

Abstract
Overexpression or amplification of the RSF1 gene has been associated with poor prognosis in various human cancers, including ovarian cancer. In previous work, RSF1 was identified as an amplified gene that facilitated the development of paclitaxel-resistant ovarian cancer. In the present study, we further demonstrated that RSF1 expression inversely correlated with paclitaxel response in patients with ovarian cancer and the mouse xenograft model. In addition, RSF1-overexpressing paclitaxel-resistant ovarian cancer cell lines were found to express elevated levels of genes regulated by NF-κB, including some involved with the evasion of apoptosis (CFLAR, XIAP, BCL2, and BCL2L1) and inflammation (PTGS2). In addition, ectopic expression of RSF1 using Tet-off inducible SKOV3 cells significantly enhanced NF-κB–dependent gene expression and transcriptional activation of NF-κB. An RSF1 knockdown using short hairpin RNAs suppressed these same pathways. Moreover, pretreatment with NF-κB inhibitors or downregulation of NF-κB–regulated gene expression considerably enhanced paclitaxel sensitivity in RSF1-overexpressing OVCAR3 and/or RSF1-induced SKOV3 cells. A coimmunoprecipitation assay revealed that RSF1 interacts with NF-κB and CREB-binding protein, a ubiquitous coactivator for NF-κB. Recruitment of RSF1 to the NF-κB binding element in the PTGS2 and XIAP promoters was demonstrated by the chromatin immunoprecipitation assay. Furthermore, hSNF2H, a well-known binding partner of RSF1, was partially involved in the interaction between RSF1 and NF-κB. Taken together, these data suggest that RSF1 may function as a coactivator for NF-κB, consequently augmenting expression of genes necessary for the development of chemoresistance in ovarian cancer cells.
Introduction
Ovarian cancer is the leading cause of death among gynecologic malignancies. Most patients present with advanced stages (stages III–IV) due to the absence of clinically effective screening methods (1). In these cases, combined treatment with surgery and chemotherapy is necessary. First-line chemotherapy with platinum drugs and taxanes yields a response rate of more than 80%; however, most patients will have a recurrence (2). The recurrent cancers are frequently drug resistant and are fatal in the majority of women. Chemotherapeutic resistance is one of the most important prognostic factor for ovarian cancer (3).
RSF1, which is known as a histone-binding protein that interacts with hSNF2H (SMARCA5), has been demonstrated to play an important role in chromatin remodeling and transcriptional regulation (4). Increasing evidence suggests that the RSF1 gene is amplified and/or overexpressed in various cancers, including ovarian (5), breast (6), bladder (7), esophageal (8), lung (9), colon cancer (10), and head and neck cancers (11). Elevated levels of RSF1 are correlated with poor prognosis (12). Inhibition of RSF1 was reported to reduce proliferation of cancer cells (13), suggesting an important role for RSF1 amplification and/or overexpression in the maintenance of cell survival and growth. However, the molecular role of RSF1 in cancer development and progression remains poorly understood. Previously, we found that RSF1 is frequently expressed and upregulated in ovarian cancer cells (6). In addition, RSF1 has a role in mediating disease progression and aggressive clinical behavior in patients with ovarian cancer (6, 14). It has also been demonstrated that RSF1 contributes to paclitaxel resistance, and the formation of the RSF1/hSNF2H complex is required for inducing this phenotype (15). Interaction network analysis using RSF1-regulated genes identified several molecular hubs in the functional network that may contribute to drug resistance, including NF-κB and Akt (15). RSF1, also known as hepatitis B X-antigen-associated protein (HBXAP), was originally identified as a factor interacting with the hepatitis B virus (HBV)-X (HBX) protein (16). HBXAP/RSF1 was found to increase HBV transcription in an HBX protein-dependent manner (17), and it was suggested at that time that HBXAP/RSF1 regulates the transcriptional activity of NF-κB (18, 19).
NF-κB transcription factors are involved in disparate processes such as inflammation (20), growth and development (21), and drug resistance (22). Many human cancers, including ovarian cancer, possess high levels of constitutive NF-κB activity, which can be further elevated by some anticancer drugs and radiation (23). Indeed, an oncoproteomic analysis study revealed that the NF-κB pathway is activated in recurrent ovarian carcinoma (24, 25). Activated NF-κB seems to trigger a series of molecular reactions, including upregulation of antiapoptotic protein-encoding genes (26) that induce cancer chemoresistance. High NF-κB activity has been identified in drug-resistant cancer cells, and ectopic expression of NF-κB can block anticancer drug-induced apoptosis (27-30).
Materials and Methods
Materials
RPMI-1640 medium, FBS, penicillin (100 U/mL), and streptomycin sulfate (100 (μg/mL) are from Life Technologies. The MTT, dimethyl sulfoxide (DMSO), RNase A, leupeptin, aprotinin, phenylmethylsulfonylfluoride (PMSF), dithiothreitol, and Triton X-100 were purchased from Sigma-Aldrich Co. CREB-binding protein (CBP), PTGS2, CFLAR, BCL2, BCL2L1, NF-κB p65 subunit, PARP, and β-actin antibodies were purchased from Santa Cruz Biotechnology, Inc. XIAP antibody was purchased from BD Biosciences. RSF1 and hSNF2H antibodies were purchased from Upstate. The RNA Extraction Kit was purchased from Intron Biotechnology. The Luciferase Assay Kit was purchased from Promega. pNF-κB–Luc reporter plasmid was purchased from BD Biosciences. Lipofectamine was purchased from Invitrogen. RSF1, PTGS2, CFLAR, XIAP, BCL2, BCL2L1, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) oligonucleotide primers were purchased from Bioneer Technology. Paclitaxel was purchased from A.G. Scientific, Inc. The IκB inhibitor Bay 11-7082 and the proteasome inhibitor MG132 were obtained from Calbiochem. PureProteome protein A magnetic beads were purchased from Millipore.
Cell cultures
Ovarian cancer cells, including SKOV3, OVCAR3, and A2780 cells, were originally purchased from American Type Culture Collection. The cell lines were authenticated by morphology, growth characteristics, and DNA fingerprinting. The short tandem repeat DNA profiling was performed using the AmplFLSTR Identifiler PCR Amplification Kit at Korean Cell Line Bank. Paclitaxel-resistant ovarian cancer cell lines (SKOV3TR and OVCAR3TR) were generated by selecting the viable SKOV3 and OVCAR3 cells 3 months after exposure to paclitaxel (0.25−0.5 μmol/L). Cells were cultured in RPMI-1640 supplemented with 5% FBS, penicillin (100 U/mL), and streptomycin sulfate (100 μg/mL; Life Technologies). Paclitaxel, MG132, and Bay 11-7082 were dissolved in DMSO. Further dilutions were performed in cell culture media. DMSO was used as a vehicle control throughout the study.
Gene expression and patient data
Patients in the Cambridge Translational Cancer Research Ovarian Study 01(CTCR–OV01) had been recruited from 2002 to 2004 and had histologically confirmed advanced (stages III and IV) epithelial ovarian cancer. All tissues had been biopsied before start of neoadjuvant chemotherapy. Patients had been randomly assigned to undergo either three cycles of carboplatin (AUC 7) or paclitaxel (175 mg/m2). Treatment response had been estimated using serum CA125 levels after three cycles of single-agent treatment. Treatment-sensitive patients were defined as those experienced more than a 50% decrease in serum CA125 levels (5). The gene expression data from biopsies was generated by using Affymetrix GeneChip human genome U133A 2.0 platform and available from the National Center for Biotechnology Information Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo; accession ID, GSE15622).
