

Abstract
Introduction
Fluticasone furoate (FF) is a novel inhaled corticosteroid with 24-h activity. FF is in development as a once-daily treatment for asthma as monotherapy and in combination with vilanterol (VI), a long-acting β2 agonist. Corticosteroids can have systemic effects on hypothalamic-pituitary-adrenal (HPA) axis function, potentially resulting in cortisol suppression.
Objectives
To assess the effect of FF/VI compared with placebo on the HPA axis by evaluating 24-h weighted mean serum cortisol levels in adolescent and adult patients with persistent asthma.
Methods
One hundred eighty-five patients with >12 weeks history of asthma were randomised in a 4:4:4:1 ratio to one of two once-daily FF/VI treatments (100/25 μg or 200/25 μg), placebo or an active control group that received inhaled placebo plus one prednisolone 10 mg capsule daily for the last 7 days of the study. Twenty-four-hour serum and urinary cortisol was measured at baseline and on day 42.
Results
Non-inferiority in 24-h weighted mean serum cortisol after 6 weeks of treatment with once-daily FF/VI at either strength was shown. Treatment ratios [95% confidence interval (CI)] to placebo for FF/VI 100/25 μg [0.99 (0.87–1.12)] or FF/VI 200/25 μg [0.97 (0.86–1.10)] indicated non-inferiority of both FF/VI doses to placebo as the lower limit of the 95% CI was greater than the predefined 0.8. Prednisolone substantially reduced 24-h weighted mean serum cortisol [treatment ratio to placebo 0.34 (0.28–0.41)]. FF/VI was well-tolerated, and no safety concerns were identified.
Conclusions
FF/VI was found to be non-inferior to placebo on HPA axis function, with no indication of significant cortisol suppression after 42 days.
Keywords: asthma, cortisol, fluticasone furoate, hypothalamic-pituitary-adrenal axis, inhaled corticosteroid, long-acting beta2 agonist, vilanterol
Introduction
Inhaled corticosteroids (ICSs) are the standard first-line therapy for the management of asthma 1. Once-daily regimens can simplify treatment and offer increased convenience to patients, potentially improving adherence 2 and therefore asthma control 3. Combination therapies incorporating a long-acting β2 agonist (LABA) with an ICS are effective in asthma when inadequately controlled despite low/medium-dose ICS therapy 1. Fluticasone furoate (FF), a novel ICS structurally distinct from fluticasone propionate (FP) 4, is in development as a monotherapy and has been shown to be effective in phase 2b trials 5–7. FF has an ester derived from 2-furoic acid at the C-17α position that replaces the simpler propionate ester 4. This feature of FF confers both greater affinity for, and longer retention in, respiratory tissues than FP 8. Vilanterol (VI) is an inhaled LABA with inherent 24-h activity 9. The combination of FF and VI (FF/VI) represents a novel, once-daily dosing ICS/LABA combination and is in development for treatment of asthma and chronic obstructive pulmonary disease 10.
Endogenous cortisol is responsible for several important functions within the body; its level is regulated by a feedback system, involving the hypothalamus, pituitary and adrenal glands, known as the HPA axis. Corticosteroids, including ICS, have the potential to induce dose-dependent systemic effects on the HPA axis 11,12. Cortisol suppression has been observed in asthma patients with normal HPA axis function at baseline receiving high doses of ICS 13. Therefore, the measurement of 24-h serum cortisol (SC), a sensitive method for assessing adrenocortical activity, was selected to evaluate cortisol suppression in a dose-dependent manner 14. The primary objective of this phase 3a study was to examine the effect of two doses of inhaled FF/VI (100/25 μg and 200/25 μg) on HPA axis function in patients with persistent asthma. Oral prednisolone was included as an active control to ensure that the assay was sufficiently sensitive to detect a treatment effect on SC.
Methods
Patients and screening
Patients aged from 12 to 65 years were eligible if they had a history of asthma as defined by the National Institute of Health 15 for at least 12 weeks prior to screening. Eligible patients had a best forced expiratory volume in 1 s (FEV1) of ≥50% predicted normal value and either demonstrated FEV1 reversibility of at least 12% and 200 mL with inhaled salbutamol, or historical documentation of FEV1 reversibility or positive methacholine/histamine challenge test within 12 months prior to screening and could not have used ICS within 4 weeks of screening. All patients were provided with albuterol/salbutamol inhalation aerosol at the prescreening assessment for symptomatic relief; other short-acting β2 agonists were not permitted from that point onwards. Non-steroidal controller medications such as leukotriene modifiers were permitted. Exclusion criteria and prohibited medications are delineated in the Online Supplement.
