

Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression and protein synthesis. To characterize functions of miRNAs and to assess their potential applications, we carried out an integrated multi-omics analysis to study miR-145, a miRNA that has been shown to suppress tumor growth. We employed gene expression profiling, miRNA profiling and quantitative proteomic analysis of a pancreatic cancer cell line. In our transcriptomic analysis, overexpression of miR-145 was found to suppress the expression of genes that are implicated in development of cancer such as ITGA11 and MAGEA4 in addition to previously described targets such as FSCN1, YES1 and PODXL. Based on miRNA profiling, overexpression of miR-145 also upregulated other miRNAs including miR-124, miR-133b and miR-125a-3p, all of which are implicated in suppression of tumors and are generally co-regulated with miR-145 in other cancers. Using the SILAC system, we identified miR-145-induced downregulation of several oncoproteins/cancer biomarkers including SET, RPA1, MCM2, ABCC1, SPTBN1 and SPTLC1. Luciferase assay validation carried out on a subset of downregulated candidate targets confirmed them to be novel direct targets of miR-145. Overall, this multi-omics approach provided insights into miR-145-mediated tumor suppression and could be used as a general strategy to study the targets of individual miRNAs.
Introduction
MicroRNAs (miRNAs) are short (18–24 nt) non-coding RNAs that play roles in post-transcriptional gene regulation. Mature miRNAs associate with the RNA-induced silencing complex and bind to the 3′-untranslated region (3′UTR) of mRNAs. This leads to repression of translation and/or degradation of transcripts, influencing diverse biological processes ranging from gametogenesis and embryonic development to tissue repair and aging.1–4 Evidence for a link between miRNAs and development of cancers is accumulating.5,6 Dysregulation of miRNAs has been associated with multiple hallmarks of cancer including proliferation, abnormal migration and aberrant angiogenesis.7–9 Depending on their effects on tumor growth, miRNAs can be categorized as oncogenic miRNAs or tumor suppressor miRNAs. Identifying targets of these miRNAs and unraveling their networks can improve our understanding of cancers and potentially lead to novel therapeutic strategies.
miR-145 has low to absent expression in several cancer types and is most abundant in cells of mesenchymal origin.10 Over-expressing miR-145 therapeutically has hindered tumor growth in model systems.11 For example, downregulation of miR-145 has been reported in leukemia, lymphoma, craniopharyngioma and various human solid tumors, including pancreatic, colon, esophageal, lung, breast, prostate, liver and bladder cancers.12–20 In some cancer cell lines, p53 can upregulate transcription and post-transcriptional maturation of miR-145 in response to DNA damage.21,22 In xenograft models, the reconstitution of miR-145 was demonstrated to inhibit tumor growth of pancreatic, hepatic and endometrial cancer.14,23,24 Given that miRNAs can exert their effects in biologic networks by regulating a large number of targets, it is necessary to identify miR-145 targets in a global fashion.
Different approaches have been employed to identify miRNA targets such as gene expression microarrays, RNA cross-linking immunoprecipitation and bioinformatics-based prediction algorithms, among others. One of the major limitations of transcriptomics-based methodologies is that changes in protein abundance are missed. Bioinformatics approaches generate many false-positive predictions of miRNA targets. Given the known effects of miRNAs on translation, quantitative proteomics serves as a complementary technology to measure the impact of miRNAs on intracellular protein levels.25,26 To identify the targets of miR-145 and thus to identify the mechanisms of tumor suppression by miR-145, we carried out stable isotope labeling by amino acids in cell culture (SILAC)-based proteomic profiling along with transcriptomic analysis to quantify the global changes subsequent to miR-145 overexpression in a pancreatic cancer cell line, MiaPaCa-2. Integrating the quantitative results from mRNA microarrays, miR microarrays and SILAC revealed previously unreported targets of miR-145 as well as provided novel insights into the tumor suppressive mechanisms of miR-145.
Results and discussion
Impact of miR-145 on mRNAs
miRNA-mediated post-transcriptional regulation of gene expression can result in the destabilization of target mRNA transcripts and hence decreased transcript abundance. Identifying the number and extent of mRNA transcripts that are downregulated by a miRNA in this fashion provides a foundation to understand miRNA-gene networks. Unlike bioinformatic predictions of miRNA targets, which often predict hundreds of transcripts targeted by each miRNA, the extent of experimental transcriptomic changes caused by miRNA overexpression are commonly lower by an order of magnitude.27 To obtain a global view of transcriptomic changes, we carried out mRNA microarray analysis 48 hours after transfection of miR-145 into MiaPaCa-2 cells. Fold-changes of 18522 genes were quantified (Table S1, ESI†). Most of the detected transcripts did not change significantly (Fig. 1A). Expression of 73 genes was downregulated ≥1.5-fold after miR-145 transfection while that of 11 genes was upregulated ≥1.5-fold (false discovery rate (FDR) < 1%, Table S2, ESI†). Notably, of the 73 downregulated genes, 9 mRNAs were previously described as targets of miR-145 including PODXL, ABRACL (C6orf115), NDUFA4, FSCN1, GMFB, AP1G1, YES1, TMEM9B and CBFB.28–30 Among the genes identified as miR-145 targets in our analyses, there are some that could explain miR-145 tumor suppressive activity. For instance, podocalyxin-like 1 (PODXL), is a transmembrane glycoprotein which is functionally associated with multiple cancers including breast, colon and pancreatic cancers.31–33 Fascin 1 (FSCN1) is involved in the assembly of polymerized actin and forms filipodia critical for cell motility and cancer migration.34,35 In addition to these miR-145 targets, we observed two mRNAs linked to cancer development that were downregulated by miR-145. Melanoma antigen family A 4 (MAGEA4) is a cancer/testis antigen whose germ-line expression is reactivated in cancers and belongs to the family proteins that may block binding of p53 to DNA.36,37 Overexpression of MAGEA4 has been previously noted in hepatocellular carcinoma.38 Importantly, cytotoxic T cell-mediated immunotherapy against MAGEA4 has shown efficacy in treatment of Hodgkin lymphoma.39 The other molecule linked to cancer that was downregulated by miR-145 is integrin alpha 11 (ITGA11), which is overexpressed in non-small cell lung cancer and is known to enhance tumorigenicity by dysregulating tumor-stromal interaction.40
Fig. 1.
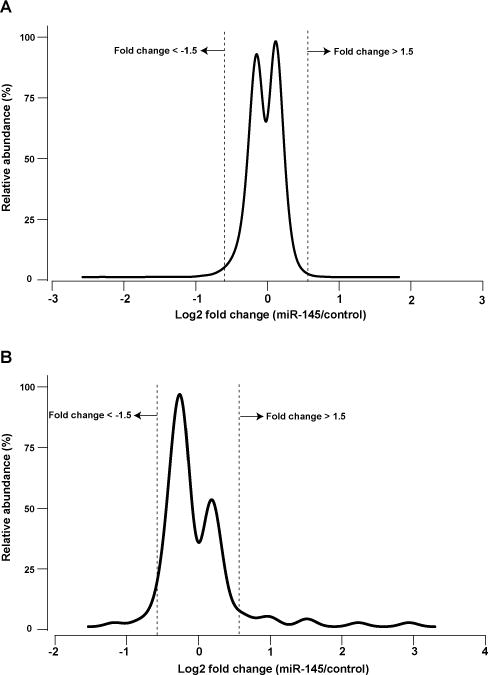
miR-145-mediated transcriptomic alterations. (A) Fold change distribution of 18 522 transcripts 48 h after miR-145 transfection in pancreatic cancer cell line MiaPaCa-2. (B) Fold change distribution of 851 miRNAs 48 h after miR-145 transfection in pancreatic cancer cell line MiaPaCa-2. As shown in A and B, majority of the transcriptome do not change significantly after miR-145 transfection.