Animal xenograft studies
Five- to 6-week-old female BALB/c athymic nude mice (Orient-Bio Inc.) were inoculated subcutaneously with OVCAR3 cells (1 × 106) resuspended in Matrigel (BD Biosciences). When the tumors reached 80 to 100 mm3 in volume (approximately 14 days), the animals were randomly divided into four groups consisting of at least three mice per group (day 0). Intratumoral administration of RSF1 short hairpin RNA (shRNA 10 μg/kg) or control shRNA (10 μg/kg) was followed by intraperitoneal injections of paclitaxel (10 mg/kg) or same volume of vehicle. shRNA and paclitaxel were injected everyday for 14 days. The length and width of the tumor mass as well as the mouse weight were measured every 3 days and tumor volume was calculated with the equation 1/2 (length × width2). At the end of the experiment the animal were sacrificed, and the tumor weights (g) were measured. Animal treatment and maintenance were conducted in accordance with guidelines approved by the Animal Care and Use Committee of Kyung Hee University (Seoul, South Korea).
RSF1 inducible cell clones
The full-length RSF1 tagged with a V5 epitope tag at the COOH-terminal was cloned into the Tet-off expression vector, pTRE-hygro (Clontech). Parental SKOV3 cells were first transfected with a tetracycline-controlled transactivator (tTA) expression vector, and the tTA stable clones were selected by G418 (300 μg/mL). The inducible RSF1 vector was then introduced into the SKOV3–tTA cells. Stable transfectants were selected in 150 μg/mL hygromycin and then subsequently maintained in culture medium containing 300 μg/mL G418, 150 μg/mL hygromycin, and 2 μg/mL doxycycline.
Cytotoxicity test
Cytotoxicity was estimated using the MTT assay. Cells were seeded in 96-well plates at a density of 5 × 103 cells per well and incubated for 24 hours. To examine the growth inhibitory effect of paclitaxel, cells were treated with paclitaxel for 48 hours. The optical density at 540 nm was determined using a microplate spectrophotometer (Molecular Devices) to determine the cell viability.
RNA isolation and real-time RT-PCR
Total RNA was prepared using Easy Blue kits (Intron Biotechnology), according to the manufacturer's instructions. Total RNA (1 μg) was reverse transcribed into first-strand cDNA (Amersham Pharmacia Biotech) following the manufacturer's procedure. The synthesized cDNA was used as a template for PCR amplification. Real-time PCR was performed using a Thermal Cycler Dice Real Time PCR System (Takara Bio Inc.). The primers used for SYBR Green real-time reverse transcription (RT)-PCR were listed in Supplementary Table S1. Mean cycle threshold (Ct) of the gene of interest was calculated from triplicate measurements and normalized to the mean Ct of a control gene, GAPDH.
Western blot analysis
The cells were washed with ice-cold PBS and extracted in protein lysis buffer (Intron Biotechnology). The protein concentration was determined by the Bradford assay. The cell lysates were mixed with an equal volume of 5× SDS sample buffer, boiled for 4 minutes and then separated on 10% to 12% SDS–PAGE. After electrophoresis, the proteins were transferred to polyvinylidene difluoride (PVDF) membranes. The membrane was immunoblotted using specific primary antibodies at 4°C overnight. After washing, the signals were detected with horseradish peroxidase–conjugated secondary antibody for 1 hour and visualized using the ECL chemiluminescent system (Amersham Pharmacia Biotech). Following three washes in TBS-T, immunopositive bands were visualized by enhanced chemiluminescence and exposed to Image Quant LAS-4000 (GE Healthcare Life Sciences).
Gene knockdown using siRNA and shRNA
Two shRNAs for RSF1 were purchased from Sigma-Aldrich Co. hSNF2H-specific siRNAs and control siRNA were obtained from Bioneer Technology. The cells were transfected with shRNA or siRNA using lipofectamine (Invitrogen) according to the manufacturer's suggested protocol. Briefly, cells were plated in 6-well culture dishes and allowed to attach and grow for 24 hours before transfection. Each transfection mixture was prepared by mixing the shRNA or siRNA with lipofectamine in serum-free Opti-Modified Eagle Medium (Invitrogen). The mixtures were incubated for 15 minutes at room temperature. The transfection mixture was slowly added to the cells, which were allowed to recover for an additional 24 hours before experimental treatments.
Luciferase assay
The cells were transfected with the NF-κB–Luc reporter plasmid (Clontech) using lipofectamine (Invitrogen) in accordance with the manufacturer's instructions. Following incubation with the transfection reagent for 24 hours, cells were lysed, and the luciferase activity was assessed using the dual luciferase assay system (Promega) and luminometer (PerkinElmer Cetus).
Immunoprecipitation assay
Cells were lysed in a high-salt buffer. Equal amounts of lysates were immunoprecipitated with 1 μg of the RSF1, NF-κB p65, or CBP antibody and 30 μL of protein A agarose using constant rotation overnight. Beads were washed with 10 mmol/L HEPES (pH 7.9), 1 mmol/L EDTA, 150 mmol/L NaCl, and 1% Nonidet P-40 twice with 400 μL each. The precipitated proteins were eluted with 30 μL of SDS–PAGE sample buffer and boiled for 10 minutes. The eluted proteins were electrophoresed by 10% SDS–PAGE, transferred to PVDF membranes, and probed with anti-RSF1, CBP, NF-κB p65 subunit, and hSNF2H antibodies. The signals were visualized using an ECL chemiluminescent system (Amersham Pharmacia Biotech). Following three washes in TBS-T, immunopositive bands were visualized by enhanced chemiluminescence and exposed to Image Quant LAS-4000 (GE Healthcare Life Sciences).
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) was performed using a ChIP Assay Kit, according to the manufacturer's protocol (Upstate). Chromatin solutions were precipitated using anti-p65 for 12 to 16 hours at 4°C. Antibody–protein complexes were isolated by immunoprecipitation with blocked protein A agarose and washed extensively. DNA was extracted with phenol/chloroform, precipitated with ethanol and analyzed by PCR. The immunoprecipitated sample DNA was compared with the input DNA The primers used to amplify a distinct region of the PTGS2 (COX2) promoter with putative NF-κB-binding sites were 5′-CGA TGC TAA ACG ACG TCA CAT TGT GCA-3′ and 5′-CTC CAG AGC AGA ATG AGC TAC AGA CAT-3′. The primers used to amplify a distinct region of the XIAP promoter were 5′-CCC ATG CAC ACT TCC CAA CT-3′ and 5′-TTC TCC CCC ATG GAT GTC CCA T-3′. PCRs were performed for 25 to 30 cycles using a standard protocol. The amplified products were subjected to 2% agarose gel electrophoresis and visualized by ethidium bromide staining and UV irradiation.
Statistical analysis
Statistical data are presented as the mean ± SD of three individual experiments preformed in triplicate. Statistical analysis was carried out using the Student t test or one-way ANOVA, and the level of significance was set at a P value of <0.05.
Results
RSF1 expression and paclitaxel response in patients with ovarian cancer and the mouse xenograft model
Through previous work, we determined that knockdown of RSF1 sensitized ovarian cancer cells to paclitaxel, but not carboplatin, and RSF1 was upregulated in paclitaxel-resistant ovarian cancer cells and primary ovarian carcinoma tissues (15). To further evaluate the clinical significance of RSF1 expression for paclitaxel response, a correlation between RSF1 expression and paclitaxel response was investigated in patients with ovarian cancer and mouse xenograft models (Fig. 1). We evaluated microarray expression data from a prospective clinical trial (CTCR-OV01) in which patients were randomized to paclitaxel monotherapy. Patients who showed paclitaxel resistance had a significantly higher expression of RSF1 (P=0.02) compared with those who showed no paclitaxel response (Fig. 1A). In addition, intratumoral administration of RSF1 shRNA resulted in a significant improvement in paclitaxel sensitivity in ovarian cancer xenograft-bearing mice (Fig. 1B and Supplementary. Fig. S1). These data suggest that RSF1 can be a target to improve paclitaxel sensitivity of drug-resistant tumor in vivo.
