deficits by lowering TNF-α generation, as well as documenting the time course of TNF-α elevation.
Immunofluorescence
To evaluate the impact of mTBI at the cellular level, particularly in relation to the described amelioration of cognitive deficits imparted by lowering TNF-α generation, immunohistochemical analyses were undertaken at 72 h post injury. These focused on two key brain areas ipsilateral to injury: the cerebral cortex, as the area closest to impact, and the dentate gyrus, a region of the hippocampal formation considered to contribute to the formation of new episodic memory [52,53], the spontaneous exploration of novel environments, and other mnemonic functions [53,54].
Illustrated in Figures 3A and 4A are brain regions (cerebral cortex and dentate gyrus, respectively) displaying immunofluorescence associated with (i) NeuN, a neuronal nuclear protein that is widely used as a marker of adult neurons, and with (ii) BID, a proapoptotic Bcl-2 protein. Quantification of NeuN staining revealed a neuronal loss in both the cortex [F(3,13) = 7.198, p < 0.005, Figure 3B] and dentate gyrus [F(3,15) = 5.641, p < 0.05, Figure 4B]. Post hoc analyses revealed that the mTBI alone group was different from all other groups (p < 0.05) in both brain regions and was reduced by 42.5% and 22.3% versus sham values in cortex and dentate gyrus, respectively. Correlated with this was an elevation in apoptotic cell number, as revealed from BID staining in the cortex [F(3,13) = 23.067, p < 0.0001, Figure 3C] and in dentate gyrus [F(3,13) = 6.301, p < 0.05, Figure 4C]. Likewise, post hoc analyses demonstrated that the mTBI group was different from all other groups (p < 0.0001, p < 0.05, respectively; and 2.76- and 1.91-fold compared to their respective sham values). In addition and illustrated in Figures 5A and 6A, mTBI-challenged mice had an elevation in astrocyte number (3.37- and 1.39-fold, respectively), as revealed by GFAP staining, within the cortex [F(3,13) = 37.641, p < 0.0001, Figure 5B] and dentate gyrus [F(3,13) = 13.284, p < 0.001, Figure 6B]. The administration of 3,6′-dithiothalidomide 1 h post injury ameliorated all mTBI-induced changes in neuron, BID, and astrocyte number as, notably, no differences were found between the mTBI + 3,6′-dithiothalidomide and the sham groups. Finally, no changes were evident between any groups (sham, mTBI, and mTBI + drug) in the total cell numbers, as revealed from DAPI staining, within the cortex and dentate gyrus [F(3,15) = 1.009, NS, Figure 5C; F(3,15) = 2.251, NS, Figure 6C].
Figure 3.

Neuronal loss and apoptosis is induced by mTBI in cerebral cortex ipsilateral to injury and mitigated by 3,6′-dithiothalidomide. At 72 h post injury, cerebral cortex ipsilateral to mTBI was assessed for cellular changes. (A) and (B) A decline in neuronal number indicative of neuronal loss (NeuN - green) was evident post mTBI (p < 0.01). Treatment with 3,6′-dithiothalidomide at 1 h post-injury prevented such a change. (A) and (C) An elevation in BID (a marker for apoptosis - red) was evident within mTBI brains (p < 0.001). No changes in apoptotic cell death were found in animals that were treated with 3,6′-dithiothalidomide (as compared to sham animals). Within (A) (representative sections within the cerebral cortex), the bar is equal to 20 μm in length.
Figure 4.

Neuronal loss and apoptosis is induced by mTBI in the dentate gyrus ipsilateral to injury and mitigated by 3,6′-dithiothalidomide. At 72 h post injury, the dentate gyrus of the hippocampus ipsilateral to mTBI was evaluated for cellular changes. (A) and (B) Neuronal loss (NeuN - green) was found post mTBI (p < 0.05). Treatment with the 3,6′-dithiothalidomide at 1 h post-injury prevented this loss. (A) and (C) An increase in BID (a marker for apoptosis in red) was evident in the mTBI brains (p < 0.01). No change in apoptotic cell death was apparent in animals treated with 3,6′-dithiothalidomide (as compared to sham animals). Within (A) (representative sections within the dentate gyrus), the bar is equal to 100 μm in length.
Figure 5.

mTBI induces an elevation in astrocyte number in ipsilateral cerebral cortex that is inhibited by 3,6′-dithiothalidomide. At 72 h post injury, cerebral cortex ipsilateral to mTBI was assessed for cellular changes. (A) and (B) Astrocyte number (GFAP - red) was increased post mTBI (p < 0.001). Treatment with 3,6′-dithiothalidomide at 1 h post-injury prevented this. (A) and (C) No difference in total number of cells was evident between groups, as revealed from DAPI (blue) staining. Within (A) (representative sections within the cerebral cortex), the bar is equal to 100 μm in length.
Figure 6.