Study design and treatments
This phase 3 study (GSK study number HZA106851; http://www.clinicaltrials.gov registration number NCT01086410) was conducted at 16 centres in three countries (Germany, Poland, United States) between 25 March 2010 and 24 September 2010. The study was approved by local ethics review committees and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Written informed consent was obtained from each patient prior to the performance of any study-specific procedures.
This was a randomised, multicentre, double-blind, parallel-group, double-dummy, placebo-controlled study. An active control (prednisolone 10 mg) arm was included in order to verify assay sensitivity (see Fig. 1 for study schema and timelines). The length of the treatment period was 42 days. Eligible patients were randomised 4:4:4:1 to receive one of four treatment regimens (Fig. 1). All patients were instructed to take one inhalation from the inhaler once daily in the evening (5–11 pm) every day of the treatment period and one capsule on each of the last seven mornings (days 36–42). All inhaled treatments were administered via a dry powder inhaler. Placebo inhalers and capsules were identical in appearance to the active treatments. The central randomisation schedule was generated by the sponsor using a validated, computerised system (RandAll, GlaxoSmithKline, Stevenage, UK), and patients were randomised using Registration and Medication Ordering System (GlaxoSmithKline).
Figure 1.
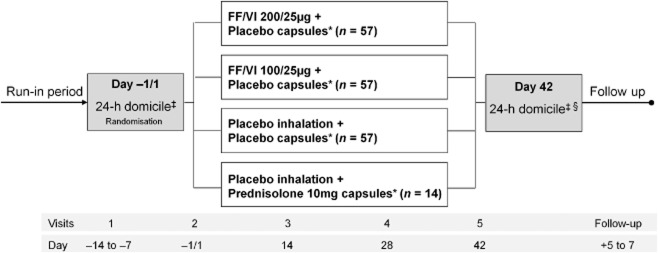
Schematic illustrating study design. Note: FF/VI 100/25 μg and fluticasone furoate (FF)/vilanterol (VI) 200/25 μg are nominal doses, representing emitted doses of FF and VI via the dry powder inhaler of 92/22 μg and 184/22 μg, respectively. *All inhaled dosing once daily in the evening. All oral capsules taken in the morning on the last 7 days of study only. ‡24-h serum cortisol and urine cortisol sampling. §Pharmacokinetic sampling.
Patients visited the clinic for assessment at baseline (day −1/1) and on days 14, 28 and 42 of the study, and were followed up 5–7 days after the last clinic visit. Adherence to study treatments and appropriate inhaler use were assessed at each visit. Treatment compliance was determined by reviewing the dose counter on the inhaler and by counting capsules. At baseline and on day 42, patients were domiciled at the clinic overnight for 24-h serial SC and urine cortisol (UC) sampling.
Outcome measurements
The primary end point was 0- to 24-h weighted mean SC at the end of the 42-day treatment period relative to baseline. Secondary pharmacodynamic (PD) end points, also measured on day 42, were area under the curve (AUC(0–24)) SC, trough SC concentration and 24-h urinary free cortisol excretion (day 42). In addition, pharmacokinetic (PK) parameters [AUC, maximum plasma concentration (Cmax) and time taken to reach Cmax (tmax)] for FF and VI were evaluated from plasma concentration time data.
Twenty-four-hour SC levels were assessed at baseline and day 42, with cortisol sampling at 0, 2, 4, 9, 12, 14, 16, 20, 22 and 24 h. A complete 24-h urine collection was also made at these visits. SC was assayed by liquid chromatography tandem mass spectrometry (LC-MS/MS). PK samples were taken on day 42 at 0, 5, 10, 15 and 30 min, and 1, 2, 4, 9, 12, 16, 20 and 24 h. Plasma samples from patients receiving FF/VI were analysed for FF and VI separately using solid phase extraction followed by high-performance LC-MS/MS. The lower limit of quantification (LLQ) of FF was 10 pg/mL. The LLQ for VI was 10 pg/mL except where only a 100-μL plasma aliquot was available, in which case the LLQ was 20 pg/mL. PK parameters were calculated by standard non-compartmental analysis using WinNonLin Pro v5.2 (Pharsight Corporation, St Lous, MO, USA).
Safety
Safety and tolerability were assessed by monitoring adverse events (AEs) and serious AEs (SAEs), vital signs, haematological and biochemical parameters, and oropharyngeal examinations. FEV1 was measured and recorded at each clinic visit to assess asthma stability. In addition, patient-recorded (diary card) peak expiratory flow and 24-h rescue use was monitored for the same reason. AEs were coded using the Medical Dictionary for Regulatory Activities.
