

Abstract
Recent studies have identified new roles for mitochondria in the regulation of autoinflammatory processes. Emerging data suggests that the release of danger signals from mitochondria in response to stress and infection promotes the formation of the inflammatory signaling platform known as inflammasomes. Activation of inflammasomes by damaged mitochondria results in caspase-1-dependent secretion of the inflammatory cytokines IL-1β and IL-18, and an inflammatory form of cell death referred to as pyroptosis. Here, we review recently described mechanisms that have been proposed to be involved in mitochondria-mediated regulation of inflammasome activation and inflammation. In addition, we highlight how aberrant regulation of mitochondria-induced inflammasome activation centrally contributes to the inflammatory process that are responsible for obesity and associated metabolic diseases.
Keywords: Mitochondria, NLRP3, inflammasome, metabolic disease
Historical perspective on the origin and importance of mitochondria
In the early 1960s it was proposed that the presence of mitochondria in cells were the result of evolutionary symbiosis, which is often referred to as endosymbiosis [1]. Several studies have argued that mitochondria were originally separate bacteria that ended up in a symbiotic relationship within mammalian cells during the course of evolution [2]. The mitochondrion is a major organelle required for numerous functions within the cell and is essential for survival. One of the earliest functions attributed to mitochondria is involvement in the glycolysis/TCA cycle/electron transport chain (see Glossary) that generates energy in the form of ATP (Figure 1A). Defects in the electron transport chain (transport system that generates the major amount of energy through oxidative phosphorylation) are usually detrimental to the host demonstrating the importance of mitochondria. One such example is cyanide poisoning that inhibits the ability of mitochondria to use oxygen to generate ATP [3]. Thus it was not surprising when in the late 1990s scientists discovered a role for mitochondria in promoting cell death (Figure 1B). Contrary to previous held beliefs that the loss of mitochondrial function and ATP produced induced cell death, it is now well established that apoptosis is actually an active process that requires energy. Several recent studies have identified a dynamic role for mitochondria in cell death, especially the role of mitochondria and mitochondrial products in the execution of an inflammatory form of cell death that is mediated by Nod-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome activation referred to as pyroptotic cell death. In this manuscript we will review the crucial role played by mitochondria in the regulation and activation of the NLRP3 inflammasome, discuss recently described modulators of these pathways and lastly detail metabolic disorders that result from mitochondrial dysfunction in context of the NLRP3 inflammasome.
Figure 1. Mitochondria are critical for generation of ATP and regulating apoptosis.
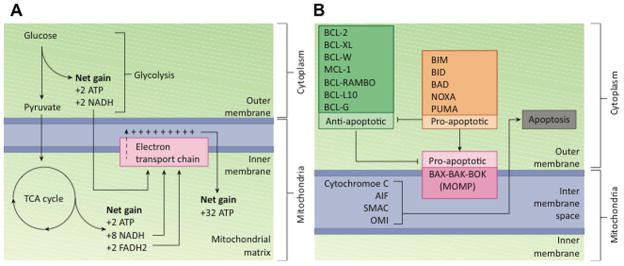
A. Energy is stored in the form of ATP in the mitochondria. Three major metabolic pathways convert glucose into ATP; Glycolysis, the TCA cycle and the Electron transport chain. Glycolysis is the first step in which one glucose molecule is converted into two pyruvate, two ATP and two NADH molecules in the cytoplasm. The TCA cycle takes place in the mitochondrial matrix, and generates two ATP, 8 NADH and two FADH2 molecules. The Electron transport chain generates 32 ATP molecules by utilizing the NADH and FADH2 generated in the previous steps.
B. BCL-2 family members are centrally involved in mitochondrial apoptosis. Activated BAX, BAK and BOK form pores in the mitochondrial membrane. Anti-apoptotic BCL-2 family members BCL-2, BCL-xL, BCL-W, MCL-1, BCL-RAMBO, BCL-L10 and BCL-G have also been shown to inhibit mitochondrial apoptosis by inhibiting BAX, BAK or BOK and subsequent mitochondrial outer membrane permeabilization (MOMP). Anti-apoptotic BCL family members BIM, BID, BAD, NOXA and PUMA on the other hand have been identified to promote apoptosis by either promoting translocation of BAX to the mitochondrial membrane or inhibiting the function of anti-apoptotic BCL proteins. Translocation of BAX, BAD and BOK results in MOMP and release of apoptotic effectors such as cytochrome C, AIF, SMAC and OMI, all of which induce apoptosis.