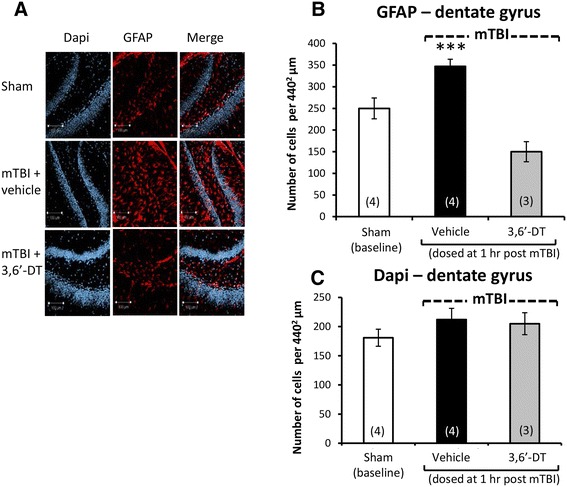
mTBI induces an elevation in astrocyte number in ipsilateral dentate gyrus that is inhibited by 3,6′-dithiothalidomide. At 72 h post injury, dentate gyrus ipsilateral injury was assessed for cellular changes. (A) and (B) Astrocyte number (GFAP - red) was elevated post mTBI (p < 0.001). Treatment with 3,6′-dithiothalidomide at 1 h post-injury inhibited this. (A) and (C) No difference in total number of cells was apparent between groups, as evaluated by DAPI (blue) staining. Within (A) (representative sections within the dentate gyrus), the bar is equal to 100 μm in length.
In conclusion, the early administration (1 h post injury) of a single dose of the TNF-α synthesis inhibitor 3,6′-dithiothalidomide inhibited cellular changes induced by mTBI in two key brain regions evaluated, cerebral cortex and dentate gyrus.
Discussion
TNF-α has been implicated in the pathogenesis of a wide number of neurological disorders that develop both acutely, as in TBI and stroke, and chronically, as in Alzheimer’s disease and Parkinson’s disease [29-36,40-42,55,56]. The current study confirms the rapid generation and release of TNF-α in a mouse closed head 50 g weight drop mTBI model, emulating a concussive head injury in humans, which led to neuronal loss and specific cognitive deficits. The inhibition of TNF-α synthesis blocked the mTBI-induced rise in brain TNF-α and protected against neuronal loss and cognitive deficits with a therapeutic window of 12 h. These results underline a role for TNF-α as a key regulator of cascades leading to neuronal loss and cognitive impairment in mTBI and highlights TNF-α as an amenable drug target for future mTBI treatment.
In light of (i) the high incidence of mTBI (approximately 600 per 100,000 people); (ii) the increased risk of dementia resulting from mTBI, particularly in the older people [15]; (iii) the upregulation of pathways leading to chronic neurodegenerative disorders induced by mTBI [12,20,57,58]; (iv) the long-term care, suffering, and economic debt associated with mTBI patients [59]; and (v) the lack of any available therapeutic [60], it is important to understand the mechanisms that underlie head injury.
TNF-α is a well-characterized protein that regulates numerous cellular processes, including inflammation and cell death as well as cellular differentiation and survival, by binding to and activation of two cognate receptors: TNF-α receptor 1 (TNFR1) (p55) and TNFR2 (p75) [29-31,61].
TNFR1 is expressed ubiquitously, including neurons, astrocytes, and microglia throughout the brain. With its intracellular death domain, it contributes to neuronal dysfunction and death and primarily is activated by soluble TNF-α [62]. TNFR2, on the other hand, is principally expressed on hematopoietic cells but also is present on other cell types, including neurons, has been associated with cell survival [61,63-65] and chiefly responds to membrane-bound TNF-α [66,67]. The engagement of homotrimeric TNF-α (either soluble or membrane bound) to either receptor can activate three major signaling pathways: an apoptotic cascade initiated via the TNF-α receptor-associated death domain, a nuclear factor kappa B (NFκB) signaling a pro-survival pathway implemented via NFκB-mediated gene transcriptional actions, and a c-Jun N-terminal kinase (JNK) cascade involved in cellular differentiation and proliferation that is generally proapoptotic [38,68]. In large part, the contrasting pro-survival versus death-induced actions of TNF-α plausibly rely on which TNF-α receptor subtype is activated, the target cell types involved and their expression ratio of TNFR1/2 and associated coupling proteins, and the temporal concentrations of available soluble and membrane-bound TNF-α [64]. However, cross talk between the different signaling pathways and the degree and duration of neuroinflammation combine in determining the eventual physiological consequences of TNF-α receptor activation [69]. Consequent to the diverse actions of TNF-α and the influence of the brain microenvironment in which they occur, it is not always clear under which conditions TNF-α promotes beneficial versus deleterious neuronal effects. This explains the sometimes contradictory literature in the TNF-α neuroscience field [29-31,36,38,55,69] and its involvement in cascades promoting neuronal dysfunction and loss in both acute and long-term neurodegenerative disorders. In the present study, no differences were evident across the sham and mTBI groups in relation to the broad measure of ‘well being’ or in the evaluation of body temperature, anxiety-related behavior, and motor activity, which is in accord with previous results in rodents [51] and humans [70]. Although indistinguishable across a wide number of measures, importantly, deficits in cognitive performance were apparent in mTBI mice in accordance with past studies in mice [24,44-46] and humans [71]. In evaluating potential mechanisms responsible for these cognitive changes, a mTBI-triggered inflammatory cascade mediated by the generation of proinflammatory cytokines appears likely [72]. In this regard, the proinflammatory cytokine TNF-α is considered essential for both initiating and regulating an inflammatory response to trauma, and early transient elevations in brain mRNA expression of TNF-α as well as rises in IL-1β and IL-6 have been described in rodent closed head TBI, and associated adverse events [33,35,73]. In the current study, a time-dependent elevation in brain TNF-α protein levels was apparent in mTBI-challenged mice that peaked at 12 h and declined to baseline by 18 h. In line with this, elevated brain protein levels of TNF-α, IL-1β, and IL-6 have been reported in rodent mTBI models as well as within human CSF within hours of injury [74-78], as they have in other neurological disorders [79-81]. Inhibiting such an elevation in brain TNF-α in this model allowed the evaluation of the role of this transient TNF-α rise in neuronal cell loss, neuroinflammation, and cognitive deficits known to accompany mTBI.