Figure 1.
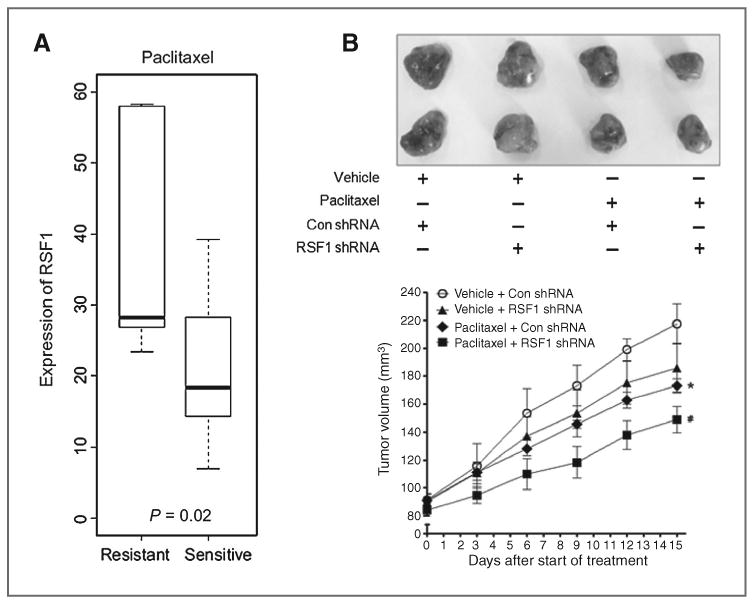
RSF1 expression and response to paclitaxel in patients with ovarian cancer and mouse xenograft models. A, expression level of RSF1 in patients with ovarian cancer in the CTCR-OV01 trial cohort. P values were determined by the Student t test. Horizontal lines, medians, boxes extend from the 25th to 75th percentiles, and error bars extend to the 10th and 90th percentiles. B, intratumoral administration of RSF1 shRNA (10 μg/kg) or control shRNA (Con shRNA; 10 μg/kg) was followed by intraperitoneal injections of paclitaxel (10 mg/kg) or same volume of vehicle. shRNA and paclitaxel were injected everyday for 14 days. Tumor sizes, relative mean volume ± SD. *, P < 0.05 compared with the vehicle treatment group with control shRNA. #, P < 0.05 compared with the paclitaxel treatment group with RSF1 shRNA.
Enhanced expression of NF-κB–dependent genes in paclitaxel-resistant ovarian cancer cells
To probe whether RSF1 expression is correlated with NF-κB–dependent gene expression in ovarian cancer cells, we performed Western blots on two groups of ovarian cancer cells, parental cells (SKOV3 and OVCAR3) and their established paclitaxel-resistant sublines (SKOV3TR and OVCAR3TR). As we had previously reported, RSF1 protein levels were significantly higher in paclitaxel-resistant cells than their parental cells (15). Importantly, RSF1-overexpressing paclitaxel-resistant cells were found to express high levels of NF-κB–dependent genes involved in the evasion of apoptosis (CFLAR, XIAP, BCL2, and BCL2L1) and inflammation (PTGS2; Fig. 2A). In a parallel experiment, real-time RT-PCR was used to determine whether the increased expression of proteins from NF-κB–dependent antiapoptotic and inflammatory genes was correlated with levels of the corresponding mRNA (Fig. 2B). As with proteins, the mRNA levels of NF-κB–dependent genes were higher in all three of the RSF1-overexpressing paclitaxel-resistant sublines compared with the respective parental cells. These results suggest that elevated levels of RSF1, which is likely associated with ovarian cancer paclitaxel resistance, is correlated with increased levels of NF-κB–regulated genes in ovarian cancer cells.
Figure 2.
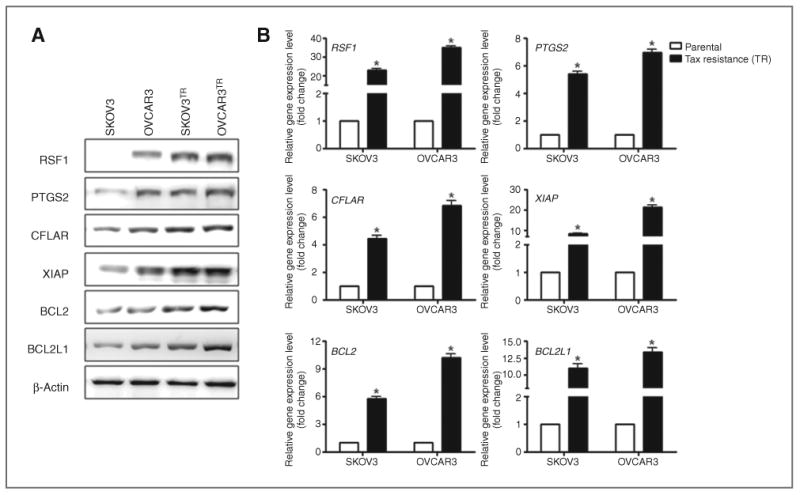
Expression of NF-κB–dependent genes in paclitaxel-resistant ovarian cancer cells. A, total protein was isolated from SKOV3, OVCAR3, SKOV3TR, and OVCAR3TR cells. Levels of RSF1, CFLAR, BCL2, BCL2L1, and PTGS2 protein were analyzed by Western blotting using specific antibodies. Expression levels were normalized with β-actin. Data are representative of three different experiments. B, total RNA was isolated from SKOV3, OVCAR3, SKOV3TR, and OVCAR3TR cells. mRNA expression of RSF1, CFLAR, BCL2, BCL2L1, and PTGS2 was analyzed by real-time RT-PCR. Data were normalized to GAPDH mRNA expression. The data are representative of three different experiments and are shown as the mean ± SD. *, P < 0.05 compared with the parental cells group.
RSF1 expression enhances NF-κB–dependent gene expression in ovarian cancer cells
Next, we asked whether induction of RSF1 expression enhanced the protein and mRNA levels of the antiapoptotic and inflammatory genes we probed previously. We used Tet-off RSF1 inducible SKOV3 ovarian cancer cells because they did not harbor RSF1 amplification or express a detectable level of endogenous RSF1 (13). Western blot analysis of lysates from these cells demonstrated that RSF1 protein was detected 12 and 24 hours after induction (Fig. 3A). Following induction of RSF1 in these SKOV3 cells, elevated protein levels of NF-κB–dependent genes were detected as well. RSF1 expression also increased the mRNA expression of NF-κB–regulated genes (Fig. 3B). To further investigate the role of RSF1 expression in NF-κB–dependent genes regulation, we transfected OVCAR3 cells with two independent shRNAs having satisfactory knockdown efficiency to reduce levels of RSF1. OVCAR3 cells are known to harbor a high level of RSF1 amplification and overexpression. As shown in Fig. 3C and D, reduced RSF1 expression significantly decreased the expression of inflammatory and antiapoptotic factors at both protein and mRNA levels.
Figure 3.