Severe asthma exacerbations were defined as an occurrence of deterioration of asthma requiring use of systemic corticosteroids for at least 3 days, or inpatient hospitalisation, or emergency department visit because of asthma. Patients were withdrawn from the study because of lack of efficacy in the event of a severe asthma exacerbation; however, severe asthma exacerbations were not recorded as an AE unless they met the definition of a SAE.
Statistical analysis
A sample size of 130 (40 patients per treatment group and 10 in the prednisolone arm) was estimated to be required to provide 90% power to demonstrate non-inferiority of FF/VI to placebo if there is no true difference between the treatment groups. Non-inferiority would be demonstrated if the lower limit of the two-sided 95% confidence interval (CI) for the geometric mean ratio of each strength of FF/VI and placebo was greater than 0.8. Furthermore, with 10 patients receiving prednisolone, the study would have 90% power to detect a 30% decrease in 0- to 24-h weighted mean SC relative to placebo. Sample size estimates were calculated from between-subject standard deviation data derived from previous studies, and patient recruitment was carried out on the basis of an assumed 20% dropout rate and 10% rate of exclusion from the primary end-point analysis.
The intent-to-treat (ITT) population consisted of all patients who were randomised to treatment and who received at least one dose of study drug and was the primary population for safety measures. The SC population comprised those patients in the ITT population who did not have protocol deviations considered to affect the SC end point and whose serum samples were not considered to have confounding factors that would affect interpretation of results. Data from the SC population were used for the primary end point and other SC end points. Details of the UC and PK populations are summarised in the Online Supplement.
The 0- to 24-h SC weighted mean for each patient was calculated by dividing the AUC over the 24-h period by the sample collection time interval. The primary end-point analysis was subsequently performed on the ratio from baseline of 0- to 24-h weighted mean SC in the SC population. An analysis of covariance model with log2-transformed baseline SC, sex, age, region and treatment group as terms was used to perform comparisons of ratio to baseline between each active treatment group and placebo, and between the prednisolone and placebo groups. Secondary analyses of SC AUC(0–24) and SC trough were performed. Trough SC concentration was defined as the minimum value of SC measured over the 24-h period. All programming was performed in SAS, version 8 or later (SAS Institute Inc., Cary, NC, USA).
Results
Study population
Of 239 patients screened, 185 were randomised and received at least one dose of study treatment (Fig. 2). Most (96%) patients completed the study. Demographics and baseline characteristics were similar across treatment groups (Table 1); however, there was a higher proportion of females in the FF/VI 100/25 μg group (55%) than in the other treatment groups (40–47%). Adolescents formed 10% of the ITT population.
Figure 2.
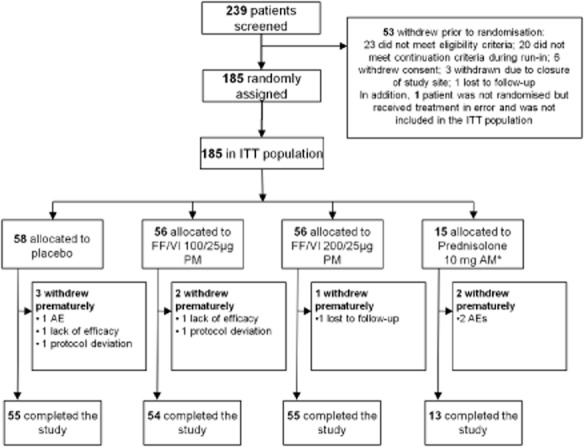
CONsolidated Standards of Reporting Trials (CONSORT)/patient flow diagram. *Placebo inhaler plus prednisolone 10 mg once-daily/morning Days 36–42. AE, adverse event; AM, morning; FF, fluticasone furoate; ITT, intent-to-treat; PM, evening; VI, vilanterol.
Table 1.
Patient baseline demographics and screening lung function (intent-to-treat population)
| Placebo (n = 58) | FF/VI 100/25 (n = 56) | FF/VI 200/25 (n = 56) | Prednisolone (n = 15) | Total (N = 185) | |
|---|---|---|---|---|---|
| Age, years | 36.1 (15.42) | 34.4 (15.63) | 34.0 (13.74) | 37.5 (14.19) | 35.1 (14.82) |
| Age <18 years, n (%) | 5 (9) | 5 (9) | 7 (13) | 1 (7) | 18 (10) |
| Female gender, n (%) | 27 (47) | 31 (55) | 23 (41) | 6 (40) | 87 (47) |
| Screening lung function | |||||
| Pre-bronchodilator FEV1 (L) | 2.78 (0.73) | 2.90 (0.83) | 2.92 (0.76) | 3.03 (1.02) | 2.88 (0.79) |
| Pre-bronchodilator % predicted FEV1 | 77.0 (11.88) | 79.9 (12.58) | 77.5 (13.22) | 78.6 (13.17) | 78.2 (12.57) |
Values are mean (SD) unless otherwise stated.