To define the relationship between the mTBI-induced elevation in TNF-α and cognitive impairment evident 7 days later, 3,6′-dithiothalidomide was administered 1, 12, and 18 h following mTBI, extending our initial concentration-dependent studies of the compound in this same mTBI model [44]. Notably, mTBI-induced impairments in both the Y-maze and NOR paradigms were blocked by a single drug dose either at 1 or 12 h post injury, the peak of TNF-α generation in brain, but were not mitigated when administration was delayed to 18 h, thereby defining a treatment window of opportunity.
To evaluate the basis of the mTBI-induced cognitive impairment, brain regions ipsilateral to the side of injury were evaluated at 72 h, as this time coincides with the substantial occurrence of markers of neuronal apoptosis [24,82]. Assessment of the cerebral cortex, the area closest to the site of impact, and dentate gyrus of the hippocampus was performed, as dysfunction in the former and latter might explain the decline in performance in visual memory evaluated by NOR [83] and in spatial learning as assessed by the Y-maze [48], respectively. Neuronal loss (NeuN), an increase neuronal apoptosis (BID), and an elevation in astrocyte number (GFAP) were evident in both brain regions, which is in accord with prior studies in this mTBI model describing elevations in apoptotic proteins (p53, c-Jun, and Bcl-2) as well as TUNEL-positive and silver stain-impregnated degenerating neurons [82,84], as well as other animal models of brain injury [20,60,85,86]. Importantly, early post injury treatment with 3,6′-dithiothalidomide fully prevented these changes. In line with this, this same agent has recently been reported to ameliorate neuroinflammation and alleviate cognitive deficits arising from intracerebral administration of LPS or amyloid-β peptide [79,87,88]. 3,6′-Dithiothalidomide is also reported to attenuate inflammatory markers, Alzheimer’s disease pathology, and behavioral deficits evident in aged Alzheimer transgenic mice [79,80], as well as mitigate neuroinflammation and apoptosis within the penumbra of focal ischemic stroke in mice [81]. Additionally, 3,6′-dithiothalidomide has recently been described to lower TNF-α and cerebral aneurysm formation and progression to rupture in mice [89,90].
Taken together, these studies support an important role for TNF-α in neuroinflammation and the modulation of neuronal function and viability across a broad range of neurological disorders. Consequent to the availability of both biological and small molecular weight TNF-α inhibitors in preclinical and clinical research, there is growing evidence that whereas physiological TNF-α levels are critical in normal brain physiology [37,38,55], excess TNF-α plays a key role in brain dysfunction [29-31,69]. In relation to the former, among a host of functions in brain, TNF-α serves as a gliotransmitter that, when secreted from glial cells surrounding synapses, can regulate synaptic communication between neurons as well as neuronal networks [36-38]. With respect to the latter, TNF-α reductions achieved with the clinical TNF-α binding protein etanercept, when administered i.p. following fluid percussion injury-induced TBI, attenuated TBI-induced contusion, ischemia, and resulting motor and cognitive deficits [91]. As in our studies, this brain TNF-α lowering approach also mitigated TBI-induced elevations in [91]. Albeit that these animal studies utilized a far higher etanercept dose than achievable in humans [91], in an open-label analysis of 12 TBI patients given perispinal etanercept up to more than 10 years following injury, motor impairment and spasticity were reported significantly reduced [55], supporting both clinical and translational relevance. Additionally, in rat studies using a TBI weight drop paradigm with some parallels to our studies in mice, immediate i.v. administration of a TNF-α binding protein or the competitive nonselective phosphodiesterase inhibitor, pentoxifylline, that lowers TNF-α at a transcriptional level, has been reported to mitigate mTBI-induced brain edema at 24 h and neurological dysfunction evaluated up to 14 days [75]. Finally, in other animal models that include ischemic and spinal cord injury, thalidomide has been reported to effectively reduce inflammation and improve the injury outcome when administered either before [92], immediately after [93], or within an hour of injury [94] in doses that varied between 20 and 300 mg/kg.