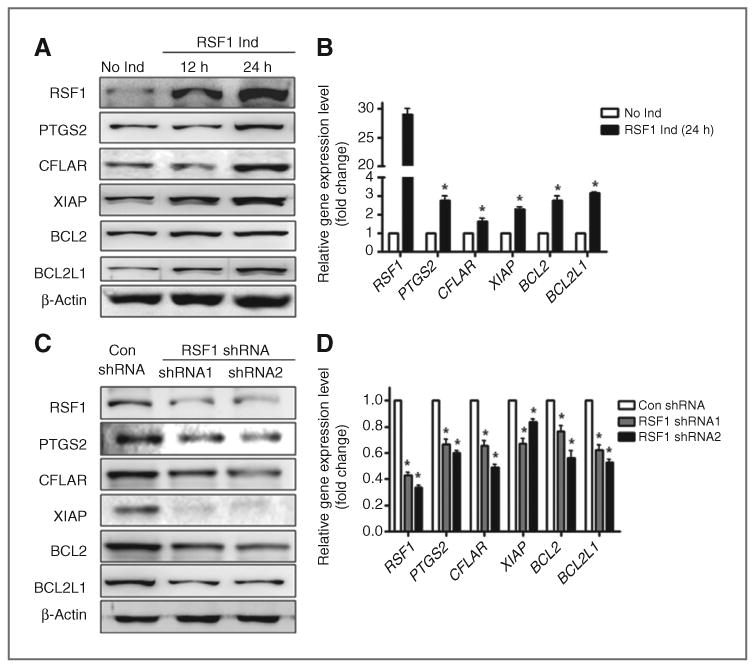
Effect of RSF1 expression on NF-κB–dependent genes expression in ovarian cancercells. A and B, SKOV3 cells with a Tet-off RSF1 induction system were used. A, levels of RSF1, CFLAR, BCL2, BCL2 L1, and PTGS2 were analyzed by Western blotting with specific antibodies. Expression levels were normalized to β-actin. Data are representative of three different experiments. B, the mRNA expression of RSF1, CFLAR, BCL2, BCL2L1, and PTGS2 was analyzed by real-time RT-PCR. Data were normalized to GAPDH mRNA expression. C and D, OVCAR3 cells were transfected with specific RSF1 shRNA1, 2, or control shRNA. C, levels of RSF1, CFLAR, BCL2, BCL2 L1, and PTGS2 were analyzed by Western blotting with specific antibodies. Expression levels were normalized to β-actin. D, the mRNA expression of RSF1, CFLAR, BCL2, BCL2L1, and PTGS2 was analyzed by real-time RT-PCR. Data were normalized to GAPDH mRNA expression. Data are representative of three different experiments and are shown as the mean ± SD. *, P < 0.05 compared with the RSF1 no induction group or the control shRNA group.
NF-κB activation by RSF1 is associated with paclitaxel resistance in ovarian cancer
To investigate whether RSF1 regulates transcriptional activity of NF-κB, we performed a luciferase assay using an NF-κB response element–derived reporter (pNF-κB–Luc). As shown in Fig. 4A, RSF1-induced cells showed markedly increased luciferase activity compared with RSF1-noninduced cells. In contrast, reducing RSF1 using shRNAs led to a significant decrease in the transcriptional activity of NF-κB (Fig. 4B). However, we observed no significant change in NF-κB mRNA levels after RSF1 induction (Supplementary Fig. S2). Thus, these data suggest that RSF1 functions as a positive regulator for NF-κB transcriptional activity.
Figure 4.
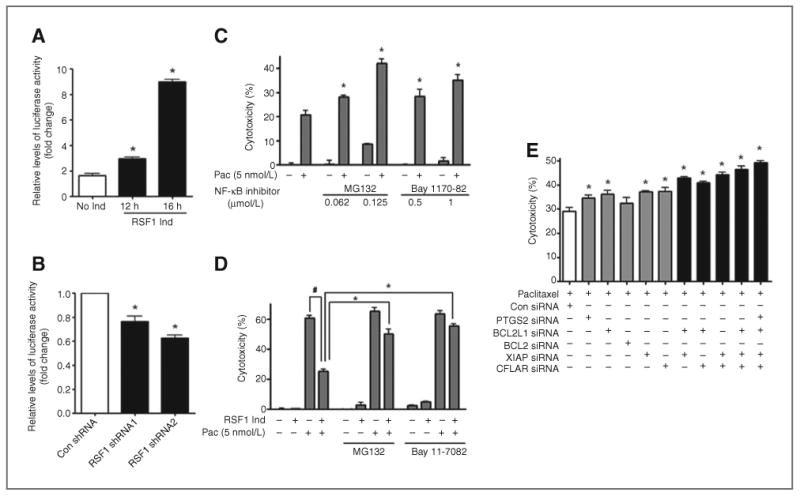
Involvement of RSF1-induced NF-κB activation in paclitaxel resistance. A, SKOV3 cells with a Tet-off RSF1 induction system were used. The cells were transiently transfected with a pNF-κB–Luc vector, and luciferase activity was assessed. B, OVCAR3 cells were transiently cotransfected with RSF1 shRNAs and a pNF-κB–Luc vector, and luciferase activity was assessed. Data are representative of three different experiments and are shown as the mean ± SD. *, P < 0.05 compared with the RSF1 no induction group or the control shRNA group. C, OVCAR3 cells were pretreated with the IκBα inhibitor, Bay 11-7082 (0.5 or 1 μmol/L), or the proteasome inhibitor, MG132 (0.062 or 0.125 μmol/L), for 1 hour and then treated with paclitaxel (5 nmol/L). After 48 hours, cytotoxicity was examined using an MTT assay. *, P < 0.05 compared with the group untreated with an inhibitor. #, P < 0.05 compared with the paclitaxel treatment group with no RSF1 induction. D, SKOV3 cells with a Tet-off RSF1 induction system were used. The cells were treated with paclitaxel (5 nmol/L) in the presence or absence of Bay 11-7082 (1 μmol/L) or MG132 (0.125 μmol/L) for 48 hours. Cytotoxicity was examined using an MTT assay. *, P < 0.05 compared with a paclitaxel-treated but inhibitor-untreated group. E, OVCAR3 cells were transfected with specific siRNA (CFLAR, XIAP, BCL2 and BCL2L1, and PTGS2) or control siRNA. After 24 hours, cells were treated with paclitaxel (5 nmol/L). After 48 hours, cytotoxicity was determined using an MTT assay. *, P < 0.05 compared with the control siRNA transfected group. Data are representative of three different experiments and are shown as the mean ± SD.
Using NF-κB pathway inhibitors Bay 11-7082 and MG132, we next investigated whether NF-κB activation by RSF1 is associated with RSF1-mediated paclitaxel resistance. Treating RSF1-overexpressing OVCAR3 cells with the NF-κB inhibitor before paclitaxel treatment was associated with a dramatic increase of cell death (Fig. 4C). We also observed that RSF1 induction markedly inhibits SKOV3 cell death by paclitaxel, but pretreating the cells with an NF-κB inhibitor significantly reduced the RSF1-induced paclitaxel resistance of the SKOV3 cells (Fig. 4D). Furthermore, to determine whether NF-κB–dependent genes are potentially involved in the paclitaxel resistance of ovarian cancer cells, we investigated paclitaxel-induced cell death following transfection of siRNAs to knockdown XIAP, CFLAR, BCL2, BCL2L, PTGS2. As shown in Fig. 4E, the knockdown of any of the five genes led to a mild increase in cytotoxic activity. In addition, simultaneous targeting of the genes by selected siRNA combinations showed enhanced cytotoxicity compared with the single siRNA treatment. These data suggest that many of antiapoptotic and prosurvival genes regulated by the NF-κB pathway may work together in RSF1-induced chemoresistance of ovarian cancer cells.