FEV1, forced expiratory volume in 1 s; FF, fluticasone furoate; SD, standard deviation; VI, vilanterol.
Mean compliance with inhaled treatment was similar across study groups ranging from 98.8% to 101.4%; most patients in each treatment group (74–86%) were between 95% and 105% compliant. Capsule compliance was also high across all treatment groups: 95–100% of patients randomised to the prednisolone arm took seven or more capsules as required.
PD assessment
SC data are from the SC population (n = 173). Non-inferiority of FF/VI compared with placebo was demonstrated for the primary end point of 0- to 24-h weighted mean SC as the lower limit of the CI was greater than 0.8 for both dosing regimens (Table 2). In the prednisolone group, however, SC was substantially lowered relative to placebo (Table 2). Adjusted treatment ratios for each of the three treatment groups compared with placebo are shown in Fig. 3.
Table 2.
Ratio from baseline of serum cortisol 0- to 24-h weighted mean, day 42 (serum cortisol population)
| Treatment comparison | LS geometric means | Treatment ratio | 95% CI | |
|---|---|---|---|---|
| Active | Placebo | |||
| FF/VI 100/25 PM vs placebo | 0.98 | 0.99 | 0.99 | 0.87, 1.12 |
| FF/VI 200/25 PM vs placebo | 0.96 | 0.99 | 0.97 | 0.86, 1.10 |
| Prednisolone AM vs placebo | 0.33 | 0.99 | 0.34 | 0.28, 0.41 |
AM, morning; CI, confidence interval; FF, fluticasone furoate; LS, least squares; PM, evening; VI, vilanterol.
Figure 3.
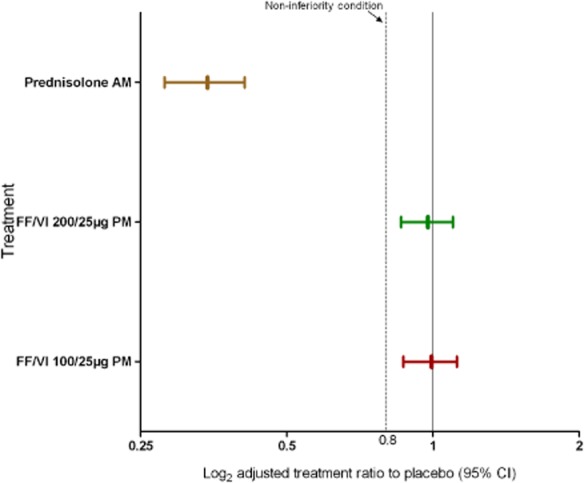
Day 42 adjusted treatment ratios to placebo of weighted mean serum cortisol in treatment groups receiving 200/25 μg fluticasone furoate (FF)/vilanterol (VI), 100/25 μg FF/VI and prednisolone, with 95% confidence interval (CI) (serum cortisol population). AM, morning; PM, evening.
SC concentration time profiles at baseline (day −1/1) were similar across treatment groups (Fig. 4A). On day 42 (Fig. 4B), SC concentrations over 24 h were notably lower in the prednisolone group compared with the placebo and FF/VI groups.
Figure 4.
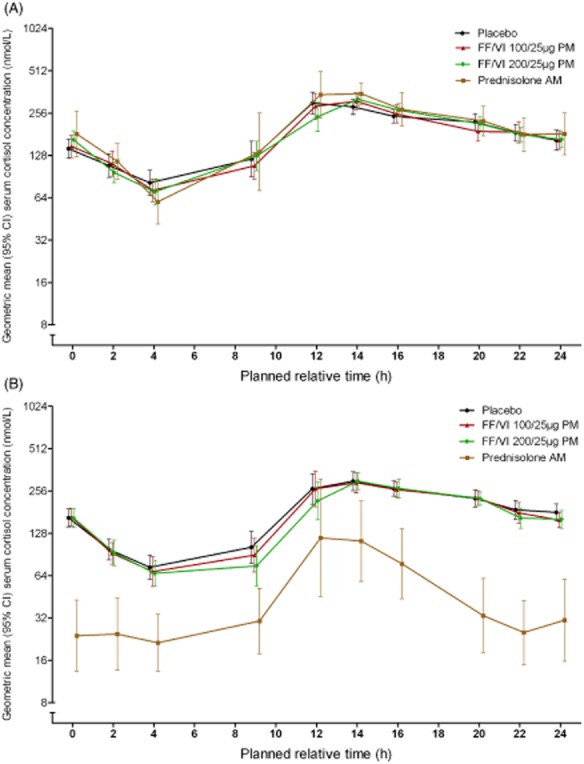
Day −1/1 (A) and day 42 (B) 0- to 24-h geometric mean serum cortisol (SC) concentration in treatment groups receiving fluticasone furoate (FF)/vilanterol (VI) 100/25 μg, FF/VI 200/25 μg, placebo and prednisolone (SC population). AM, morning; CI, confidence interval; PM, evening.