Regulation of miRNAs by miR-145
It is documented in various cancers that the expression of miR-145 changes in parallel with that of certain other miRNAs.13,20,28,41–46 To evaluate how miR-145 might act via other miRNAs as a tumor suppressor in pancreatic cancer, we carried out miRNA profiling using microarrays to analyze the global change of miRNAs 24 and 48 hours after miR-145 transfection. The correlation between differential expression of miRNAs at 24 and 48 hours was high (r = 0.92); we focused on the analysis of the 48 hour experiment to integrate with other omics datasets that were all at the 48 hours. Totally, fold changes of 851 miRNAs were quantified, and the expression of 120 miRNAs changed significantly (FDR 5%, Fig. 1B and Table S3, ESI†). Seven miRNAs increased > 2-fold while only let-7e decreased > 2-fold (FDR < 3.0 × 10−6, Table S4, ESI†). Upregulation of miR-124 (8.7-fold), miR-133b (5.2-fold) and miR-125a-3p (3.0-fold) might be related to the observed effect of miR-145 as a tumor suppressor, and their expression often changed concomitantly with that of miR-145 in other cancers as discussed below (Table S4, ESI†). For instance, miR-124 is also known as a multifaceted tumor suppressor miRNA. In glioblastoma multiforme stem cells, miR-124 has been shown to cause cell cycle arrest and induce differentiation.47 In cholangiocarcinoma, cancer cell migration and invasion were inhibited by miR-124 overexpression.48 In esophageal squamous cell carcinoma, where miR-133b shares FSCN1 as a target with miR-145, inhibition of cancer cell growth and invasion was observed in miR-133b overexpression.28 In non-small cell lung cancer and gastric cancer, downregulation of miR-125a-3p correlates with clinical cancer invasion in adjacent lymph nodes.49,50 This suggests a potential role of miR-125a-3p in inhibiting migration of cancer cells. Taken together, miR-145 upregulates an ensemble of miRNAs, including three that have previously been reported as tumor suppressors; adding to potential mechanisms contributing to the tumor suppressive properties of miR-145 in cancers.
Impact of miR-145 on the proteome
Another important mechanism for miRNA-mediated regulation of targets is to repress protein synthesis that can occur with or without alteration of mRNA transcript abundance.51 In other words, measuring protein abundance not only reflects the ultimate impact of miRNAs on translation but also complements what transcriptomic analysis alone is not able to reveal. We decided to use quantitative proteomics to characterize proteome dynamics subsequent to miR-145 overexpression. The strategy for our SILAC proteomics study in the MiaPaCa-2 pancreatic cancer cell line is depicted in Fig. 2. The cells labeled with heavy amino acids were transfected with miR-145 while those cultured in the regular (light) medium were transfected with a scrambled RNA control. Forty-eight hours after trans-fection, the cells were harvested and proteins were extracted. We mixed lysates from heavy and light samples and subjected the samples to four different fractionation methods — in-gel digestion, strong cation exchange, off-gel peptide fractionation and off-gel protein fractionation. Via liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis on an LTQ-Orbitrap Velos mass spectrometer, we identified ~ 20 000 peptides corresponding to 2905 proteins at a 1% FDR (Table S5 and Fig. S1, ESI†). Ninety percent (2605) of these proteins were quantifiable (Fig. 3A). Membranous and nuclear proteins comprised 13.3% and 15.0%, respectively, of the total. While the majority of the proteome remained unchanged, 160 (6.1%) proteins were down-regulated ≥ 1.5-fold and 43 (1.7%) proteins were upregulated ≥ 1.5-fold. The representative lists of the most regulated proteins are given in Table S6 (ESI†). Based on TarBase 6.0, a database of experimentally validated miRNA targets, six of the downregulated proteins had been previously identified as miR-145 targets including FSCN1, SWAP70, YES1, TPM3, AP1G1 and PODXL.28,52 In addition, based on SILAC quantitation, we identified several novel miR-145 targets where the mRNA 3′UTR sequences contained perfect complementarity to the miR-145 seed sequence. This included proteins encoded by the SET, RPA1, MCM2, ABCC1, SPTLC1, SPTBN1, EP300, HAT1, DIAPH1, PRPSAP2 and RBM22 genes. The protein SET (SET) binds and inhibits protein phosphatase 2A and is known as an oncoprotein which is overexpressed in multiple cancers such as pancreatic cancer, chronic lymphocytic leukemia, B-cell non-Hodgkin lymphoma and Wilms’ tumor.53–55 Knockdown of SET inhibits tumor growth in these cancers cells.54,56 Replication protein A (RPA1) is a single-strand DNA binding protein, which is crucial for DNA replication. Expression of RPA1 is positively correlated with tumor grade and survival in patients with astrocytic tumors.57 Higher expression of RPA1 is also linked to esophageal cancer and associated with lymph node metastasis.58 Minichromosome maintenance protein 2 (MCM2) is a subunit of the hetero-hexameric DNA replication machinery, which is tightly regulated through cell cycle. It serves as a histopathological cancer bio-marker in pancreaticobiliary cancer, colon cancer, retinoblastoma and lung cancer.59–62 ATP-binding cassette subfamily C member 1 (ABCC1), also known as multidrug resistance-associated protein 1, is a neutral and anion efflux pump on plasma membranes that confers chemoresistance in leukemias and solid cancers.63 Expression of ABCC1 is also increased in pancreatic cancer.64 Serine palmitoyltransferase 1 (SPLTC1) participates in sphingolipid metabolism, and its activity is elevated in endometrial cancer tissue.65 To validate these novel miR-145 candidate targets along with known targets, we employed luciferase-3′UTR assays to examine the effect of miR-145 overexpression on these targets, several of which showed significant repression (miR-145/control): FSCN1, DIAPH1, PRPSAP2, RPA1, RBM22, SET, SPTLC1 and SPTBN1 (Fig. 3B). Deleting the miR-145 seed sequence from some of these plasmids could rescue the luciferase activity. On the other hand, one of the proteins upregulated by miR-145 was claudin-11, which is a critical component of tight junctions. We observed a 2.4-fold claudin-11 protein increase and postulated that miR-145 could inhibit invasiveness of cancer cells through the tumor suppressive network including upregulated claudin-11 and suppressed FSCN1 among others. We employed a matrigel invasion assays to functionally examine the effects of miR-145 on cell migration of MiaPaCa-2 pancreatic cancer cells. Forty-eight hours after transfection of miR-145/scrambled RNA control, we observed that miR-145-overexpressing pancreatic cancer cells were less capable of penetrating the matrigel membrane (Fig. 4).