RSF1 interacts with NF-κB in chemoresistant ovarian cancer cells
A previous study suggested that HBXAP colocalizes with NF-κB in the nuclei of 293T cells (18). In this study, we examined whether RSF1 and NF-κB interact with each other in ovarian cancer cells using a coimmunoprecipitation assay. As shown in Fig. 5A, inducing RSF1 expression increased the interaction between NF-κB and RSF1. Interaction between RSF1 and NF-κB was also observed in OVCAR3 cells overexpressing endogenous RSF1, but not in A2780 cells, which barely express RSF1. An association between RSF1 and NF-κB was also apparent in a reverse immunoprecipitation using NF-κB p65 antibody followed by immunoblotting with a RSF1 antibody (Fig. 5B). In addition, RSF1 was found to interact with CBP, which is a ubiquitous coactivator for NF-κB in RSF1-induced SKOV3 and OVCAR3 cells (Fig. 5C). Importantly, increased interaction between RSF1 and NF-κB was observed in RSF1-overexpressing SKOV3TR and OVCAR3TR cells (Fig. 6A), suggesting the biologic significance of the interaction in chemoresistant ovarian cancer cells. We next performed a ChIP assay to further confirm the mechanism by which RSF1 regulates PTGS2 and XIAP expression using transcriptional activation of NF-κB. As shown in Fig. 6B, RSF1 binds to an NF-κB element in the PTGS2 and XIAP promoters of SKOV3TR cells. High binding of CBP to an NF-κB element was also observed. These data indicate that a RSF1 and CBP complex may increase PTGS2 and XIAP transcription by enhancing NF-κB binding to the promoters in chemoresistant ovarian cancer cells.
Figure 5.
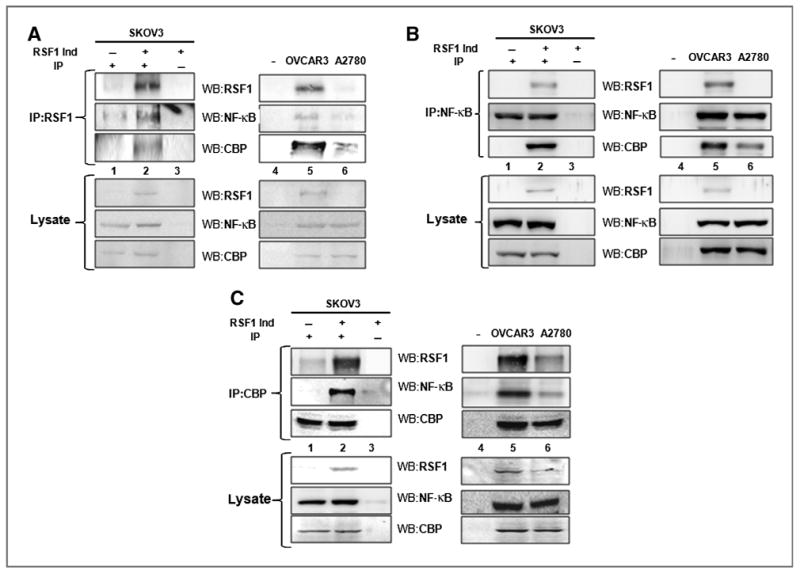
Interaction between RSF1 and NF-κB in ovarian cancer cells. A, the protein lysate was used to pull-down RSF1 using an anti-RSF1 antibody. Samples were analyzed by Western blotting using anti-RSF1, NF-κB p65 subunit, and CBP-specific antibodies. B, the reverse immunoprecipitation (IP) was also performed using the anti–NF-κB p65 subunit antibody followed by Western blotting using anti-RSF1, NF-κB, and CBP-specific antibodies. C, the reverse immunoprecipitation was also performed using an anti-CBP antibody followed by Western blotting using anti-RSF1, NF-κB p65 subunit, and CBP-specific antibodies. Lane 1, immunoprecipitation with indicated antibody in RSF1-noninduced SKOV3 cells; lane 2, immunoprecipitation with indicated antibody in RSF1-induced SKOV3 cells; lane 3, immunoprecipitation with control immunoglobulin G (IgG) in RSF1-induced SKOV3 cells; lane 4, no cells with control IgG; lane 5, immunoprecipitation with indicated antibody in RSF1-overexpressing OVCAR3 cells; and lane 6, immunoprecipitation with indicated antibody in A2780 cells, which barely express RSF1. Data are representative of three different experiments.
Figure 6.
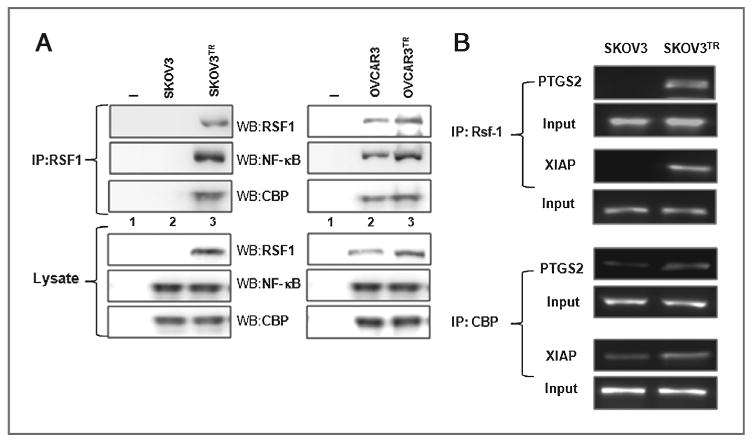
Binding of RSF1 to the NF-κB element in the PTGS2 and XIAP promoters in paclitaxel-resistant ovarian cancer cells. A, the protein lysate was used to pull-down RSF1 using an anti-RSF1 antibody. Samples were analyzed by Western blotting using anti-RSF1, NF-κB p65 subunit, and CBP-specific antibodies. Lane 1, no cells with control IgG; lane 2, immunoprecipitation with indicated antibody in parental cells (SKOV3 and OVCAR3); and lane 3, immunoprecipitation with indicated antibody in established paclitaxel-resistant sublines (SKOV3TR and OVCAR3TR). B, RSF1 and CBP binding to the PTGS2 or XIAP promoter was determined by the ChIP assay. Cross-linked chromatin fragments were immunoprecipitated with an antibody against RSF1 or CBP, and the purified DNA was amplified by PCR using primers specific for PTGS2 or XIAP promoters. The promoter was not detected when normal IgA was used or antibody was omitted from the immunoprecipitation (IP) step. Data are representative of three different experiments.
hSNF2H is involved in the interaction between RSF1 and NF-κB in chemoresistant ovarian cancer cells
Previously, we showed that downregulation of hSNF2H or disruption of the hSNF2H and RSF1 interaction enhanced paclitaxel sensitivity in tumor cells with elevated RSF1 levels (15). Thus, we investigated whether hSNF2H is associated with the interaction between NF-κB and RSF1 in chemoresistant ovarian cancer cells. A coimmunoprecipitation assay revealed that NF-κB interacts with hSNF2H as well as CBP and RSF1 in RSF1-induced SKOV3 and OVCAR3 cells (Fig. 7A). In addition, we investigated whether hSNF2H mediates the interaction between RSF1 and the NF-κB subunit p65 using hSNF2H siRNA As shown in Fig. 7B and C, reducing hSNF2H expression suppressed the interaction between RSF1 and NF-κB in RSF1-induced SKOV3 cells and paclitaxel-resistant SKOV3 and OVCAR3TR cells.
Figure 7.