Findings for the secondary end point of AUC(0–24) SC were similar to the primary end point (data not shown). Day 42 SC trough treatment ratio was decreased with all active treatments relative to placebo. The treatment ratios (95% CI) from baseline were 0.31 (0.21–0.46) for prednisolone, 0.89 (0.69–1.15) for FF/VI 100/25 μg and 0.76 (0.59–0.97) for FF/VI 200/25 μg.
In the UC population (N = 161), there were no statistically significant differences in day 42 24-h UC excretion ratio between either of the FF/VI groups and placebo, but there was between the prednisolone group and placebo [0.43 (95% CI 0.28–0.66); P < 0.001].
PK assessments
Data are from 54 (FF/VI 100/25 μg group) and 56 (FF/VI 200/25 μg group) patients who provided samples for PK analysis. In total, 1249 FF PK samples and 1265 VI PK samples were analysed.
FF was rapidly absorbed into plasma (Cmax was achieved at a median time of 30 min following dosing with FF/VI; Table 3). FF Cmax and AUC(0–t) increased in a dose-related manner. In the FF/VI 100/25 μg group, systemic exposure to FF was limited for most of the 0- to 24-h dose interval. In the FF/VI 200/25 μg group, FF could be quantified in more samples and for a longer period of time than in the FF/VI 100/25 μg group. Twenty-four-hour LLQ data for day 42 are provided in Supporting Information Table S1. VI was rapidly absorbed (median Cmax 5 min) and rapidly eliminated from plasma.
Table 3.
Day 42 0–24-h plasma concentration of FF and VI geometric mean (pharmacokinetics population)
| Parameter geometric mean (95% CI) | Cmax [pg/mL] | Median tmax [h] (range) | AUC(0–t) [pg.h/mL] |
|---|---|---|---|
| FF | |||
| 100/25 μg | 19.4 (16.8–22.4) | 0.50 (0.03–8.97) | 58.8 (37.7–91.8) |
| 200/25 μg | 33.0 (28.6–38.1) | 0.50 (0.07–4.00) | 221.7 (152.7–321.9) |
| VI | |||
| 100/25 μg | 71.1 (54.2–93.2) | 0.083 (0.00–2.00) | 22.6 (16.0–32.1) |
| 200/25 μg | 89.9 (71.7–112.8) | 0.083 (0.00–16.00) | 28.9 (20.0–41.6) |
AUC(0–t), area under the curve between time 0 and t; CI, confidence interval; Cmax, maximum plasma concentration; FF, fluticasone furoate; tmax, time taken to reach Cmax; VI, vilanterol.
Safety assessment
Data are from the ITT population (N = 185). The proportion of patients reporting any AE during the treatment period was 38% for FF/VI 200/25 μg, 41% for FF/VI 100/25 μg, 28% for placebo and 33% for prednisolone (Table 4). Four patients had AEs deemed to be treatment-related. Three patients had AEs that led to study withdrawal (gastroenteritis in the placebo group; facial palsy and varicose vein in the prednisolone group), none of which were considered treatment-related. No AEs were reported during the post-treatment period. No SAEs were reported during any period of the study. The most frequently reported on-treatment AE was headache. Five patients experienced a total of six AEs of special interest (see Online Supplement for the list of AEs considered to be of special interest). Four patients in the FF/VI groups had local steroid effects (dysphonia, oral candidiasis, throat irritation). Cardiovascular effects were experienced by one patient in the placebo group (increased blood pressure) and one in the FF/VI 200/25 μg group (palpitations). None of these AEs led to study withdrawal (Table 4).
Table 4.