The single dose of 3,6′-dithiothalidomide used in the current study (28 mg/kg in mouse) compares favorably with former studies of thalidomide (20 to 300 mg/kg) and equates to a dose of 150 mg in a 65 kg human (2.3 mg/kg), following normalization of body surface area in accord with FDA guidelines [95]. Prior cellular [43,79] and animal studies [80,81] indicate that 3,6′-dithiothalidomide (albeit administered systemically by the i.p. route, as in the current study) is more potent in reducing TNF-α elevations than thalidomide and that it enters the brain but does not appear to be soporific [44,79,80]. In light of recent studies suggesting that thalidomide analogues can express more potent anti-inflammatory action with less neurotoxicity than the parent compound [96,97], the development of new and well-tolerated small molecular weight TNF-α inhibitors that can be administered orally may be of great clinical potential. Past studies evaluating genetically engineered mice that either lack TNF-α or its receptors have suggested a ‘Jekyll and Hyde’ scenario in which elevated TNF-α is detrimental during the acute phase after a TBI incident, but a part of the regenerative processes during the later chronic post-injury phase [42,98,99]. More recent studies in which the two individual receptors, TNFR1 (p55) and TNFR2 (p75), have been separately deleted suggest that each may have a distinct time-dependent function in TBI [40,41]. TNFR1 knockout mice possessed a smaller contusion volume and a clearly improved neurobehavioral performance for up to 4 weeks following a controlled cortical impact TBI, as compared with wild-type mice, whereas TNFR2 knockout mice demonstrated significant worsening post injury [42]. This implicates TNFR1 involvement in the immediate deleterious actions associated with acute TNF-α release following an injury and an involvement of TNFR2 in later tissue repair.
In summary, our studies suggest that the administration of a TNF-α synthesis inhibitor, 3,6′-dithiothalidomide, within the initial 12-h window of a mTBi event, may be therapeutically valuable at a time when elevated TNF-α interacts with TNFR1 to drive the development of neuroinflammation, neuronal dysfunction, and apoptosis. But such a therapeutic strategy should best be acute to allow later potentially beneficial actions of TNF-α mediated via TNFR2. Our results, together with other studies [33,36,39-41,44,56,74-78,97], underscore the potential of TNF-α as a potential therapeutic target in TBI and other neurological disorders.
Conclusion
This study implicates TNF-α in the delayed neuronal cell death and gliosis that occurs within the brain following mTBI, which leads to cognitive deficits. It additionally indicates that pharmacologically limiting the elevation of TNF-α within 12 h of the mTBI event markedly reduces such secondary damage and leads to improved cognitive outcome measures. Such a window provides an opportunity for translational studies in mTBI that is more difficult to define for other neurological disorders [100]. Building on the growing literature on the role of TNF-α in the initiation and perpetuation of the neuroinflammation that can drive the progression of acute and chronic neurological disorders [29-31,36,55,56,68,69,101,102], the present study underscores the value of targeting of TNF-α as a treatment strategy for TBI and the development of new and well-tolerated oral small molecular weight TNF-α inhibitors and related approaches as clinical treatment options.
Acknowledgements
This study was supported in part by the Intramural Research Program, National Institute on Aging, NIH, Baltimore, USA; by Tel-Aviv University, Tel-Aviv, Israel; and by a grant from the Ministry of Science and Technology, Taiwan (MOST103-2321-B-038-002 to JY Wang). None of the authors have any competing interests in the manuscript.
Footnotes
Nigel H Greig and Chaim G Pick contributed equally to this work.
Competing interests
The small molecule TNF-α synthesis inhibitor 3,6′-dithiothalidomide was originally synthesized within the laboratory of NHG and colleagues within the Intramural Research Program of NIA, NIH. Whereas NHG is a co-inventor of the agent, neither he nor any of the authors hold any rights to the agent. All authors declare that they are without any financial and non-financial competing interests (political, personal, religious, ideological, academic, intellectual, commercial, or any other) in relation to this article.
Authors’ contributions
RB, DT, VR, and WL undertook ‘hands-on’ experimental studies generating data used within the manuscript, preliminary data on which studies were then optimized, and synthetic chemistry to provide compounds used within the studies. RD, DT, JYW, VR, WL, BJH, NHG, and CGP analyzed immunohistochemical, ELISA, behavioral, or chemical characterization data essential to the studies within the manuscript. CGP, BJH, and NHG conceived and planned the studies. RB, DT, JYW, BJH, CGP, and NHG prepared the manuscript. All authors have read the manuscript in full and are in agreement with the data and its interpretation.
Contributor Information
Renana Baratz, Email: renanaba@post.tau.ac.il.
David Tweedie, Email: tweedieda@grc.nia.nih.gov.
Jia-Yi Wang, Email: jywang2010@tmu.edu.tw.
Vardit Rubovitch, Email: Rubovitc@post.tau.ac.il.
Weiming Luo, Email: luowe@mail.nih.gov.
Barry J Hoffer, Email: bjh82@case.edu.
Nigel H Greig, Email: greign@grc.nia.nih.gov.
Chaim G Pick, Email: pickc@post.tau.ac.il.