Fig. 2.
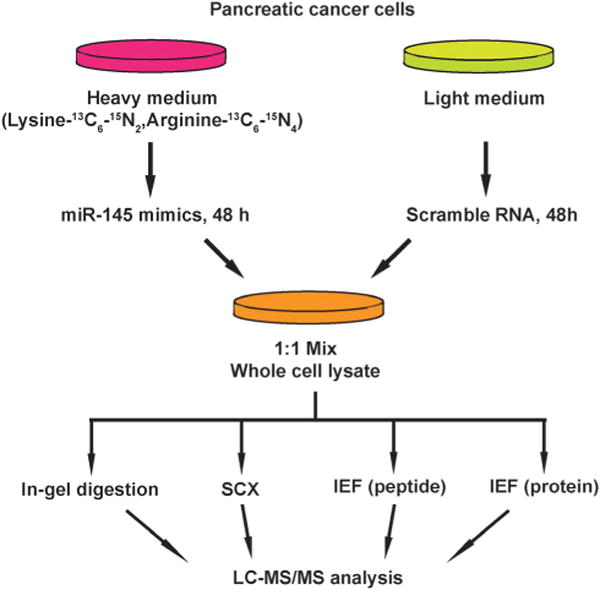
Strategy of proteomic analysis for identifying miR-145 targets. miR-145 mimic was transfected into MiaPaCa-2 cells that were labeled with heavy amino acids while scrambled RNAs were transfected into unlabeled cells. Proteins from both groups of cells were extracted 48 hours after transfection and fractionated through four methods: in-gel digestion, strong cation exchange chromatography, off-gel peptide and protein pI-based separations. Fractionated peptides were then analyzed on an LTQ-Orbitrap Velos mass spectrometer.
Fig. 3.
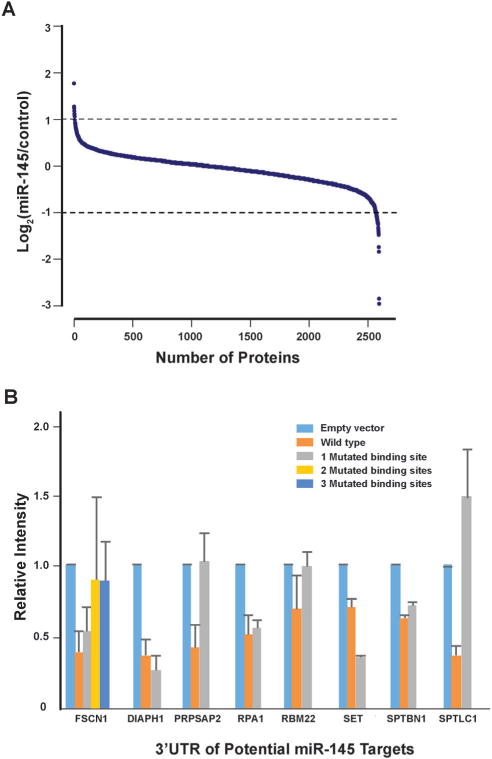
miR-145-mediated proteomic changes and luciferase assays for novel miR-145 targets. (A) SILAC quantitation of proteomic changes induced by miR-145 overexpression. (B) Luciferase assays to evaluate the effect of miR-145 overexpression on protein targets identified from proteomic experiments. Error bars represent the standard error.
Fig. 4.
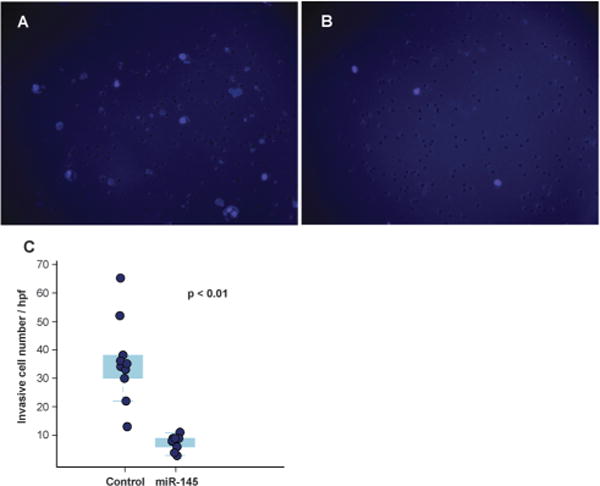
Decreased invasiveness of pancreatic cancer cells mediated by miR-145 in matrigel invasion assays. Cell nuclei of MiaPaCa-2 were stained with DAPI and are visualized as light purple dots by fluorescence microscopy. (A) Forty-eight hours after scrambled RNA transfection. (B) Forty-eight hours after miR-145 transfection. (C) Quantitation of invasive cancer cells depicted as a box-dot plot.
Integration of transcriptome and proteome and effect of miR-145 target sites on mRNA/protein levels
To elucidate miRNA-mediated regulation on gene expression, two molecular mechanisms have been proposed, including transcript destabilization and translational repression.66–68 In transcript destabilization, miRNAs and the RNA-induced silencing complex base-pair with mRNAs, leading to degradation of the regulated transcripts and subsequent downregulation of protein abundance. In translational repression, however, the engagement between miRNAs and mRNAs aborts translational initiation or halts translational elongation, causing decreased protein abundance without a concordant transcript degradation. To interrogate the contribution of each mechanism, we integrated the transcriptomic and proteomic changes caused by miRNA-145 and examined the correlation between these two datasets. There were 2525 transcript/protein pairs in this study, and the overall correlation between transcript and protein fold changes was low (r = 0.21, p < 0.01) (Fig. 5). This level of correlation was close to other miRNA-transfection studies and lower than that reported by Schwanhausser et al. which was focused on unperturbed mammalian cells.66,68–70 Of these 2525 pairs were 13 known miR-145 targets according to TarBase 6.0, and the concordant mRNA/protein fold-changes were observed in 9 out of these 13 pairs: FSCN1, YES1, GMFB, APH1A, CBFB, CLINT1, AP1G1, NIPSNAP1, PODXL. On the other hand, these 2525 transcript/protein pairs contained 1110 pairs that had discordant mRNA and protein changes, i.e. the transcript level was upregulated while the protein abundance was downregulated or vice versa. In all discordant pairs, we noticed enrichment of molecules in the EIF2 signaling and protein ubiquitylation pathways (p = 9.2 × 10−31 and 1.26 × 10−11, respectively) with the majority of these pathway components being suppressed at the protein level (Table S4 and S5, ESI†). It is known that the “seed sequence” of a miRNA, i.e. nucleotides 2–7 at the 5′ ends, is important for its targeting specificity.71 The seed sequence of a miRNA can base-pair with different regions of an mRNA (5′UTR, coding sequence (CDS) or 3′UTR) in different complementarity: such as “6mer”, “7mer-A1”, “7mer-m8” or “8mer” target site matches.72 In our study, seed sequence matches were enriched in miR-145-downregulated mRNA transcripts (odds ratio 1.69, p = 4.2 × 10−10) and miR-145-downregulated proteins (odds ratio 1.25, p = 0.007). Therefore, to compare the global effects of miRNA-145 targeting different regions of transcripts, we examined miR-145-mediated transcriptomic and proteomic alterations by stratifying the analysis into these three mRNA regions. Overall, among the 2525 mRNA/protein pairs, there were 78 genes with miR-145 target sites in their 5′UTRs, 1149 genes with miR-145 target sites in their coding regions (CDS) and 636 genes with miR-145 target sites in their 3′UTRs. 312 genes have miR-145 target sites both in CDS and 3′UTR. The presence of miR-145 target sites in CDS or 3′UTR is correlated with downregulation of transcript and protein levels (Table 1). Furthermore, we evaluated the transcriptomic and proteomic changes related to different types of miR-145 target sites in 3′UTR, including 8mer, 7mer-m8, 7mer-A1, and 6mer sequences. We found that 8mer matching correlated with downregulation of transcript and protein levels (p = 4.7 × 10−12 and 4.7 ×10−5, respectively) (Fig. 6) whereas other types of target sites only affected transcript levels but not protein levels (Table 1). The correlation between transcript and protein abundance in genes containing 8mer matches was slightly higher than that observed for all transcripts (r = 0.28, p < 0.01). More than 70% (69/95) of genes with 8mer target site(s) in their 3′UTRs exhibited downregulated protein levels, and two thirds (46/69) of them also had a coordinate down-regulation of transcript abundance, i.e. genes with 8mer target site(s) in their 3′UTR tended to have concordant downregulated mRNA/protein levels (p = 4.5 × 10−7). A similar trend has been reported by several other groups, corroborating that transcript destabilization plays an important role in 8mer matching.66–69
Fig. 5.