Summary of adverse events (AEs) reported in each of the four treatment groups (intent-to-treat population)
| Placebo (n = 58) | FF/VI 100/25 μg (n = 56) | FF/VI 200/25 μg (n = 56) | Prednisolone (n = 15) | |
|---|---|---|---|---|
| AEs, n (%) | ||||
| On-treatment | 16 (28) | 23 (41) | 21 (38) | 5 (33) |
| Treatment-related AEs | 1 (2) | 1 (2) | 2 (4) | 0 |
| AEs leading to permanent discontinuation of study drug or withdrawal from study | 1 (2) | 0 | 0 | 2 (13) |
| Most frequent on-treatment AEs, n (%)* | ||||
| Headache | 5 (9) | 15 (27) | 9 (16) | 2 (13) |
| Back pain | 0 | 1 (2) | 2 (4) | 1 (7) |
| Nasopharyngitis | 1 (2) | 2 (4) | 1 (2) | 0 |
| Oropharyngeal pain | 2 (3) | 1 (2) | 0 | 0 |
| Sinus headache | 0 | 1 (2) | 2 (4) | 0 |
| Arthralgia | 0 | 2 (4) | 0 | 0 |
| Cough | 2 (3) | 0 | 0 | 0 |
| Nausea | 1 (2) | 0 | 0 | 1 (7) |
| Rhinitis allergic | 0 | 2 (4) | 0 | 0 |
| Sinusitis | 0 | 2 (4) | 0 | 0 |
| AEs of special interest (on- and post-treatment), n (%) | ||||
| Any local steroid effect | 0 | 2 (4) | 2 (4) | 0 |
| Dysphonia | 0 | 1 (2) | 1 (2) | 0 |
| Oropharyngeal candidiasis | 0 | 0 | 1 (2) | 0 |
| Throat irritation | 0 | 1 (2) | 0 | 0 |
| Any cardiovascular effect | 1 (2) | 0 | 1 (2) | 0 |
| Increase in blood pressure | 1 (2) | 0 | 0 | 0 |
| Palpitations | 0 | 0 | 1 (2) | 0 |
AEs occurring only in the prednisolone group are not shown. These were as follows: dizziness, exertional dyspnoea, facial palsy, fatigue, hypotension, insomnia, varicose vein [n = 1 (7%) patient for each].
FF, fluticasone furoate; VI, vilanterol.
Asthma stability measures were monitored for safety. Lung function remained stable over the course of the study in the placebo and prednisolone groups; improvements in predose FEV1 of 335 mL and 218 mL were observed at day 42 for FF/VI 100/25 μg and FF/VI 200/25 μg, respectively. One patient in the FF/VI 100/25 μg group experienced a severe asthma exacerbation concurrent with sinusitis and was withdrawn due to lack of efficacy. The patient did not require hospitalisation, and the event, which was not classified as an AE, resolved following treatment with prednisone.
No clinically significant haematological or clinical chemistry findings were observed during this study, and no statistically significant differences in adjusted mean change from baseline between the active treatment groups and placebo were observed for any vital sign measure. Mean values for vital sign parameters were similar across all treatment groups.
Discussion
The main objective of the study was to examine the effect of inhaled FF/VI on the HPA axis through the assessment of 24-h mean SC levels. No statistically significant differences in 0- to 24-h weighted mean SC between either FF/VI treatment group and placebo were found. Substantially lower 24-h cortisol levels after 7 days of once-daily prednisolone 10 mg (active control treatment) indicate that the measurements used were able to detect a treatment effect on HPA axis function. Secondary analyses of AUC(0–24) SC and 24-h UC were similar to and supported the primary end point findings. Trough SC was numerically lower in the FF/VI groups compared with placebo. Evaluation of endogenous cortisol concentrations is complicated because of the presence of circadian rhythm with highest cortisol concentrations in the morning and trough concentrations around bedtime. Therefore, single time-point measures are considered insensitive measures of HPA axis function, and the numerically lower values seen for trough SC were not considered clinically significant in the context of the 24-h data.
The systemic side effects of ICS have been extensively investigated 11,16. The use of corticosteroids, by all modes of administration, has the potential to induce adrenal suppression by affecting the HPA axis' regulation of cortisol, particularly at high doses 17. Such systemic effects of ICS are dependent on the amount of drug absorbed into the systemic circulation and also the drug's PK and PD properties 11,12. Previous studies have shown no significant suppression of 24-h UC with FF after 8 weeks of treatment with doses less than 800 μg 5–7. The results of the present study, together with previous evidence that the addition of VI does not affect systemic exposure to FF 18, suggest that the findings from these FF studies should be applicable to FF/VI.
In addition, the PK findings described here are consistent with previous studies of FF/VI 18,19. FF median Cmax was achieved around 30 min post-dose with both FF/VI doses. The high first-pass metabolism and limited systemic exposure to FF at therapeutic doses may help explain the lack of significant cortisol suppression seen at FF doses up to 600 μg 5–7.
The study used a robust methodology to assess non-inferiority of therapeutic doses of FF/VI on HPA axis function compared with placebo. The incorporation of domiciled 24-h clinic visits at baseline and on day 42 enabled plasma, urinary and PK samples to be collected continuously over a 24-h period, which is particularly important for HPA axis assessment because of the diurnal variation in cortisol levels 14. Multiple methods of assessing treatment compliance were applied, including dose and capsule counting, and a treatment diary. PD, PK and FEV1 findings correspondingly suggested very good compliance with both inhaled and capsule treatments. Although the study was not designed to assess efficacy, improvements in FEV1 were observed in the FF/VI groups, but not in those who received placebo inhalers.