References
- 1.Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC. The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation. 2007;22:341–53. [PubMed] [Google Scholar]
- 2.Setnik L, Bazarian JJ. The characteristics of patients who do not seek medical treatment for traumatic brain injury. Brain Inj. 2007;21(1):1–9. doi: 10.1080/02699050601111419. [DOI] [PubMed] [Google Scholar]
- 3.Faul M, Xu L, Wald M, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- 4.McGarry LJ, Thompson D, Millham FH, Cowell L, Snyder PJ, Lenderking WR, et al. Outcomes and costs of acute treatment of traumatic brain injury. J Trauma. 2002;53(6):1152–9. doi: 10.1097/00005373-200212000-00020. [DOI] [PubMed] [Google Scholar]
- 5.McAllister TW. Neurobehavioral sequelae of traumatic brain injury: evaluation and management. World Psychiatry. 2008;7(1):3–10. doi: 10.1002/j.2051-5545.2008.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, et al. Centers for Disease Control and Prevention (CDC). Surveillance for traumatic brain injury-related deaths–United States, 1997–2007. MMWR Surveill Summ. 2011;60(5):1–32. [PubMed] [Google Scholar]
- 7.Yu S, Kaneko Y, Bae E, Stahl CE, Wang Y, van Loveren H, et al. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009;1287:157–63. doi: 10.1016/j.brainres.2009.06.067. [DOI] [PubMed] [Google Scholar]
- 8.Iverson GL. Outcome from mild traumatic brain injury. Curr Opinion Psychiatry. 2005;18:301–7. doi: 10.1097/01.yco.0000165601.29047.ae. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber S, Barkai G, Gur-Hartman T, Peles E, Tov N, Dolberg OT, et al. Long-lasting sleep patterns of adult patients with minor traumatic brain injury (mTBI) and non-mTBI subjects. Sleep Med. 2008;9:481–7. doi: 10.1016/j.sleep.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case–control studies, EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl 2):S28–35. doi: 10.1093/ije/20.Supplement_2.S28. [DOI] [PubMed] [Google Scholar]
- 11.Mayeux R, Ottman R, Maestre G, Ngai C, Tang MX, Ginsberg H, et al. Synergistic effects of traumatic head-injury and apolipoprotein-epsilon-4 in patients with Alzheimers-disease. Neurology. 1995;45:555–7. doi: 10.1212/WNL.45.3.555. [DOI] [PubMed] [Google Scholar]
- 12.Tweedie D, Rachmany L, Rubovitch V, Zhang Y, Becker KG, Perez E, et al. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol Dis. 2013;54:1–11. doi: 10.1016/j.nbd.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4(134):134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K. Traumatic brain injury and risk of dementia in older veterans. Neurology. 2014;83(4):312–9. doi: 10.1212/WNL.0000000000000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 2014 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 16.Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging. 2013;34(5):1397–411. doi: 10.1016/j.neurobiolaging.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31(12):596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJ, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma. 2014;31(2):135–58. doi: 10.1089/neu.2013.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finnie JW, Blumbergs PC, Manavis J, Vink R. Pattern of cerebrospinal immediate early gene c-fos expression in an ovine model of non-accidental head injury. J Clin Neurosci. 2013;20:1759–61. doi: 10.1016/j.jocn.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, et al. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement. 2014;10(1 Suppl):S62–75. doi: 10.1016/j.jalz.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barkhoudarian G, Hovda DA, Giza C. The molecular pathophysiology of concussive brain injury. Clin Sports Med. 2011;30:33–48. doi: 10.1016/j.csm.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104. doi: 10.1002/msj.20104. [DOI] [PubMed] [Google Scholar]
- 23.Finnie JW, Blumbergs PC. Traumatic brain injury. Vet Pathol. 2002;39(6):679–89. doi: 10.1354/vp.39-6-679. [DOI] [PubMed] [Google Scholar]
- 24.Tweedie D, Milman A, Holloway HW, Li YZ, Harvey BK, Shen H, et al. Apoptotic and behavioral sequelae of mild brain trauma in mice. J Neurosci Res. 2007;85:805–15. doi: 10.1002/jnr.21160. [DOI] [PubMed] [Google Scholar]
- 25.Stoica BA, Byrnes KR, Faden AI. Cell cycle activation and CNS injury. Neurotox Res. 2009;16:221–37. doi: 10.1007/s12640-009-9050-0. [DOI] [PubMed] [Google Scholar]
- 26.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opinion Crit Care. 2002;8:101–5. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury - an inflammatory disease? Brain Res Rev. 2005;48:388–99. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 29.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frankola KA, Greig NH, Luo W, Tweedie D. Targeting TNF-α to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS Neurol Disord Drug Targets. 2011;10(3):391–403. doi: 10.2174/187152711794653751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark IA, Alleva LM, Vissel B. The roles of TNF in brain dysfunction and disease. Pharmacol Ther. 2010;128(3):519–48. doi: 10.1016/j.pharmthera.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma. 2010;27:497–507. doi: 10.1089/neu.2009.1120. [DOI] [PubMed] [Google Scholar]