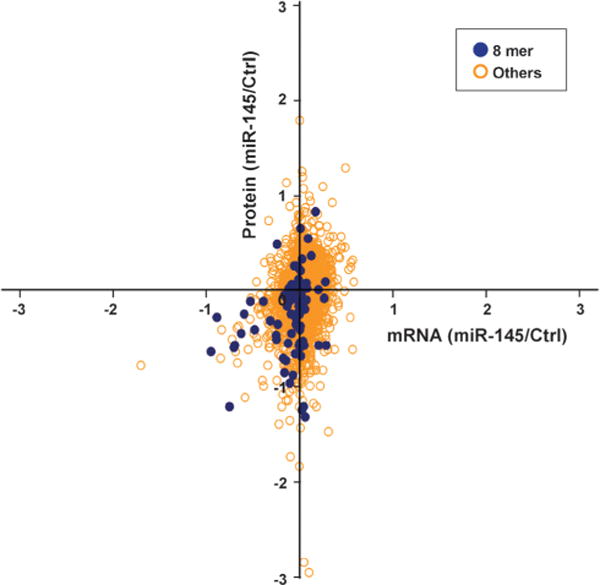
Integrated transcriptomic and proteomic analysis for miR-145 effect in pancreatic cancer cells. Quantitation results from mRNA gene expression microarrays and SILAC were intersected to show that “8mer” type of miR-145 binding predominantly caused downregulation of transcript and protein levels. All other types did not show any obvious pattern.
Table 1.
Locations and types of miR-145 target sites and downregulation of transcripts/proteins
| Location of miR-145 target site | ANOVA F test P value
|
|
|---|---|---|
| mRNA downregulation Protein downregulation | ||
| 5′-Untranslated region | 0.55 | 0.82 |
| Coding sequence | 9.26×10−5* | 1.06×10−3* |
| 3′-Untranslated region | <2.00×10−16* | 4.17×10−5* |
| 8mer | 4.71×10−12* | 4.73×10−5* |
| 7mer-m8 | 1.24×10−7* | 0.15 |
| 7mer-A1 | <2.00×10−16* | 0.26 |
| 6mer | 3.65×10−7* | 0.23 |
ANOVA: analysis of variance.
Statistical significance.
Fig. 6.
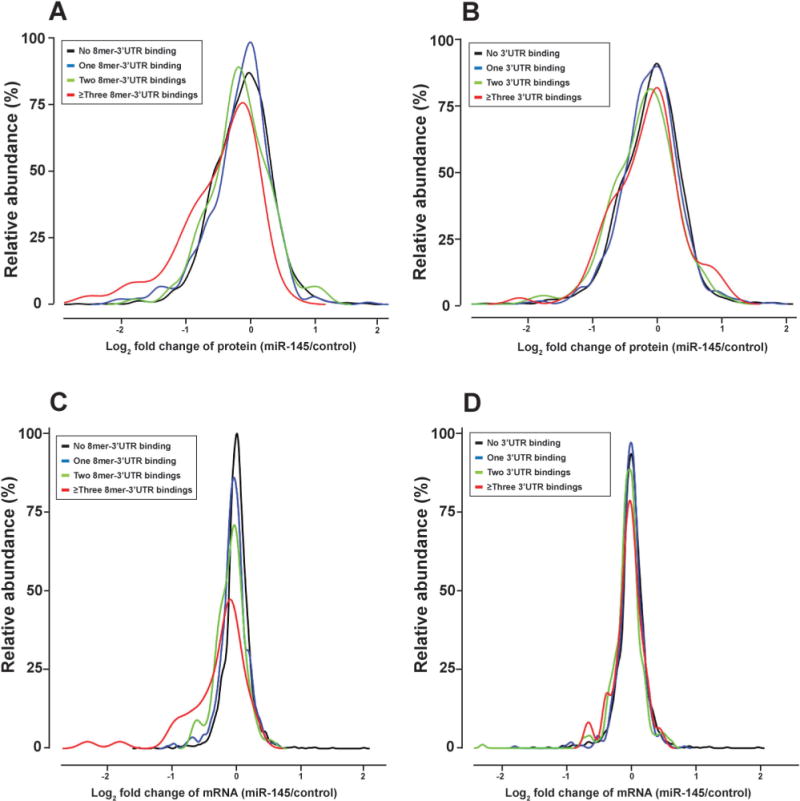
Different types of miR-145 targeting in 3′ untranslated regions (3′UTRs) of transcripts and their impact on protein and mRNA fold changes. (A) Relationship between protein fold changes and 8mer binding sites in 3′UTRs. (B) Relationship between protein fold changes and all types of binding in 3′UTR. (C) Relationship between mRNA fold changes and 8mer binding in 3′UTR. (D) Relationship between mRNA fold change and all types of binding in 3′UTRs. Types of binding in 3′UTR include 8mer, 7mer-A1, 7mer-m8 and 6mer. Overall, 8mer binding of miR-145 in the 3′UTR correlates better with downregulation of protein and transcript levels that other types.