Metabolic pathways, mitochondria and the generation of energy
To fuel the cellular processes that are required to support life, food must be transformed into ATP. Three major metabolic pathways are involved in the stepwise generation of ATP; Glycolysis, the Tricarboxylic acid (TCA) cycle (also known as the Krebs cycle or citric acid cycle) and the Electron transport chain (also known as oxidative phosphorylation) (Figure 1A). Glycolysis, where glucose is converted into two pyruvate molecules, takes place in the cytoplasm. This process yields two ATP and two NADH molecules [4]. The pyruvates released in this step are shuttled into the TCA cycle, which takes place in the mitochondrial matrix. The TCA cycle generates two ATP, eight NADH and two FADH2 molecules [5]. The NADH and FADH2 molecules are then used in the electron transport chain, located in the inner membrane of mitochondria, to generate additional ATP [6]. In total, 32 ATP molecules are generated by the electron transport chain system. Therefore one glucose molecule generates a total of 34 ATP molecules within the mitochondria. Even though electron transport chain is an efficient ATP generating process, harmful reactive oxygen species (ROS) are generated as by-products of these reactions. Certain genetic conditions that cause imbalances in the cell metabolic pathways can result in the accumulation of ROS, which can induce cell damage and ultimately death.
The role of mitochondria in cell death
Cell death regulated by mitochondria-associated proteins is termed ‘mitochondrial apoptosis’ (Figure 1B). The B cell lymphoma (BCL) protein family includes members that are both pro- and anti-apoptotic molecules, and regulate cell death by controlling mitochondrial outer membrane permeabilization (MOMP). The BCL-2 family members BAX, BAK and BOK are known to be pro-apoptotic and promote MOMP [7–9]. The BH3-only protein BID assists BAX activation and oligomerization on the mitochondrial outer membrane to promote MOMP [10]. BCL-2 was one of the first mitochondria-associated proteins shown to regulate apoptosis by inhibiting BAX-mediated initiation of MOMP [11]. Alongside BCL-2, family members known for their anti-apoptotic functions include BCL-XL, BCL-W, MCL-1, BCL-RAMBO BCL-L10 and BCL-G [12]. Furthermore, BH3-only proteins such as BIM, BID, BAD, NOXA and PUMA promote mitochondrial apoptosis by inhibiting BCL-2, or directly inducing MOMP [12]. Thus, members of BCL-2 family tightly regulate mitochondrial apoptosis. Apart from BCL-2 family members, accumulation of ROS and increased calcium mobilization are also known to damage mitochondria through MOMP [13]. The final outcome of MOMP is the release of effector molecules from mitochondria that induce apoptosis through the activation of executioner caspases. The major effectors that mediate mitochondrial apoptosis as a result of MOMP are cytochrome c, apoptosis inducing factor (AIF), second mitochondria-derived activator of caspase (SMAC) and OMI (serine protease also known as HtrA2) [14]. All of these proteins are normally located within the intermembrane space of mitochondria, and when released into the cytoplasm they induce caspase-dependent apoptosis. While cytochrome c and AIF directly induce apoptotic protease activating factor 1 (APAF-1) oligomerization to promote formation of the apoptosome, SMAC and OMI act indirectly by sequestering inhibitor of apoptosis (IAP) proteins to release APAF-1 [14, 15]. Although caspase-dependent apoptosis is the main pathway for mitochondrial apoptosis, cell death can occur independent of caspases once mitochondrial function is perturbed [16].
Recent studies have now shown that cell death pathways are interconnected with inflammation, especially with the flurry of reports on caspase-8 promoting inflammasome activation [18].
Nod-like receptors and inflammasomes
Nod-like receptors (NLRs) are an evolutionary conserved family of receptors that reside in the cytoplasm and recognize pathogen and danger associated molecular patterns (PAMPS and DAMPs). Of the 22 NLRs in humans and 34 NLRs in mice, only a handful have been formally characterized to date [19]. While NLRs can engage several signaling pathways upon ligand recognition, certain NLRs including NLRP1b, NLRP2, NLRP3, NLRC4, NLRP6, NLRP7 and NLRP12 form multimeric protein complexes comprising of adaptor apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1, termed inflammasomes [18, 20, 21]. However, it should be noted that the precise roles for NLRP2, NLRP6, NLRP7 and NLRP12 in inflammasome complex formation still remain incompletely characterized and thus future studies are needed to define their specific roles in inflammasome activation. Assembly of the inflammasome triggers autocleavage and activation of the caspase-1. Biologically active caspase-1 then processes the proinflammatory cytokines pro-IL-1β and pro-IL-18 into their bioactive mature forms. In addition, caspase-1 activation also provokes an inflammatory pyroptotic cell death.