- 33.Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-α inhibitor and an effective neuroprotectant. J Neuroimmunol. 1997;72:169–77. doi: 10.1016/S0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- 34.Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci. 2009;14:3795–813. doi: 10.2741/3489. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, You Z, Kim HH, Hwang SK, Khuman J, Guo S, et al. Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma. 2010;27:1037–46. doi: 10.1089/neu.2009.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuttolomondo A, Pecoraro R, Pinto A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: a review of the evidence to date. Drug Des Devel Ther. 2014;8:2221–38. doi: 10.2147/DDDT.S67655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Yin Q, Wu H. Structural basis of signal transduction in the TNF receptor superfamily. Adv Immunol. 2013;119:135–53. doi: 10.1016/B978-0-12-407707-2.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santello M, Volterra A. TNFα in synaptic function: switching gears. Trends Neurosci. 2012;35(10):638–47. doi: 10.1016/j.tins.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Bermpohl D, You Z, Lo EH, Kim HH, Whalen MJ. TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J Cerebral Blood Flow Metabol. 2007;27:1806–18. doi: 10.1038/sj.jcbfm.9600487. [DOI] [PubMed] [Google Scholar]
- 40.Longhi L, Ortolano F, Zanier ER, Perego C, Stocchetti N, De Simoni MG. Effect of traumatic brain injury on cognitive function in mice lacking p55 and p75 tumor necrosis factor receptors. Acta Neurochir Suppl. 2008;102:409–13. doi: 10.1007/978-3-211-85578-2_80. [DOI] [PubMed] [Google Scholar]
- 41.Longhi L, Perego C, Ortolano F, Aresi S, Fumagalli S, Zanier ER, et al. Tumor necrosis factor in traumatic brain injury: effects of genetic deletion of p55 or p75 receptor. J Cereb Blood Flow Metab. 2013;33(8):1182–9. doi: 10.1038/jcbfm.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, et al. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96:8721–6. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu X, Giordano T, Yu QS, Holloway HW, Perry T, Lahiri DK, et al. Thiothalidomides: novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J Med Chem. 2003;46:5222–9. doi: 10.1021/jm030152f. [DOI] [PubMed] [Google Scholar]
- 44.Baratz R, Tweedie D, Rubovitch V, Luo W, Yoon JS, Hoffer BJ, et al. Tumor necrosis factor-alpha synthesis inhibitor, 3,6′-dithiothalidomide, reverses behavioral impairments induced by minimal traumatic brain injury in mice. J Neurochem. 2011;118:1032–42. doi: 10.1111/j.1471-4159.2011.07377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zohar O, Schreiber S, Getslev V, Schwartz JP, Mullins PG, Pick CG. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience. 2003;118:949–55. doi: 10.1016/S0306-4522(03)00048-4. [DOI] [PubMed] [Google Scholar]
- 46.Milman A, Rosenberg A, Weizman R, Pick CG. Mild traumatic brain injury induces persistent cognitive deficits and behavioral disturbances in mice. J Neurotrauma. 2005;22:1003–10. doi: 10.1089/neu.2005.22.1003. [DOI] [PubMed] [Google Scholar]
- 47.Hogg S. A review of the validity and variability of the elevated plus-maze as an animal model of anxiety. Pharmacol Biochem Behav. 1996;54:21–30. doi: 10.1016/0091-3057(95)02126-4. [DOI] [PubMed] [Google Scholar]
- 48.Dellu F, Mayo W, Cherkaoui J, Lemoal M, Simon H. A 2-trial memory task with automated recording - study in young and aged rats. Brain Res. 1992;588:132–9. doi: 10.1016/0006-8993(92)91352-F. [DOI] [PubMed] [Google Scholar]
- 49.Dix SL, Aggleton JP. Extending the spontaneous preference test of recognition: evidence of object-location and object-context recognition. Behav Brain Res. 1999;99:191–200. doi: 10.1016/S0166-4328(98)00079-5. [DOI] [PubMed] [Google Scholar]
- 50.Meisser C. Object recognition in mice: improvement of memory by glucose. Neurobiol Learning Memory. 1997;67:172–5. doi: 10.1006/nlme.1996.3755. [DOI] [PubMed] [Google Scholar]
- 51.Edut S, Rubovitch V, Schreiber S, Pick CG. The intriguing effects of ecstasy (MDMA) on cognitive function in mice subjected to a minimal traumatic brain injury (mTBI) Psychopharmacol. 2011;214:877–89. doi: 10.1007/s00213-010-2098-y. [DOI] [PubMed] [Google Scholar]
- 52.Amaral D, Scharfman H, Lavenex P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies) Prog Brain Res. 2007;163:3–22. doi: 10.1016/S0079-6123(07)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saab BJ, Georgiou J, Nath A, Lee FJ, Wang M, Michalon A, et al. NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron. 2009;63(5):643–56. doi: 10.1016/j.neuron.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 54.Perederiy JV, Westbrook GL. Structural plasticity in the dentate gyrus- revisiting a classic injury model. Front Neural Circuits. 2013;7:17. doi: 10.3389/fncir.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs. 2012;26(12):1051–70. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- 56.Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des. 2008;14:3574–89. doi: 10.2174/138161208786848739. [DOI] [PubMed] [Google Scholar]