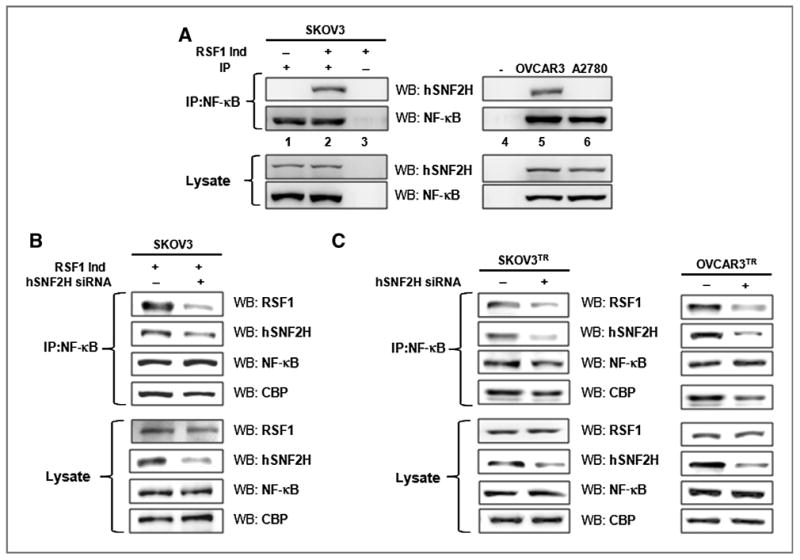
Involvement of hSNF2H in interaction between RSF1 and NF-κB. A, an anti-p65 antibody was used to pull-down associated proteins from a protein lysate. Samples were analyzed by Western blotting using anti-RSF1, hSNF2H, NF-κB p65 subunit, and CBP-specific antibodies. Lane 1, immunoprecipitation (IP) with indicated antibody in RSF1-noninduced SKOV3 cells; lane 2, immunoprecipitation with indicated antibody in RSF1-induced SKOV3 cells; lane 3, immunoprecipitation with control IgG in RSF1-induced SKOV3 cells; lane 4, no cells with control IgG; lane 5, immunoprecipitation with indicated antibody in RSF1-overexpressing OVCAR3 cells; and lane 6, immunoprecipitation with indicated antibody in A2780 cells, which barely express RSF1. B and C, RSF1-induced SKOV3, SKOV3TR, and OVCAR3TR cells were transfected with hSNF2H siRNA (50 nmol/L) or control siRNA, and protein lysate was used to perform an immunoprecipitation with an anti–NF-κB antibody. Samples were analyzed by Western blotting using anti-RSF1, hSNF2H, NF-κB, and CBP-specific antibodies. Data are representative of three different experiments.
Discussion
Chemoresistance is the major clinical obstacle for the treatment of patients with ovarian cancer, and a better understanding of the molecular mechanisms involved in this cellular protection is required to develop more successful therapies. Paclitaxel has been used as a front-line chemotherapeutic agent for several types of human cancer, including ovarian cancer. However, the drug frequently induces resistance, and the molecular mechanisms of resistance to paclitaxel remain poorly understood. Mounting evidence indicates that PI3K/Akt and the NF-κB signaling pathways are involved in development of cancer chemoresistance by regulating the expression of genes critical for cell survival, thereby evading apoptosis (27–29). Indeed, paclitaxel has been demonstrated to induce constitutive NF-κB activation and lead to chemoresistance in several human cancer cells, including ovarian cancer (31–33).
Previously, we found that RSF1 was upregulated in paclitaxel-resistant ovarian cancer cell lines (15), and RSF1 immunoreactivity in primary ovarian carcinoma tissues correlated with in vitro paclitaxel resistance. In addition, ectopic expression of RSF1 significantly enhanced paclitaxel resistance of ovarian cancer cells. Furthermore, cDNA microarray analysis followed by interaction network analysis revealed that RSF1 altered expression of several genes and was associated with several molecular hubs, including NF-κB, ERK (extracellular signal–regulated kinase), and Akt. However, the precise molecular mechanisms of how RSF1 contributed to ovarian cancer resistance remained poorly understood. Therefore, in our more recent work described here, we focused on the pathways regulated by RSF1 as a way to explore the potential mechanism by which RSF1 induces chemoresistance. Among the molecular hubs suggested by the previous finding (15), NF-κB has gained more attention due to its importance in the regulation of survival and antiapoptotic genes, which may contribute to paclitaxel resistance. In addition, it is notable that HBXAP/RSF1 has been shown to regulate the transcriptional activity of NF-κB (17, 18). Therefore, it was reasonable to hypothesize that inhibition of NF-κB or pathways regulated by NF-κB may be potential targets of RSF1 in chemoresistant ovarian cancer cells.
With recent advances in drug resistance, it has become evident that defects in control of cell death and apoptosis are strongly associated with chemoresistance (34, 35). The inhibitors of apoptosis proteins (IAP), including XIAP, suppress apoptosis induced by both extrinsic and intrinsic pathways through direct inhibition of effector caspases (caspases 3, 6, 7, and 9), whereas the antiapoptotic members of the BCL2 family antagonize the function of the proapoptotic members, such as BAX. Indeed, upregulation of the antiapoptotic protein BCL2 and/or downregulation of proapoptotic BAX have been correlated with increased chemoresistance (36, 37). In addition, inhibitors of caspases such as CFLAR (FLIP) and XIAP were implicated as determinants of the chemosensitivity of ovarian cancer (38, 39). NF-κB induces expression of some antiapoptotic BCL2 family proteins such as BCL2 and BCL2L1 (BCLX) as well as inhibitors of caspases like CFLAR (FLIP) and XIAP (40, 41). In the present study, we found a strong correlation between high levels of RSF1 in ovarian cancer and increased expression of NF-κB–dependent genes involved in evasion of apoptosis (CFLAR, XIAP, BCL2, and BCL2L1) and inflammation (PTGS2). In addition, we found that RSF1 stimulates the transcriptional activity of NF-κB but not mRNA expression of the NF-κB. These data suggest that RSF1 induces survival and antiapoptotic gene expression by regulating the transcriptional activity of NF-κB in ovarian cancer cells. Details of the molecular mechanism through which RSF1 regulates this transcriptional activity are poorly understood. Huang and colleagues suggested that HBXAP regulates NF-κB–mediated gene activation in normal 293T cells by influencing the DNA binding activity of NF-κB (18). Additional studies are needed to examine whether RSF1 increases the transcriptional activity of NF-κB by enhancing DNA binding of NF-κB in chemoresistant ovarian cancer cells.
Our observation that RSF1 can stimulate the transcriptional activity of NF-κB is in agreement with other findings that HBXAP/RSF1, both alone and in the presence of HBX, increases NF-κB transcription in human liver cancer HepG2 cells (17). In this study, we further tested the hypothesis that RSF-driven increases in NF-κB–mediated gene expression might confer paclitaxel resistance in ovarian cancer. Inhibition of the NF-κB pathway using NF-κB inhibitors and siRNA of NF-κB–dependent genes significantly increased paclitaxel sensitivity of RSF1-overexpressing OVCAR3 cells and/or RSF1-induced SKOV3 cells. These data suggest that involvement of the NF-κB pathway in RSF1-induced paclitaxel resistance in ovarian cancer cells. Furthermore, coimmunoprecipitation assays revealed interaction of RSF1 with NF-κB in RSF1-overexpressing ovarian cancer cells and paclitaxel-resistant cells.