NLRP3 is the best-characterized inflammasome, mainly because of its ability to be activated by a wide variety of ligands. Activation of the NLRP3 inflammasome requires two signals; a priming signal that is necessary for the upregulation of NLRP3 and pro-IL-1β followed by an activation signal that prompts NLRP3 to assemble the inflammasome complex [22]. Several DAMPs such as ATP, alum hydroxide, silica crystals, urea crystals, nigericin and active infections with bacteria, viruses and fungi activate the NLRP3 inflammasome [23]. How NLRP3 recognizes these different ligands and whether a common signal converges downstream of PAMPs and DAMPs to activate NLRP3 has been a longstanding question in the field. Some of the proposed common events downstream from PAMPs and DAMPs are mitochondrial dysfunction and ROS generation [24, 25]. Several reports propose distinct mechanisms that explain the central role of mitochondria in the activation of the NLRP3 inflammasome. These studies provide mechanistic insight to explain how both mitochondrial dysfunction and gain-of-function mutations in NLRP3 can result in similar metabolic disorders in patients.
Mitochondria, inflammation and regulation of the NLRP3 inflammasome
The role of mitochondria in inflammation was initially suggested from studies that discovered a role for the mitochondrial antiviral signaling protein (MAVS) in eliciting antiviral interferon responses during viral infections [26–28]. With the discovery of the NLRP3 inflammasome and pyroptotic cell death in the last decade, the involvement of mitochondria in these pathways has similarly evolved. Several distinct pathways emanating from mitochondria, and in particular mitochondrial dysfunction, have been proposed to modulate NLRP3 inflammation activation (Figure 2). Even though these pathways are shown as individual separate pathways in Figure 2 and discussed below likewise, it is important to note that these pathways are interconnected and might not be mutually exclusive.
Figure 2. Mitochondria at the center of NLRP3 inflammasome activation.

NLRP3 inflammasomes are activated by a myriad of stimuli that ranges from virus, bacteria, and fungi to danger associated molecular patterns (DAMPs) such as ATP, nigericin and alum. Once activated, NLRP3 forms a multimeric protein complex with associated speck-like protein containing a CARD (ASC) and caspase-1 (CASP1) termed the inflammasome. Caspase-1 is activated in the inflammasome complex, which cleaves pro-IL1β and pro-IL-18 into their bioactive mature forms. Mitochondria are central regulators of NLRP3 inflammasome activation. Mitochondrial reactive oxygen species (ROS), Ca2+ overload, reduced NAD+, cardiolipin, mitofusin, mitochondrial antiviral signaling protein (MAVS) and mitochondrial DNA (mtDNA) have all been shown to promote NLRP3 inflammasome activation. NOD2/RIPK2-dependent mitophagy and LC3B/Beclin 1-mediated autophagy are involved in the clearance of damaged mitochondria, and thus negatively regulate NLRP3 inflammasome activation.
Mitochondrial ROS
Mitochondria are the major source of cellular ROS. The electron transport chain in the mitochondrial inner membrane is critically involved in the generation of energy, where oxygen acts as an electron acceptor. When the electron transport chain breaks down, ROS can accumulate to toxic levels within cells. Several studies have shown that ATP- and monosodium urate (MSU) crystal-induced ROS production activates inflammasomes [29, 30]. ROS generated during phagocytosis of silica and asbestos particles activated NLRP3 inflammasome formation in macrophages [31]. Conversely, treatment of macrophages with the ROS inhibitors N-acetyl-L-cystine [32] or (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate (APDC) [31] can inhibit silica and asbestos-induced NLRP3 inflammasome activation. Furthermore, inhibition of mitochondrial complex-I by rotenone or complex-III by antimycin A induces robust ROS production by mitochondria [33, 34]. This enhanced ROS production is sufficient to drive NLRP3 inflammasome activation, suggesting mitochondrial ROS as a direct activator of the NLRP3 inflammasome [25].
ROS activation is not an absolute requirement for activation of all NLRP3 inflammasomes. In particular, stimulation of macrophages with linezolid (from the oxazolidinone class of antibiotics) or infection of macrophages with influenza and encephalomyocarditis viruses does not require ROS for activation of the NLRP3 inflammasome [35, 36]. Additionally, whether ROS production is a prerequisite or just a consequence of inflammasome activation remains obscure. Future studies looking at a time lapse release/production of ROS following stimulation of macrophages during NLRP3 inflammasome activation at a single cell level will be instrumental in defining the dynamics and requirement of ROS production.