- 57.Tweedie D, Rachmany L, Rubovitch V, Lehrmann E, Zhang Y, Becker KG, et al. Exendin-4, a glucagon-like peptide-1 receptor agonist prevents mTBI-induced changes in hippocampus gene expression and memory deficits in mice. Exp Neurol. 2013;239:170–82. doi: 10.1016/j.expneurol.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nat Rev Neurol. 2014;10(3):156–66. doi: 10.1038/nrneurol.2014.15. [DOI] [PubMed] [Google Scholar]
- 59.Ryu WH, Feinstein A, Colantonio A, Streiner DL, Dawson DR. Early identification and incidence of mild TBI in Ontario. Can J Neurol Sci. 2009;36(4):429–35. doi: 10.1017/S0317167100007745. [DOI] [PubMed] [Google Scholar]
- 60.Moppett IK. Traumatic brain injury: assessment, resuscitation and early management. Br J Anaesth. 2007;99:18–31. doi: 10.1093/bja/aem128. [DOI] [PubMed] [Google Scholar]
- 61.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 62.Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U S A. 1998;95(2):570–5. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22(7):RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22(8):3025–32. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279(31):32869–81. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 66.Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83(5):793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 67.Grell M. Tumor necrosis factor (TNF) receptors in cellular signaling of soluble and membrane-expressed TNF. J Inflamm. 1995–1996;47(1–2):8–17 [PubMed]
- 68.Park KM, Bowers WJ. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal. 2010;22(7):977–83. doi: 10.1016/j.cellsig.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tweedie D, Sambamurti K, Greig NH. TNF-α inhibition as a treatment strategy for neurodegenerative disorders: new drug candidates and targets. Curr Alzheimer Res. 2007;4:378–85. doi: 10.2174/156720507781788873. [DOI] [PubMed] [Google Scholar]
- 70.Ponsford J, Cameron P, Fitzgerald M, Grant M, Mikocka-Walus A. Long-term outcomes after uncomplicated mild traumatic brain injury: a comparison with trauma controls. J Neurotrauma. 2011;28:937–46. doi: 10.1089/neu.2010.1516. [DOI] [PubMed] [Google Scholar]
- 71.Ghajar J, Ivry RB. Cognitive and Neurobiological Research Consortium. The predictive brain state: timing deficiency in traumatic brain injury? Neurorehabil Neural Repair. 2008;22(3):217–27. doi: 10.1177/1545968308315600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Israelsson C, Bengtsson H, Kylberg A, Kullander K, Lewén A, Hillered L, et al. Distinct cellular patterns of upregulated chemokine expression supporting a prominent inflammatory role in traumatic brain injury. J Neurotrauma. 2008;25(8):959–74. doi: 10.1089/neu.2008.0562. [DOI] [PubMed] [Google Scholar]
- 73.Israelsson C, Wang Y, Kylberg A, Pick CG, Hoffer BJ, Ebendal T. Closed head injury in a mouse model results in molecular changes indicating inflammatory responses. J Neurotrauma. 2009;26(8):1307–14. doi: 10.1089/neu.2008.0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion: influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42:177–85. doi: 10.1016/0165-5728(93)90008-M. [DOI] [PubMed] [Google Scholar]
- 75.Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF-α and IL-6 by brain tissue. J Cereb Blood Flow Metab. 1994;14:615–9. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- 76.Gourin CG, Shackford SR. Production of tumor necrosis factor-alpha and interleukin-1beta by human cerebral microvascular endothelium after percussive trauma. J Trauma. 1997;42(6):1101–7. doi: 10.1097/00005373-199706000-00020. [DOI] [PubMed] [Google Scholar]
- 77.Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95(1–2):115–25. doi: 10.1016/S0165-5728(98)00273-2. [DOI] [PubMed] [Google Scholar]
- 78.Ross SA, Halliday MI, Campbell GC, Byrnes DP, Rowlands BJ. The presence of tumour necrosis factor in CSF and plasma after severe head injury. Br J Neurosurg. 1994;8(4):419–25. doi: 10.3109/02688699408995109. [DOI] [PubMed] [Google Scholar]
- 79.Tweedie D, Ferguson RA, Fishman K, Frankola KA, Van Praag H, Holloway HW, et al. Tumor necrosis factor-α synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation. Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J Neuroinflammation. 2012;9:106. doi: 10.1186/1742-2094-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gabbita SP, Srivastava MK, Eslami P, Johnson MF, Kobritz NK, Tweedie D, et al. Early intervention with a small molecule inhibitor for tumor necrosis factor-α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2012;9:99. doi: 10.1186/1742-2094-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoon JS, Lee JH, Tweedie D, Mughal MR, Chigurupati S, Greig NH, et al. 3,6′-Dithiothalidomide improves experimental stroke outcome by suppressing neuroinflammation. J Neurosci Res. 2013;91(5):671–80. doi: 10.1002/jnr.23190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tashlykov V, Katz Y, Volkov A, Gazit V, Schreiber S, Zohar O, et al. Minimal traumatic brain injury induce apoptotic cell death in mice. J Mol Neurosci. 2009;37(1):16–24. doi: 10.1007/s12031-008-9094-2. [DOI] [PubMed] [Google Scholar]