Although RSF1 has been classified as a transcriptional activator in HepG2 liver cancer (17) and in ovarian cancer cells (on the basis of this work presented here), it is noteworthy that HBXAP/RSF1 also acts as a transcriptional repressor for NF-κB under different conditions. For example, upon TNF-α stimulation, HBXAP/RSF1 alone repressed the TNF-α-induced transcription activity of NF-κB in HepG2 cells, whereas HBXAP became a super coactivator in the presence of HBX (17). In human embryonic kidney 293T (HEK293T) cells, HBXAP/RSF1 repressed the transcriptional activity of NF-κB both in the absence and presence of TNF-α stimulation (17, 18). These observations indicate that HBXAP/RSF1 acts as either a transcriptional activator or repressor depending on the cell type and/or the interactions with a transcription coactivator/mediator such as HBX. In this regard, we speculated that RSF1 may interact with other transcription coactivators to stimulate NF-κB transcription in ovarian cancer cells. NF-κB–dependent gene expression involves a growing family of proteins termed transcriptional coactivators, which most likely function by facilitating or bridging the sequence-specific activators to the basal transcriptional machinery and altering chromatin structure. In the work described here, we found that RSF1 interacts with NF-κB and its ubiquitous coactivator CBP. It is generally accepted that the p65 component of NF-κB binds to the coactivator CBP and its structural homolog p300, resulting in enhanced transactivation (42, 43). For example, phosphorylation of p65 by protein kinase A stimulates NF-κB–dependent gene expression by enhancing the interaction of p65 with CBP (44). Interestingly, a possible role for CBP in chemoresistance has been reported. p300-regulated β-catenin signaling and NF-κB–specific transcriptional activity have been reported to induce ABCB1(MDR1) and BCL2L1 gene expression, which facilitate chemoresistance in breast tumor cells (45). In addition, inhibition of CBP/catenin interactions leads to eradication of drug-resistant primary leukemia in combination with conventional therapy (46). Overexpression of the CBP/p300-interacting transactivator (CITED2) in cisplatin-resistant ovarian cancer cells has also been reported (47). Hypoxia-induced tumor growth and resistance can be repressed by disrupting the interaction between HIF1a and the p300/CBP coactivator (48). In this study, no significant differences in CBP expression were observed between drug-resistant and parental cells (Supplementary Fig. S3). However, we demonstrated that RSF1 interacts with the ubiquitous NF-κB coactivator CBP as well as NF-κB. In addition, ChIP assays revealed that RSF1 and CBP are located on the consensus NF-κB binding element in the XIAP and PTGS2 promoters with NF-κB in chemoresistant ovarian cancer cells. These observations indicate that RSF1 might act as a bridging factor that facilitates an interaction among NF-κB, CBP, and a basal transcriptional complex, which, therefore, regulates NF-κB–mediated antiapoptotic protein gene expression and results in chemoresistance of ovarian cancer cells. Further experimentation is required to confirm this hypothesis.
The RSF1 protein partners with hSNF2H, also known as SMARCA5, to form an ISWI (imitation switch) chromatin remodeling complex (13). In this complex, RSF1 is known to function as a histone chaperone to modulate DNA binding activity of the RSF complex, whereas hSNF2H possesses nucleosome-dependent ATPase and helicase activities for DNA unwinding. In previous studies, we demonstrated that hSNF2H plays a role in the protumor functions of RSF1 in ovarian cancer cells. hSNF2H knockdown had little effect on paclitaxel response in the control SKOV3 cells that have a very low level of endogenous RSF1 expression (15). However, downregulation of hSNF2H or disruption of the hSNF2H and RSF1 interaction enhanced paclitaxel sensitivity of tumor cells with RSF1 upregulation (15). These data suggest that hSNF2H may play a role in paclitaxel resistance probably via the interaction with RSF1. In this regard, we have investigated whether hSNF2H is associated with RSF1/NF-κB interaction in ovarian cancer cells. Knockdown studies using hSNF2H siRNA revealed that hSNF2H was only partially involved in the interaction between RSF1 and NF-κB.
In summary, we have shown that RSF1 contributes to paclitaxel resistance of ovarian cancer. RSF1 may function as a coactivator for NF-κB, consequently augmenting expression of genes necessary for the development of chemoresistance. Furthermore, for the first time, this study suggested a molecular mechanism for RSF1-induced ovarian cancer chemoresistance.
Supplementary Material
Acknowledgments
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant (J.-H. Choi) funded by the Korea government (MEST; NRF-2010-0004306 and NRF-2013R1A2A2A01067888).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Authors' Contributions: Conception and design: K-T. Lee, I.-M. Shih, J.-H. Choi
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): Y.-I. Yang, J.-H. Ahn, K.-T. Lee, J.-H. Choi
Writing, review, and/or revision of the manuscript: I.-M. Shih, J.-H. Choi
Study supervision: J.-H. Choi
References
- 1.Ozols RF, Bookman MA, Connolly DC, Daly MB, Godwin AK, Schilder RJ, et al. Focus on epithelial ovarian cancer. Cancer Cell. 2004;5:19–24. doi: 10.1016/s1535-6108(04)00002-9. [DOI] [PubMed] [Google Scholar]
- 2.Ishibashi M, Nakayama K, Yeasmin S, Katagiri A, Iida K, Nakayama N, et al. A BTB/POZ gene, NAC-1, a tumor recurrence-associated gene, as a potential target for Taxol resistance in ovarian cancer. Clin Cancer Res. 2008;14:3149–55. doi: 10.1158/1078-0432.CCR-07-4358. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 4.Sheu JJ, Guan B, Choi JH, Lin A, Lee CH, Hsiao YT, et al. Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J Biol Chem. 2010;285:38260–9. doi: 10.1074/jbc.M110.138735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shih Ie M, Sheu JJ, Santillan A, Nakayama K, Yen MJ, Bristow RE, et al. Amplification of a chromatin remodeling gene, Rsf-1/HBXAP, in ovarian carcinoma. Proc Natl Acad Sci U S A. 2005;102:14004–9. doi: 10.1073/pnas.0504195102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mao TL, Hsu CY, Yen MJ, Gilks B, Sheu JJ, Gabrielson E, et al. Expression of Rsf-1, a chromatin-remodeling gene, in ovarian and breast carcinoma. Hum Pathol. 2006;37:1169–75. doi: 10.1016/j.humpath.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 7.Chen TJ, Huang SC, Huang HY, Wei YC, Li CF. Rsf-1/HBXAP over-expression is associated with disease-specific survival of patients with gallbladder carcinoma. APMIS. 2011;119:808–14. doi: 10.1111/j.1600-0463.2011.02808.x. [DOI] [PubMed] [Google Scholar]
- 8.Fang FM, Li CF, Huang HY, Lai MT, Chen CM, Chiu IW, et al. Over-expression of a chromatin remodeling factor, RSF-1/HBXAP, correlates with aggressive oral squamous cell carcinoma. Am J Pathol. 2011;178:2407–15. doi: 10.1016/j.ajpath.2011.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Q, Dong Q, Wang E. Rsf-1 is overexpressed in non–small cell lung cancers and regulates cyclinDI expression and ERK activity. Biochem Biophys Res Commun. 2012;420:6–10. doi: 10.1016/j.bbrc.2012.02.095. [DOI] [PubMed] [Google Scholar]
- 10.Liu S, Dong Q, Wang E. Rsf-1 overexpression correlates with poor prognosis and cell proliferation in colon cancer. Tumour Biol. 2012;33:1485–91. doi: 10.1007/s13277-012-0399-y. [DOI] [PubMed] [Google Scholar]
- 11.Tai HC, Huang HY, Lee SW, Lin CY, Sheu MJ, Chang SL, et al. Associations of Rsf-1 overexpression with poor therapeutic response and worse survival in patients with nasopharyngeal carcinoma. J Clin Pathol. 2012;65:248–53. doi: 10.1136/jclinpath-2011-200413. [DOI] [PubMed] [Google Scholar]