Calcium mobilization and potential role for mitochondria
Calcium (Ca2+) is a secondary messenger that plays pivotal roles in the regulation of multiple signaling pathways within cells. Aberrant fluxes in Ca2+ can result in catastrophic cellular events and thus cells deploy multiple strategies to control intracellular Ca2+ levels. In particular, mitochondria play a pivotal role in regulating Ca2+ levels by accepting Ca2+ that is released from the endoplasmic reticulum [37]. While a controlled level of Ca2+ storage within the mitochondria modulates Ca2+ signaling, Ca2+ overload can cause mitochondrial dysfunction. A role for Ca2+ influx to the cytoplasm in NLRP3 inflammasome activation is substantiated by studies using the Ca2+-chelating agent BAPTA-AM [38, 39]. Incubation of macrophages with BAPTA-AM before co-stimulation with lipopolysaccharide (LPS) and ATP or infection with Mycobacterium abscessus inhibited NLRP3 inflammasome activation in a dose dependent manner [38, 39]. Additional studies confirmed that Ca2+ influx into the cytoplasm following ATP, ultraviolet B radiation (UBV), and cholesterol-dependent cytolysins treatment activates the NLRP3 inflammasome in LPS primed macrophages [40–42]. Furthermore, Ca2+ detection by the Ca2+-sensing receptor (CaSR) directly promotes NLRP3 inflammasome activation [43, 44]. Mounting evidence now suggests that an influx of Ca2+ into the cytoplasm is a common proximal event involved in activation of the NLRP3 inflammasome [44, 45]. A recent study suggested that K+ efflux could be upstream of Ca2+ influx during NLRP3 inflammasome activation [46], although how these two pathways may be linked is still ambiguous and will need further investigation.
Most of the studies looking at the role of Ca2+ overload suggest a direct role for Ca2+ in NLRP3 inflammasome activation, although these studies are not without caveats. Ca2+ is a secondary messenger required for various cellular functions and is abundantly present within the cellular cytoplasm. Why does this Ca2+ present within the cytoplasm not activate NLRP3 inflammasomes? Is there a Ca2+ threshold that determines activation of the NLRP3 inflammasome? One hypothesis that could possibly explain these discrepancies is that Ca2+ overload causes mitochondrial dysfunction and mitochondrial dysfunction eventually induces NLRP3 inflammasome activation. Whether Ca2+ induced NLRP3 inflammasome activation requires mitochondria remains unknown and experiments with cells lacking mitochondria could answer these questions.
Reductions in NAD+ trigger NLRP3 inflammasome assembly
Inducers of the NLRP3 inflammasome such as ATP and nigericin have been shown to cause mitochondrial damage. Moreover, ATP- and ROS-induced damage to mitochondria causes reductions in cytoplasmic levels of NAD+ [47]. Misawa et al. further showed that the reduced NAD+ induces α-tubulin dependent assembly of mitochondria that ultimately promotes recruitment of ASC and NLRP3 [47]. This study clearly demonstrated that mitochondrial dysfunction-associated reduction in NAD+ promotes NLRP3 inflammasome activation. Several other models have been described where specific molecules on mitochondria such as cardiolipin and mitofusins promote NLRP3 inflammasome assembly. Further investigations are required to delineate whether NAD+ is a specific mechanism or a precursor to these pathways.
Cardiolipin in NLRP3 inflammasome activation
Cardiolipins are lipids that facilitate optimal oxidative phosphorylation in mitochondria [48]. Interestingly, cardiolipin deficient cells are resistant to cell death [49]. Recent studies by Iyer et al. show that cardiolipins are critical modulators of NLRP3 inflammasome activation [35]. Mitochondrial destabilization induces externalization of cardiolipin to the outer membrane of the mitochondria [50]. Iyer et al. showed that cardiolipin on the mitochondrial outer membrane directly binds to the leucine rich repeats (LRR) of NLRP3 and activates the NLRP3 inflammasome [35]. Knockdown of cardiolipin synthase (CLS; the enzyme that synthesizes cardiolipin) using siRNA, although only ~50% efficient, resulted in a significant reduction in caspase-1 activation and subsequent IL-1β release [35]. In a cell free system, addition of cardiolipin is sufficient to activate the NLRP3 inflammasome and induce caspase-1 cleavage. This study was the first to show that mitochondria-associated cardiolipin is required for recruitment and activation of the NLRP3 inflammasome. Molecular insights into how cardiolipin binding and its ligand activity induce NLRP3 inflammasome activation are needed. Studying CLS-deficient cells and mice will ultimately be required to establish the direct role of cardiolipin in cell death and NLRP3 inflammasome activation.