The principal limitation of the study is its short duration (6 weeks). The study was designed to assess the short-term effects of FF/VI use on the HPA axis, and inferences cannot be drawn regarding any potential inhibitive effect of prolonged use in a real-world clinical setting. Also, the doses tested were those that are in development for therapeutic use; the effect on the HPA axis of supratherapeutic dosing was not examined in this study. The time of dosing was inexact (evening dosing was defined as an inhalation between 5 pm and 11 pm); however, the effect of this variability on the diurnal HPA axis profile is predicted to be small given the relatively flat PK profile for FF and that the natural cortisol peak is in the morning. Although, adrenocorticotropic hormone (ACTH) stimulation test was not included in this study, the use of complete 24-h SC profiling provides a robust and sensitive end point for assessment of effect on HPA axis. Finally, the exclusion criteria by which patients were ineligible if they had used oral corticosteroids within 12 weeks of the screening visit may not have been stringent enough to ensure that patients with suppressed adrenal response (within the normal range) were not enrolled; it has been shown that adrenal suppression can persist for over 6 months in a minority of patients treated with short-term, high-dose glucocorticoids 20.
The findings of this short-term study of the effects of inhaled FF/VI on HPA axis function demonstrated non-inferiority of FF/VI therapy compared with placebo for the primary end point of 0- to 24-h SC weighted mean after 42 days of treatment. The secondary 24-h serum AUC and UC results support this finding. Results for the prednisolone active control were as expected, with substantial cortisol suppression seen after 7 days of treatment. The lack of FF/VI effect on 24-h SC and UC is consistent with the low levels of FF systemic exposure observed for both doses of FF/VI. The safety and tolerability results, absence of SAEs and few treatment-related AEs are consistent with the findings of previous FF/VI studies. In conclusion, this study suggests that FF administered in combination with a LABA is not associated with suppression of mean SC at therapeutic doses.
Acknowledgments
The authors thank Courtney Crim, Tom Toler and Rodger Kempsford of GlaxoSmithKline for their contribution to the design of the study, and Jodie West of GlaxoSmithKline for assisting with the statistical analyses. Additionally, we would like to thank the study investigators, the patients and their families. Editorial support (in the form of development of draft outline, development of manuscript first draft, editorial suggestions to draft versions of the paper, assembling tables and figures, collating author comments, copy editing, fact checking, referencing, and graphic services) was provided by Ian Grieve at Gardiner-Caldwell Communications and was funded by GlaxoSmithKline. The charge for colour figures was paid by GlaxoSmithKline.
Supporting Information
Table S1. Percentage of PK samples below the lower limit of quantification (LLQ) (10 pg/mL FF, 20 pg/mL VI)* by treatment and time, study Day 42 (PK population).
References
- Global Initiative for Asthma (GINA) 2011. Global strategy for asthma management and prevention. Available at: http://www.ginasthma.com/ (accessed 10 January 2013)
- Price D, Robertson A, Bullen K, Rand C, Horne R, Staudinger H. Improved adherence with once-daily versus twice-daily dosing of mometasone furoate administered via a dry powder inhaler: a randomized open-label study. BMC Pulm Med. 2010;10:1. doi: 10.1186/1471-2466-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C, Santanello NC, Boccuzzi SJ, Wogen J, Strub P, Nelsen LM. Adherence to prescribed treatment for asthma: evidence from pharmacy benefits data. J Asthma. 2003;40:93–101. doi: 10.1081/jas-120017212. [DOI] [PubMed] [Google Scholar]
- Biggadike K, Bledsoe RK, Hassell AM, Kirk BE, McLay IM, Shewchuk LM, Stewart EL. X-ray crystal structure of the novel enhanced-affinity glucocorticoid agonist fluticasone furoate in the glucocorticoid receptor-ligand binding domain. J Med Chem. 2008;51:3349–3352. doi: 10.1021/jm800279t. [DOI] [PubMed] [Google Scholar]