miR-145-regulated miRNAs and their targets: integration of omics datasets with computational predictions
Redundant networks among mRNA targets and the miRNAs that regulate them have been observed across different model systems.73–75 Each mRNA transcript can be targeted by multiple miRNAs simultaneously, and the extent of repression from different miRNAs might be variable. This complexity presents challenges for studying the interplay between miRNAs and the transcriptome/proteome. In our study, miR-145 caused alteration of other miRNAs, which could lead to repression of their targets. For example, miR-124 and miR-133b were significantly upregulated by miR-145 (see above), and they are also predicted to target the proteins we observed in SILAC results: SPTLC1 and SPTBN1 by miR-124; ABCC1 and SPTBN1 by miR-133b. This observation led us to suspect that at least a subset of the proteomic changes might result from changes in the expression of these secondary miRNAs. To study how these miR-145-regulated miRNAs affected their targets, we obtained target predictions from TargetScan database and used TargetScan total context scores as a surrogate measure of miRNA effective binding.76 We chose the TargetScan database because it incorporates different types of target sites and its optimization of algorithms based on previous proteomics data along with inclusion of comparative genomics analysis. In principle, a lower TargetScan total context score predicts a higher miRNA binding efficacy. From the miRNA microarray results at a total FDR < 0.05, miRNAs regulated by miR-145 were divided into upregulated and downregulated groups (Fig. 7A). We correlated these miRNAs with their predicted mRNA/protein pairs, which exhibit a concordant change in our dataset. Interestingly, the mRNA/protein pairs targeted by the upregulated miRNAs had lower TargetScan total context scores than those targeted by downregulated miRNAs (p = 0.05, Fig. 7B). If only those pairs with protein fold change ≥ 1.5 were considered, the p-value was < 0.01. In other words, miRNAs upregulated by miR-145 were predicted to repress their targets’ protein abundance, suggesting that the proteomic change unexplained by miR-145 transfection could result from alteration of levels of other miRNAs.
Fig. 7.
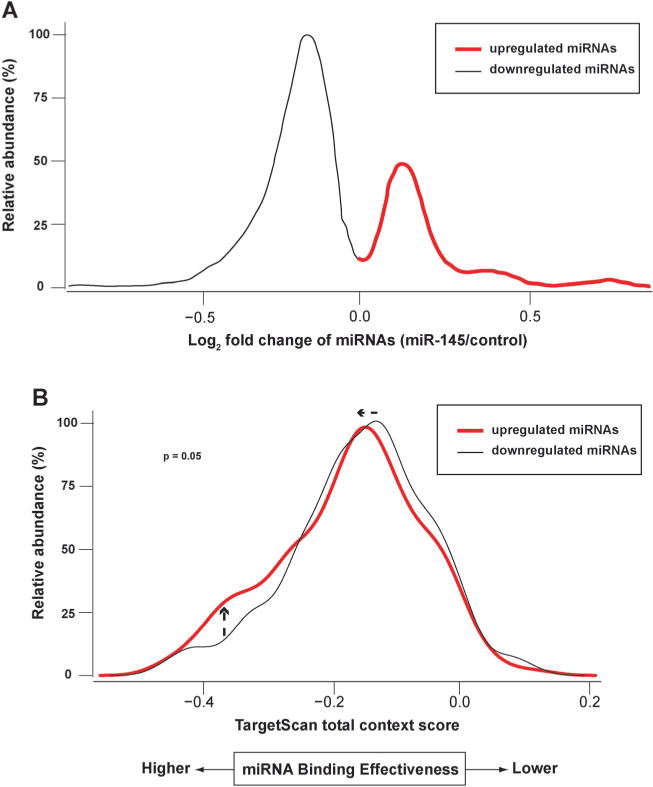
Impact of differentially regulated miRNAs on target binding. Transfection of miR-145 caused changes of other miRNAs that were assayed by miRNA microarrays. Through bioinformatics analysis, those upregulated miRNAs (red) were found to have target binding with lower TargetScan context scores (i.e. stronger binding to their targets). (A) Fold-change of miRNAs after miR-145 transfection. Only miRNAs targeting mRNA/protein pairs that showed concordant changes are depicted. (B) Upregulated miRNAs by miRNA-145 were predicted to possess stronger target binding than downregulated ones.
Conclusions
The advent of high-throughput technologies has expanded our ability to study complex biological systems. A multi-omics analysis enables even fuller characterization of biological intricacies. In this study, we took advantage of this systems approach to uncover novel aspects of miR-145-mediated pathway regulation. Through transcriptomic analysis in which we profiled mRNAs and miRNAs, we identified several unreported transcripts targeted by miR-145 along with a miRNA expression signature in multiple malignancies. To supplement bioinformatics and transcriptomic analysis, SILAC-based quantitative proteomics integrated into this study revealed novel miR-145 targets that were not revealed through transcriptomic analysis alone. In summary, novel targets and regulatory networks from this study help further our understanding about miR-145-mediated tumor suppression, and we believe this multi-omics analysis will advance biological research in other systems.
Experimental section
Cell culture, SILAC labeling and miRNA transfection
A pancreatic cancer cell line, MiaPaCa-2, was maintained in Dulbecco’s Modified Eagle’ Medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with L-glutamine, 10% FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. Growth of these cells were incubated in 5% CO2 at 37 °C and adapted to the SILAC media as described earlier.77 The heavy medium was labeled with 13C6,15N2-lysine, 13C6,15N4-arginine. Cells grown in the heavy medium were transfected with 50 nM miR-145 mimic oligonucleotides while those grown in light medium were transfected with scrambled RNA control (Dharmacon, Lafayette, CO).
Protein extraction and in-gel digestion
Cells from scrambled RNA and miR-145 transfection were harvested in 2% SDS lysis buffer, homogenized and sonicated. Protein concentration was measured using Lowry assay. Equal amount of protein from control and miR-145-transfected samples were mixed and ~ 200 μg was loaded on SDS-PAGE. The gel was stained with colloidal Coomassie blue. The protein bands were excised and were subjected to in-gel digestion, as described previously.78 Briefly, proteins were reduced using 5 mM dithiothreitol in 40 mM ammonium bicarbonate in 40% acetonitrile at 60 °C for 20 minutes and alkylated using 20 mM iodoacetamide in 40% acetonitrile. In-gel trypsin digestion was carried out at 1:20 enzyme to protein ratio at 37 °C overnight.
Strong cation exchange chromatography (SCX) and OFFGEL fractionation
Protein were reduced and alkylated with 5 mM DTT and 10 mM iodoacetamide, respectively. In-solution trypsin digestion was carried out at 1: 20 enzyme to protein ratio overnight at 37 °C. The peptides were fractionated by SCX.79 SCX fractionation was carried out on a PolySULPHOETHYL A column (PolyLC, Columbia, MD, USA) using an Agilent 1200 HPLC system containing a binary pump, UV detector and a fraction collector. Fractionation of peptides was carried out by a linear gradient between solvent A (10 mM KH2PO4, 25% Acetonitrile, pH 2.8) and solvent B (350 mM KCl in solvent A) from 8% to 50% solvent B over 60 min. UV absorbance of eluted peptides were detected at 214 nm. The peptide fractions were pooled based on chromatography profile into 24 fractions, vacuum-dried and stored at −20 °C. Peptides were reconstituted in 40 μl of 0.1% formic acid prior to LC-MS/MS analysis. Proteins as well as peptides were fractionated using OFFGEL fractionator (Agilent Technologies, Santa Clara, CA, USA). Briefly, around 100 μg of peptides/proteins in 2.4 ml of 5% glycerol buffer with 0.5% ampholytes (pH 3–10) was fractionated into 12 fractions in 24 h. Fractions were collected and stored at −20 °C.