- 83.Bussey TJ, Padain TL, Skillings EA, Winters BD, Morton AJ, Saksida LM. The touchscreen cognitive testing method for rodents: how to get the best out of your rat. Learn Mem. 2008;2008(15):516–23. doi: 10.1101/lm.987808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tashlykov V, Katz Y, Gazit V, Zohar O, Schreiber S, Pick CG. Apoptotic changes in the cortex and hippocampus following minimal brain trauma in mice. Brain Res. 2007;1130(1):197–205. doi: 10.1016/j.brainres.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 85.Wakade C, Sukumari-Ramesh S, Laird MD, Dhandapani KM, Vender JR. Delayed reduction in hippocampal postsynaptic density protein-95 expression temporally correlates with cognitive dysfunction following controlled cortical impact in mice. J Neurosurg. 2010;113(6):1195–201. doi: 10.3171/2010.3.JNS091212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han X, Tong J, Zhang J, Farahvar A, Wang E, Yang J, et al. Imipramine treatment improves cognitive outcome associated with enhanced hippocampal neurogenesis after traumatic brain injury in mice. J Neurotrauma. 2011;28(6):995–1007. doi: 10.1089/neu.2010.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Russo I, Caracciolo L, Tweedie D, Choi SH, Greig NH, Barlati S, et al. 3,6′-Dithiothalidomide, a new TNF-α synthesis inhibitor, attenuates the effect of Aβ1-42 intracerebroventricular injection on hippocampal neurogenesis and memory deficit. J Neurochem. 2012;122(6):1181–92. doi: 10.1111/j.1471-4159.2012.07846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Belarbi K, Jopson T, Tweedie D, Arellano C, Luo W, Greig NH, et al. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J Neuroinflammation. 2012;9:23. doi: 10.1186/1742-2094-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Starke RM, Chalouhi N, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J Neuroinflammation. 2014;11:77. doi: 10.1186/1742-2094-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. TNF-α induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. J Cereb Blood Flow Metab. 2013;33(10):1564–73. doi: 10.1038/jcbfm.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chio CC, Lin JW, Chang MW, Wang CC, Kuo JR, Yang CZ, et al. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J Neurochem. 2010;115(4):921–9. doi: 10.1111/j.1471-4159.2010.06969.x. [DOI] [PubMed] [Google Scholar]
- 92.Lee CJ, Kim KW, Lee HM, Nahm FS, Lim YJ, Park JH, et al. The effect of thalidomide on spinal cord ischemia/reperfusion injury in a rabbit model. Spinal Cord. 2007;45(2):149–57. doi: 10.1038/sj.sc.3101931. [DOI] [PubMed] [Google Scholar]
- 93.Koopmans GC, Deumens R, Buss A, Geoghegan L, Myint AM, Honig WH, et al. Acute rolipram/thalidomide treatment improves tissue sparing and locomotion after experimental spinal cord injury. Exp Neurol. 2009;216(2):490–8. doi: 10.1016/j.expneurol.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 94.Genovese T, Mazzon E, Esposito E, Di Paola R, Caminiti R, Meli R, et al. Effect of thalidomide on signal transduction pathways and secondary damage in experimental spinal cord trauma. Shock. 2008;30(3):231–40. doi: 10.1097/shk.0b013e318162d290. [DOI] [PubMed] [Google Scholar]
- 95.U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. July 2005 <http://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf> (viewed Nov. 19, 2014).
- 96.Mahony C, Erskine L, Niven J, Greig NH, Figg WD, Vargesson N. Pomalidomide is nonteratogenic in chicken and zebrafish embryos and nonneurotoxic in vitro. Proc Natl Acad Sci U S A. 2013;110(31):12703–8. doi: 10.1073/pnas.1307684110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vargesson N, Mahony C, Erskine L, Niven J, Greig NH, Figg WD. Reply to D’Amato et al. and Zeldis et al.: Screening of thalidomide derivatives in chicken and zebrafish embryos. Proc Natl Acad Sci U S A. 2013;110(50):E4820. doi: 10.1073/pnas.1318475110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16(3):378–84. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 99.Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, et al. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–56. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Becker RE, Greig NH, Giacobini E, Schneider LS, Ferrucci L. A new roadmap for drug development for Alzheimer's disease. Nat Rev Drug Discov. 2014;13(2):156. doi: 10.1038/nrd3842-c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ignatowski TA, Spengler RN, Dhandapani KM, Folkersma H, Butterworth RF, Tobinick E. Perispinal etanercept for post-stroke neurological and cognitive dysfunction: scientific rationale and current evidence. CNS Drugs. 2014;28:679–97. doi: 10.1007/s40263-014-0174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Morris GP, Clark IA, Zinn R, Vissel B. Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Mem. 2013;105:40–53. doi: 10.1016/j.nlm.2013.07.002. [DOI] [PubMed] [Google Scholar]