- 12.Liang PI, Wu LC, Sheu JJ, Wu TF, Shen KH, Wang YH, et al. Rsf-1/HBXAP overexpression is independent of gene amplification and is associated with poor outcome in patients with urinary bladder urothelial carcinoma. J Clin Pathol. 2012;65:802–7. doi: 10.1136/jclinpath-2012-200897. [DOI] [PubMed] [Google Scholar]
- 13.Sheu JJ, Choi JH, Yildiz I, Tsai FJ, Shaul Y, Wang TL, et al. The roles of human sucrose nonfermenting protein 2 homologue in the tumor-promoting functions of Rsf-1. Cancer Res. 2008;68:4050–7. doi: 10.1158/0008-5472.CAN-07-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson B, Trope CG, Wang TL, Shih Ie M. Expression of the chromatin remodeling factor Rsf-1 is upregulated in ovarian carcinoma effusions and predicts poor survival. Gynecol Oncol. 2006;103:814–9. doi: 10.1016/j.ygyno.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 15.Choi JH, Sheu JJ, Guan B, Jinawath N, Markowski P, Wang TL, et al. Functional analysis of 11q13.5 amplicon identifies Rsf-1 (HBXAP) as a gene involved in paclitaxel resistance in ovarian cancer. Cancer Res. 2009;69:1407–15. doi: 10.1158/0008-5472.CAN-08-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loyola A, Huang JY, LeRoy G, Hu S, Wang YH, Donnelly RJ, et al. Functional analysis of the subunits of the chromatin assembly factor RSF. Mol Cell Biol. 2003;23:6759–68. doi: 10.1128/MCB.23.19.6759-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shamay M, Barak O, Doitsh G, Ben-Dor I, Shaul Y. Hepatitis B virus pX interacts with HBXAP, a PHD finger protein to coactivate transcription. J Biol Chem. 2002;277:9982–8. doi: 10.1074/jbc.M111354200. [DOI] [PubMed] [Google Scholar]
- 18.Huang JY, Shen BJ, Tsai WH, Lee SO. Functional interaction between nuclear matrix-associated HBXAP and NF-kappaB. Exp Cell Res. 2004;298:133–43. doi: 10.1016/j.yexcr.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 19.Shamay M, Barak O, Shaul Y. HBXAP, a novel PHD-finger protein, possesses transcription repression activity. Genomics. 2002;79:523–9. doi: 10.1006/geno.2002.6717. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 21.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–99. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bentires-Alj M, Barbu V, Fillet M, Chariot A, Relic B, Jacobs N, et al. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene. 2003;22:90–7. doi: 10.1038/sj.onc.1206056. [DOI] [PubMed] [Google Scholar]
- 23.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 24.Jinawath N, Vasoontara C, Jinawath A, Fang X, Zhao K, Yap KL, et al. Oncoproteomic analysis reveals coupregulation of RELA and STAT5 in carboplatin-resistant ovarian carcinoma. PLoS ONE. 2010;5:e11198. doi: 10.1371/journal.pone.0011198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Cassidy J. Constitutive nuclear factor-kappa B mRNA, protein overexpression and enhanced DNA-binding activity in thymidylate synthase inhibitor-resistant tumour cells. Br J Cancer. 2003;88:624–9. doi: 10.1038/sj.bjc.6600753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006;25:6800–16. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 27.Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, et al. Inhibition of inhibitor of nuclear factor-kappaB phosphorylation ncreases the efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Clin Cancer Res. 2004;10:7645–54. doi: 10.1158/1078-0432.CCR-04-0958. [DOI] [PubMed] [Google Scholar]
- 28.Fraser M, Leung B, Jahani-Asl A, Yan X, Thompson WE, Tsang BK. Chemoresistance in human ovarian cancer: the role of apoptotic regulators. Reprod Biol Endocrinol. 2003;1:66. doi: 10.1186/1477-7827-1-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 30.Kim SH, Juhnn YS, Song YS. Akt involvement in paclitaxel chemoresistance of human ovarian cancer cells. Ann Ny Acad Sci. 2007;1095:82–9. doi: 10.1196/annals.1397.012. [DOI] [PubMed] [Google Scholar]
- 31.Huang Y, Fan W. IkappaB kinase activation is involved in regulation of paclitaxel-induced apoptosis in human tumor cell lines. Mol Pharmacol. 2002;61:105–13. doi: 10.1124/mol.61.1.105. [DOI] [PubMed] [Google Scholar]
- 32.Yang YI, Lee KT, Park HJ, Kim TJ, Choi YS, Shih Ie M, et al. Tectorigenin sensitizes paclitaxel-resistant human ovarian cancer cells through downregulation of the Akt and NFkappaB pathway. Carcinogenesis. 2012;33:2488–98. doi: 10.1093/carcin/bgs302. [DOI] [PubMed] [Google Scholar]
- 33.Bava SV, Puliappadamba VT, Deepti A, Nair A, Karunagaran D, Anto RJ. Sensitization of taxol-induced apoptosis by curcumin involves downregulation of nuclear factor-kappaB and the serine/threonine kinase Akt and is independent of tubulin polymerization. J Biol Chem. 2005;280:6301–8. doi: 10.1074/jbc.M410647200. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, et al. DNA damage-induced G2–M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4:993–7. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 35.Arts HJ, Van Der Zee AG, De Jong S, De Vries EG. Options for modulation of drug resistance in ovarian cancer. Int J Gynecol Cancer. 2000;10:47–52. doi: 10.1046/j.1525-1438.2000.99511.x. [DOI] [PubMed] [Google Scholar]
- 36.Eliopoulos AG, Kerr DJ, Herod J, Hodgkins L, Krajewski S, Reed JC, et al. The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and Bcl-2. Oncogene. 1995;11:1217–28. [PubMed] [Google Scholar]
- 37.Herod JJ, Eliopoulos AG, Warwick J, Niedobitek G, Young LS, Kerr DJ. The prognostic significance of Bcl-2 and p53 expression in ovarian carcinoma. Cancer Res. 1996;56:2178–84. [PubMed] [Google Scholar]
- 38.Sasaki H, Sheng Y, Kotsuji F, Tsang BK. Downregulation of X-linked nhibitor of apoptosis protein induces apoptosis in chemoresistant human ovarian cancer cells. Cancer Res. 2000;60:5659–66. [PubMed] [Google Scholar]
- 39.Kirchhoff S, Muller WW, Krueger A, Schmitz I, Krammer PH. TCR-mediated upregulation of c-FLIPshort correlates with resistance toward CD95-mediated apoptosis by blocking death-inducing signaling complex activity. J Immunol. 2000;165:6293–300. doi: 10.4049/jimmunol.165.11.6293. [DOI] [PubMed] [Google Scholar]
- 40.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 41.Lee JU, Hosotani R, Wada M, Doi R, Kosiba T, Fujimoto K, et al. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur J Cancer. 1999;35:1374–80. doi: 10.1016/s0959-8049(99)00134-3. [DOI] [PubMed] [Google Scholar]
- 42.Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–55. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 43.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sei U S A. 1997;94:2927–32. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–71. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 45.Bourguignon LY, Xia W, Wong G. Hyaluronan-mediated CD44 interaction with p300 and SIRT1 regulates beta-catenin signaling and NFkappaB-specific transcription activity leading to MDR1 and Bcl-xL gene expression and chemoresistance in breast tumor cells. J Biol Chem. 2009;284:2657–71. doi: 10.1074/jbc.M806708200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsieh YT, Gang EJ, Geng H, Park E, Huantes S, Chudziak D, et al. Integrin alpha4 blockade sensitizes drug-resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood. 2013;121:1814–8. doi: 10.1182/blood-2012-01-406272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yanagie H, Hisa T, Ogata A, Miyazaki A, Nonaka Y, Nishihira T, et al. Improvement of sensitivity to platinum compound with siRNA knockdown of upregulated genes in platinum complex-resistant ovarian cancer cells in vitro. Biomed Pharmacother. 2009;63:553–60. doi: 10.1016/j.biopha.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 48.Dubey R, Levin MD, Szabo LZ, Laszlo CF, Kushal S, Singh JB, et al. Suppression of tumor growth by designed dimeric epidithiodiketopiperazine targeting hypoxia-inducible transcription factor complex. J Am Chem Soc. 2013;135:4537–49. doi: 10.1021/ja400805b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.