Mass spectrometry
Peptide fractions were analyzed on an LTQ-Orbitrap Velos mass spectrometer coupled to Agilent 1200 series nano HPLC system. The MS1 scans were acquired in the m/z range of 350–1800 while MS2 scans were acquired in 100–2000 m/z using higher-energy collisional dissociation (HCD). Top 15 precursor ions were chosen for MS2 analysis in each duty cycle. Precursor ions with singly charged and unknown charges were excluded for MS2 analyses. Mass spectrometry raw data were searched against Human RefSeq protein database (Release 46) using Proteome Discoverer platform (Thermo, Bremen, Germany). We used Mascot and SEQUEST search algorithms with following search parameters: trypsin as a protease with maximum one allowed missed cleavage; carbamidomethylation of cysteine as a fixed modification; oxidation of methionine, acetylation of protein N-terminus and deamidation of glutamine. Mass tolerances at MS and MS/MS were set to 20 ppm and 0.1 Da, respectively. FDR of 1% was used for peptide validation. The proteomic datasets are accessible in the PRIDE Archive with the accession number: PXD001260.
Gene expression and miRNA microarray analysis
One μg total RNA was used on Agilent-014850 Whole Human Genome Microarray platform (4×44K G4112F). miR-145-transfected and control samples were analyzed in dye-swap duplicates (Pearson correlation coefficients were 0.99 and 0.99, respectively). One-hundred ng total RNA was used on Agilent’s-Human miRNA Microarray platform (V3). Samples were collected 24 and 48 h after miRNA transfection and were analyzed in duplicate. Raw data were pre-processed using the R package “Limma”.80 Briefly, for the gene expression microarray experiments, normalization within arrays was carried out using “Loess” method and normalization between arrays was carried out using the “Scale” method. For the miRNA microarray experiments, normalization between arrays was carried out with the “Quantile” method. Both microarray analyses were performed using a generalized linear model approach, coupled with empirical Bayes shrinkage of standard errors.80 Adjustment for multiple testing was achieved using the Benjamini and Hochberg method.81 Raw data and analyses were uploaded to Gene Expression Omnibus at the National Center for Biotechnology Information with the accession numbers: GSE45245 for gene expression microarray and GSE45246 for miRNA microarray.
Luciferase assays
3′UTRs of candidate miR-145 target genes were cloned into the pLightSwitch-3UTR control vector (SwitchGear Genomics, Carlsbad, CA) which contains an upstream Renilla luciferase open reading frame. Twenty-four hours before co-transfection, 3 × 104 MiaPaCa-2 cells were seeded in each well of 48-well plates. For co-transfection, 10 ng of the pLightSwitch-3UTR vector and 100 ng of the firefly luciferase-based pGL3 control vector were mixed with either miR-145 mimic (40 nM) or scrambled RNA oligos, packaged with Lipofectamine 2000 reagent (Invitrogen) to enter MiaPaCa-2 cells. Luciferase assays were carried out using the Dual-Luciferase® Reporter Assay System (Promega, San Luis Obispo, CA). The miR-145 seed sequence in 3′UTR was deleted using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent). Firefly luciferase activity was normalized to Renilla luciferase activity for each transfected well. For each experimental trial, cells were transfected in triplicate. For each 3′UTR construct, the ratio of miR-145 mimic to scrambled RNA oligos was calculated.
Invasion assays
Growth factor-reduced Matrigel invasion chambers (BD) were used according to the manufacturer’ instructions. Briefly, MiaPaCa-2 cells (5 × 104 ml−1 in DMEM + 0.1% FBS) in invasion chambers were transferred and exposed to the chemo-attractant DMEM + 5% FBS. Forty-eight hours later, noninvasive cells were wiped off using cotton swabs. Invasive cells were fixed in 10% formalin for 5 minutes, and then the Matrigel membranes were cut and mounted in DAPI to count cell nuclei.
Bioinformatics and statistical analysis
A web-based bioinformatics tool was developed in house for analyzing miRNA binding sites. All statistics analyses were performed in R 2.14 (64-bit). ANOVA and TukeyHSD multiple comparison tests were performed to compare effects of different target site matching on transcript and protein levels. To analyze luciferase assay results, Welch’s two sample t-test was used based on the different variance between control and miR-145-treated groups. The correlation between transcript and protein fold changes was calculated using Pearson’s product-moment correlation. To calculate the enrichment of miR-145 seed sequence in miR-145-downregulated transcripts/proteins, Fisher’s Exact tests were used in the contingency table analysis. To calculate the enrichment of 8mer target site matching in 3′UTRs, a Pearson’s Chi-squared test with Yate’s continuity correction was used. TargetScan Release 6.2 (http://www.targetscan.org/) was used for analysis of miRNA targeting context scores. TarBase 6.0 (http://www.microrna.gr/tarbase) was used as a curated database of known miR-145 targets. To obtain pathway enrichment analysis, we employed IPA® Core Analysis (Ingenuity System Inc. USA) and its embedded Fisher’s exact test (right-tailed).