- Bateman ED, Bleecker ER, Lötvall J, Woodcock A, Forth R, Medley H, Davis AM, Jacques L, Haumann B, Busse WW. Dose effect of once-daily fluticasone furoate in persistent asthma: a randomized trial. Respir Med. 2012;106:642–650. doi: 10.1016/j.rmed.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Bleecker ER, Bateman ED, Busse WW, Woodcock A, Frith L, House KW, Jacques L, Davis AM, Haumann B, Lötvall J. Once-daily fluticasone furoate is efficacious in asthma patients symptomatic on low-dose inhaled corticosteroids. Ann Allergy Asthma Immunol. 2012;109:353–358. doi: 10.1016/j.anai.2012.08.017. e4. [DOI] [PubMed] [Google Scholar]
- Busse WW, Bleecker ER, Bateman ED, Lötvall J, Forth R, Davis AM, Jacques L, Haumann B, Woodcock A. Fluticasone furoate demonstrates efficacy in patients with asthma symptomatic on medium doses of inhaled corticosteroid therapy: an 8-week, randomised, placebo-controlled trial. Thorax. 2012;67:35–41. doi: 10.1136/thoraxjnl-2011-200308. [DOI] [PubMed] [Google Scholar]
- Salter M, Biggadike K, Matthews JL, West MR, Haase MV, Farrow SN, Uings IJ, Gray DW. Pharmacological properties of the enhanced-affinity glucocorticoid fluticasone furoate in vitro and in an in vivo model of respiratory inflammatory disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L660–667. doi: 10.1152/ajplung.00108.2007. [DOI] [PubMed] [Google Scholar]
- Lötvall J, Bateman ED, Bleecker ER, Busse WW, Woodcock A, Follows R, Lim J, Stone S, Jacques L, Haumann B. 24h duration of the novel LABA vilanterol trifenatate in asthma patients treated with ICSs. Eur Respir J. 2012;40:570–579. doi: 10.1183/09031936.00121411. [DOI] [PubMed] [Google Scholar]
- Lötvall J, Bakke P, Bjermer L, Steinshamn S, Crim C, Sanford L, Scott-Wilson C, Haumann B. Efficacy and safety of 4 weeks' treatment with combined fluticasone furoate/vilanterol in a single inhaler given once daily in COPD: a placebo-controlled randomised trial. BMJ Open. 2012;2:e000370. doi: 10.1136/bmjopen-2011-000370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipworth BJ. Systemic adverse effects of inhaled corticosteroid therapy: a systemic review and meta-analysis. Arch Intern Med. 1999;159:941–955. doi: 10.1001/archinte.159.9.941. [DOI] [PubMed] [Google Scholar]
- Masoli M, Weatherall M, Holt S, Shirtcliffe P, Beasley R. Inhaled fluticasone propionate and adrenal effects in adult asthma: systematic review and meta-analysis. Eur Respir J. 2006;28:960–967. doi: 10.1183/09031936.06.00119305. [DOI] [PubMed] [Google Scholar]
- Clark DJ, Grove A, Cargill RI, Lipworth BJ. Comparative adrenal suppression with inhaled budesonide and fluticasone propionate in adult asthmatic patients. Thorax. 1996;51:262–266. doi: 10.1136/thx.51.3.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DI, Allen DB. Evaluation of tests of hypothalamic-pituitary-adrenal axis function used to measure effects of inhaled corticosteroids. Ann Allergy Asthma Immunol. 2007;98:118–127. doi: 10.1016/S1081-1206(10)60683-7. [DOI] [PubMed] [Google Scholar]
- National Institutes for Health (NIH) Guidelines for the Diagnosis and Management of Asthma – Expert Panel Report 3, 2007. Bethesda, MD: U.S. Department of Health and Human Services; 2007. NIH Publication No. 07-4051. [Google Scholar]
- Barnes PJ. Inhaled corticosteroids. Pharmaceuticals. 2010;3:514–540. doi: 10.3390/ph3030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmet A, Kim H, Spier S. Adrenal suppression: a practical guide to the screening and management of this under-recognized complication of inhaled corticosteroid therapy. Allergy Asthma Clin Immunol. 2011;7:13. doi: 10.1186/1710-1492-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempsford R, Allen A, Bareille P, Bishop H, Hamilton M, Cheesbrough A. The safety, tolerability, pharmacodynamics and pharmacokinetics of inhaled fluticasone furoate (FF) and vilanterol (VI) are unaffected by administration in combination. Eur Respir J. 2011;38:824s. (Suppl. 55). [abstract] [Google Scholar]
- Allen A, Bianco J, Bal J, Tombs L, Kempsford R. The absolute bioavailability of fluticasone furoate (FF) and vilanterol (VI) trifenatate following inhaled administration in combination in healthy subjects. Eur Respir J. 2011;38(Suppl. 55):3976s. abstract. [Google Scholar]
- Henzen C, Suter A, Lerch E, Urbinelli R, Schorno XH, Briner VA. Suppression and recovery of adrenal response after short-term, high-dose glucocorticoid treatment. Lancet. 2000;355:542–545. doi: 10.1016/S0140-6736(99)06290-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Percentage of PK samples below the lower limit of quantification (LLQ) (10 pg/mL FF, 20 pg/mL VI)* by treatment and time, study Day 42 (PK population).