Supplementary Material
Acknowledgments
This work was supported by an NIH roadmap grant for Technology Centers of Networks and Pathways (TCNP) (U54GM103520), NCI’ Clinical Proteomic Tumor Analysis Consortium initiative (U24CA160036) and a contract (HHSN268201000032C) from the National Heart, Lung and Blood Institute (NHLBI).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00585f
References
- 1.Hayashi K, Chuva De Sousa Lopes SM, Kaneda M, Tang F, Hajkova P, Lao K, O’Carroll D, Das P, Tarakhovsky A, Miska E, Surani M. PLoS One. 2008;3:e1738. doi: 10.1371/journal.pone.0001738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu N, Landreh M, Cao K, Abe M, Hendriks G, Kennerdell J, Zhu Y, Wang L, Bonini N. Nature. 2012;482:519–523. doi: 10.1038/nature10810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stefani G, Slack FJ. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 4.Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Koppel T, Jahantigh M, Lutgens E, Wang S, Olson E, Schober A, Weber C. Sci Signaling. 2009;2:ra81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 5.Di Leva G, Briskin D, Croce C. Upsala J Med Sci. 2012;117:202–216. doi: 10.3109/03009734.2012.660551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farazi T, Spitzer J, Morozov P, Tuschl T. J Pathol. 2011;223:102–115. doi: 10.1002/path.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voorhoeve P, Le Sage C, Schrier M, Gillis A, Stoop H, Nagel R, Liu Y, Van Duijse J, Drost J, Griekspoor A, Zlotorynski E, Yabuta N, De Vita G, Nojima H, Looijenga L, Agami R. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 8.Khew-Goodall Y, Goodall G. Nat Cell Biol. 2010;12:209–211. doi: 10.1038/ncb0310-209. [DOI] [PubMed] [Google Scholar]
- 9.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth E, Lee W, Enders G, Mendell J, Thomas-Tikhonenko A. Nat Genet. 2006;38:1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chivukula R, Shi G, Acharya A, Mills E, Zeitels L, Anandam J, Abdelnaby A, Balch G, Mansour J, Yopp A, Maitra A, Mendell J. Cell. 2014;157:1104–1116. doi: 10.1016/j.cell.2014.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kent O, Mccall M, Cornish T, Halushka M. Nucleic Acids Res. 2014;42:7528–7538. doi: 10.1093/nar/gku461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akao Y, Nakagawa Y, Kitade Y, Kinoshita T, Naoe T. Cancer Sci. 2007;98:1914–1920. doi: 10.1111/j.1349-7006.2007.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campanini M, Colli L, Paixao B, Cabral T, Amaral F, Machado H, Neder L, Saggioro F, Moreira A, Antonini S, De Castro M. Horm Cancer. 2010;1:187–196. doi: 10.1007/s12672-010-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kent O, Chivukula R, Mullendore M, Wentzel E, Feldmann G, Lee K, Liu S, Leach S, Maitra A, Mendell J. Genes Dev. 2010;24:2754–2759. doi: 10.1101/gad.1950610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michael M, O’Connor SM, Van Holst Pellekaan NG, Young G, James R. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- 16.Wu B, Xu L, Du Z, Liao L, Zhang H, Huang Q, Fang G, Li E. World J Gastroenterol. 2011;17:79–88. doi: 10.3748/wjg.v17.i1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z, Zeng H, Guo Y, Liu P, Pan H, Deng A, Hu J. J Exp Clin Cancer Res. 2010;29:151. doi: 10.1186/1756-9966-29-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iorio M, Ferracin M, Liu C, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo J, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin G, Querzoli P, Negrini M, Croce C. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 19.Ozen M, Creighton C, Ozdemir M, Ittmann M. Oncogene. 2008;27:1788–1793. doi: 10.1038/sj.onc.1210809. [DOI] [PubMed] [Google Scholar]
- 20.Song T, Xia W, Shao N, Zhang X, Wang C, Wu Y, Dong J, Cai W, Li H. Asian Pac J Cancer Prev. 2010;11:905–911. [PubMed] [Google Scholar]
- 21.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. Proc Natl Acad Sci U S A. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 23.Jia Y, Liu H, Zhuang Q, Xu S, Yang Z, Li J, Lou J, Zhang W. Oncol Rep. 2012;27:1865–1872. doi: 10.3892/or.2012.1701. [DOI] [PubMed] [Google Scholar]
- 24.Wu Y, Liu S, Xin H, Jiang J, Younglai E, Sun S, Wang H. Cancer. 2011;117:3989–3998. doi: 10.1002/cncr.25944. [DOI] [PubMed] [Google Scholar]
- 25.Chaerkady R, Pandey A. Proteomics: Clin Appl. 2007;1:1080–1089. doi: 10.1002/prca.200700284. [DOI] [PubMed] [Google Scholar]
- 26.Ong S, Blagoev B, Kratchmarova I, Kristensen D, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Yue D, Chen Y, Gao S, Huang Y. BMC Bioinf. 2010;11:476. doi: 10.1186/1471-2105-11-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kano M, Seki N, Kikkawa N, Fujimura L, Hoshino I, Akutsu Y, Chiyomaru T, Enokida H, Nakagawa M, Matsubara H. Int J Cancer. 2010;127:2804–2814. doi: 10.1002/ijc.25284. [DOI] [PubMed] [Google Scholar]
- 29.Fuse M, Nohata N, Kojima S, Sakamoto S, Chiyomaru T, Kawakami K, Enokida H, Nakagawa M, Naya Y, Ichikawa T, Seki N. Int J Oncol. 2011;38:1093–1101. doi: 10.3892/ijo.2011.919. [DOI] [PubMed] [Google Scholar]
- 30.Gregersen L, Jacobsen A, Frankel L, Wen J, Krogh A, Lund A. PLoS One. 2010;5:e8836. doi: 10.1371/journal.pone.0008836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Somasiri A, Nielsen J, Makretsov N, Mccoy M, Prentice L, Gilks C, Chia S, Gelmon K, Kershaw D, Huntsman D, Mcnagny K, Roskelley C. Cancer Res. 2004;64:5068–5073. doi: 10.1158/0008-5472.CAN-04-0240. [DOI] [PubMed] [Google Scholar]
- 32.Larsson A, Fridberg M, Gaber A, Nodin B, Leveen P, Jonsson G, Uhlen M, Birgisson H, Jirstrom K. BMC Cancer. 2012;12:282. doi: 10.1186/1471-2407-12-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dallas M, Chen S, Streppel M, Sharma S, Maitra A, Konstantopoulos K. Am J Physiol: Cell Physiol. 2012;303:C616–C624. doi: 10.1152/ajpcell.00149.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamakita Y, Matsumura F, Yamashiro S. Cell Motil Cytoskeleton. 2009;66:524–534. doi: 10.1002/cm.20356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hashimoto Y, Kim D, Adams J. J Pathol. 2011;224:289–300. doi: 10.1002/path.2894. [DOI] [PubMed] [Google Scholar]
- 36.Simpson A, Caballero O, Jungbluth A, Chen Y, Old L. Nat Rev Cancer. 2005;5:615–625. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 37.Marcar L, Maclaine N, Hupp T, Meek D. Cancer Res. 2010;70:10362–10370. doi: 10.1158/0008-5472.CAN-10-1341. [DOI] [PubMed] [Google Scholar]
- 38.Fu X, Wang H, Tan L, Liu S, Cao H, Wu M. World J Gastroenterol. 2002;8:638–643. doi: 10.3748/wjg.v8.i4.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cruz C, Gerdemann U, Leen A, Shafer J, Ku S, Tzou B, Horton T, Sheehan A, Copeland A, Younes A, Rooney C, Heslop H, Bollard C. Clin Cancer Res. 2011;17:7058–7066. doi: 10.1158/1078-0432.CCR-11-1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu C, Popova S, Brown E, Barsyte-Lovejoy D, Navab R, Shih W, Li M, Lu M, Jurisica I, Penn L, Gullberg D, Tsao M. Proc Natl Acad Sci U S A. 2007;104:11754–11759. doi: 10.1073/pnas.0703040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho W, Chow A, Au J. Eur J Cancer. 2009;45:2197–2206. doi: 10.1016/j.ejca.2009.04.039. [DOI] [PubMed] [Google Scholar]
- 42.Gao W, Shen H, Liu L, Xu J, Xu J, Shu Y. J Cancer Res Clin Oncol. 2011;137:557–566. doi: 10.1007/s00432-010-0918-4. [DOI] [PubMed] [Google Scholar]
- 43.Hu J, Guo H, Li H, Liu Y, Liu J, Chen L, Zhang J, Zhang N. PLoS One. 2012;7:e45965. doi: 10.1371/journal.pone.0045965. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Li Z, Gu X, Fang Y, Xiang J, Chen Z. Oncol Lett. 2012;3:346–350. doi: 10.3892/ol.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Motoyama K, Inoue H, Takatsuno Y, Tanaka F, Mimori K, Uetake H, Sugihara K, Mori M. Int J Oncol. 2009;34:1069–1075. doi: 10.3892/ijo_00000233. [DOI] [PubMed] [Google Scholar]
- 46.Tchernitsa O, Kasajima A, Schafer R, Kuban R, Ungethum U, Gyorffy B, Neumann U, Simon E, Weichert W, Ebert M, Rocken C. J Pathol. 2010;222:310–319. doi: 10.1002/path.2759. [DOI] [PubMed] [Google Scholar]
- 47.Silber J, Lim D, Petritsch C, Persson A, Maunakea A, Yu M, Vandenberg S, Ginzinger D, James C, Costello J, Bergers G, Weiss W, Alvarez-Buylla A, Hodgson J. BMC Med. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng B, Li Z, Chen R, Guo N, Zhou J, Zhou Q, Lin Q, Cheng D, Liao Q, Zheng L, Gong Y. FEBS Lett. 2012;586:3271–3278. doi: 10.1016/j.febslet.2012.06.049. [DOI] [PubMed] [Google Scholar]
- 49.Hashiguchi Y, Nishida N, Mimori K, Sudo T, Tanaka F, Shibata K, Ishii H, Mochizuki H, Hase K, Doki Y, Mori M. Int J Oncol. 2012;40:1477–1482. doi: 10.3892/ijo.2012.1363. [DOI] [PubMed] [Google Scholar]
- 50.Jiang L, Huang Q, Zhang S, Zhang Q, Chang J, Qiu X, Wang E. BMC Cancer. 2010;10:318. doi: 10.1186/1471-2407-10-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhattacharyya S, Habermacher R, Martine U, Closs E, Filipowicz W. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 52.Chiyomaru T, Tatarano S, Kawakami K, Enokida H, Yoshino H, Nohata N, Fuse M, Seki N, Nakagawa M. Prostate. 2011;71:1559–1567. doi: 10.1002/pros.21372. [DOI] [PubMed] [Google Scholar]
- 53.Bhutia Y, Hung S, Krentz M, Patel D, Lovin D, Manoharan R, Thomson J, Govindarajan R. PLoS One. 2013;8:e53436. doi: 10.1371/journal.pone.0053436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Christensen D, Chen Y, Oddo J, Matta K, Neil J, Davis E, Volkheimer A, Lanasa M, Friedman D, Goodman B, Gockerman J, Diehl L, De Castro CM, Moore J, Vitek M, Weinberg J. Blood. 2011;118:4150–4158. doi: 10.1182/blood-2011-04-351072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlson S, Eng E, Kim E, Perlman E, Copeland T, Ballermann B. J Am Soc Nephrol. 1998;9:1873–1880. doi: 10.1681/ASN.V9101873. [DOI] [PubMed] [Google Scholar]
- 56.Switzer C, Cheng R, Vitek T, Christensen D, Wink D, Vitek M. Oncogene. 2011;30:2504–2513. doi: 10.1038/onc.2010.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanakis D, Levidou G, Gakiopoulou H, Eftichiadis C, Thymara I, Fragkou P, Trigka E, Boviatsis E, Patsouris E, Korkolopoulou P. Hum Pathol. 2011;42:1545–1553. doi: 10.1016/j.humpath.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 58.Dahai Y, Sanyuan S, Hong L, Di Z, Chong Z. Cell Biochem Biophys. 2013;67:175–180. doi: 10.1007/s12013-013-9530-y. [DOI] [PubMed] [Google Scholar]
- 59.Ayaru L, Stoeber K, Webster G, Hatfield A, Wollenschlaeger A, Okoturo O, Rashid M, Williams G, Pereira S. Br J Cancer. 2008;98:1548–1554. doi: 10.1038/sj.bjc.6604342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanna-Morris A, Badvie S, Cohen P, Mccullough T, Andreyev H, Allen-Mersh TG. J Clin Pathol. 2009;62:325–330. doi: 10.1136/jcp.2007.054643. [DOI] [PubMed] [Google Scholar]
- 61.Kim M, Kim J, Kim J, Kim D, Yu Y. Korean J Ophthalmol. 2010;24:35–39. doi: 10.3341/kjo.2010.24.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramnath N, Hernandez F, Tan D, Huberman J, Natarajan N, Beck A, Hyland A, Todorov I, Brooks J, Bepler G. J Clin Oncol. 2001;19:4259–4266. doi: 10.1200/JCO.2001.19.22.4259. [DOI] [PubMed] [Google Scholar]
- 63.Gottesman M, Fojo T, Bates S. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 64.Chen M, Xue X, Wang F, An Y, Tang D, Xu Y, Wang H, Yuan Z, Gao W, Wei J, Zhang J, Miao Y. Oncol Rep. 2012;27:265–269. doi: 10.3892/or.2011.1475. [DOI] [PubMed] [Google Scholar]
- 65.Knapp P, Baranowski M, Knapp M, Zabielski P, Blachnio-Zabielska AU, Gorski J. Prostaglandins Other Lipid Mediators. 2010;92:62–66. doi: 10.1016/j.prostaglandins.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 66.Baek D, Villen J, Shin C, Camargo F, Gygi S, Bartel D. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 68.Guo H, Ingolia N, Weissman J, Bartel D. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bauer K, Hummon A. J Proteome Res. 2012;11:4744–4754. doi: 10.1021/pr300600r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 71.Lewis B, Shih I, Jones-Rhoades MW, Bartel D, Burge C. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 72.Bartel D. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsang J, Ebert M, Van Oudenaarden A. Mol Cell. 2010;38:140–153. doi: 10.1016/j.molcel.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ventura A, Young A, Winslow M, Lintault L, Meissner A, Erkeland S, Newman J, Bronson R, Crowley D, Stone J, Jaenisch R, Sharp P, Jacks T. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martinez N, Ow M, Barrasa M, Hammell M, Sequerra R, Doucette-Stamm L, Roth F, Ambros V, Walhout A. Genes Dev. 2008;22:2535–2549. doi: 10.1101/gad.1678608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grimson A, Farh K, Johnston W, Garrett-Engele P, Lim L, Bartel D. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harsha H, Molina H, Pandey A. Nat Protoc. 2008;3:505–516. doi: 10.1038/nprot.2008.2. [DOI] [PubMed] [Google Scholar]
- 78.Chaerkady R, Kelkar D, Muthusamy B, Kandasamy K, Dwivedi S, Sahasrabuddhe N, Kim M, Renuse S, Pinto S, Sharma R, Pawar H, Sekhar N, Mohanty A, Getnet D, Yang Y, Zhong J, Dash A, Maccallum R, Delanghe B, Mlambo G, Kumar A, Keshava Prasad TS, Okulate M, Kumar N, Pandey A. Genome Res. 2011;21:1872–1881. doi: 10.1101/gr.127951.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Y, Chaerkady R, Kandasamy K, Huang T, Selvan L, Dwivedi S, Kent O, Mendell J, Pandey A. Mol BioSyst. 2010;6:1873–1882. doi: 10.1039/c004401f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smyth G. Stat Appl Genet Mol Biol. 2004;3:3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 81.Benjamini Y, Hochberg Y, Roy J. Statist Soc Ser B (Methodological) 1995:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.