Mitofusins in NLRP3 inflammasome activation
Mitofusin 1 and 2 are mitochondrial outer membrane associated proteins that are crucial for mitochondrial fusion. Mice lacking either mitofusin 1 or mitofusin 2 are embryonic lethal, demonstrating the critical roles played by these proteins in vivo [51, 52]. In vitro studies with cells lacking either mitofusin 1 or 2 have corroborated their roles in mitochondrial fusion and loss of mitochondrial membrane potential [51, 52]. A recent study has now implicated mitofusins in the activation of the NLRP3 inflammasome during influenza and encephalomyocarditis virus (EMCV) infections [36]. Influenza and EMCV infections induce a loss of mitochondrial membrane potential, which is essential for NLRP3-mitofusin 1 and NLRP3-mitofusin 2 interactions. Biochemical analysis identified that the 4,3 hydrophobic heptad repeat (HR) region 1 of mitofusin 2 interacts with NLRP3 [36]. Knockdown of mitofusin 2 significantly reduced caspase-1 activation and IL-1β production in response to viral infections. While this study clearly suggests mitofusins as potential docking sites for NLRP3 on the mitochondria, whether mitofusins directly activate NLRP3 or merely act as a docking site for inflammasome assembly needs to be further investigated. Even though mitofusin 1 was able to equally recruit NLRP3, its role in NLRP3 inflammasome activation remains obscure, and whether NLRP3-mitofusin 1- mitofusin 2 are present in the same complex is not known. Lastly, the roles for both mitofusin 1 and mitofusin 2 need to be confirmed using cells deficient in these molecules and by generating mitofusin 1 and mitofusin 2 conditional knockout mice.
Mitochondrial DNA
Bacterial and viral RNA can also activate the NLRP3 inflammasome [53]. Similarly, self and foreign DNA in the cytoplasm have also been shown to activate the AIM2 inflammasome [54, 55]. Mitochondrial DNA is released into the cytoplasm following intrinsic cell death, resulting in inflammation [56, 57]. Following on from these reports, two separate studies have now shown that mitochondrial DNA in the cytoplasm activates the NLRP3 inflammasome [17, 58]. Nakahira et al. suggested that mitochondrial ROS is required for the release of mitochondrial DNA and activation of the NLRP3 inflammasome [59]. Shamida et al. further showed that mitochondrial death is a prerequisite for the release of oxidized mitochondrial DNA into the cytoplasm [58]. The oxidized mitochondrial DNA directly bound NLRP3 to activate the inflammasome. Further supporting a direct role for mitochondrial DNA, cells lacking mitochondrial DNA (generated by ethidium bromide treatment) were unable to provoke NLRP3 inflammasome activation [17]. Interestingly, NLRP3 is required for the release of mitochondrial DNA during inflammasome activation [17]. This suggests that mitochondrial DNA might act in a positive feedback loop to potentiate NLRP3 inflammasome activation. In contrast, a recent report demonstrated that mitochondrial apoptosis is totally dispensable for NLRP3 inflammasome activation using a series of genetically deficient mouse models [60]. Additional studies are needed to clarify different ways mitochondrial apoptosis may be involved in the activation of the NLRP3 inflammasome.
Mitochondrial antiviral signaling protein (MAVS) in NLRP3 inflammasome activation
MAVS are central adapter molecules required for transmitting interferon responses and signaling events downstream of nucleic acid sensors such as retinoic acid-inducible gene 1 (RIG-I) and melanoma differentiation-associated protein 5 (MDA-5) [26–28]. The discovery that MAVS are localized in the mitochondrial membrane implicated mitochondria in innate immune signaling [27]. MAVS form prion like aggregates on the outer membrane of mitochondria to perpetuate antiviral signaling [61], and it is the caspase activation and recruitment domains (CARDs) in MAVS that are necessary and sufficient to form these aggregates in vitro [61]. Interestingly, MAVS has also been reported to be required for NLRP3 inflammasome activation in response to non-crystalline NLRP3 activators such as ATP, nigericin and poly I:C [62]. Indeed, MAVS were required for recruitment of NLRP3 to the mitochondria, although surprisingly this recruitment did not require ASC and was solely dependent on the pyrin domain of NLRP3 [62]. Although the requirement of MAVS for NLRP3 inflammasome activation in response to poly I:C has been corroborated [63, 64], several groups have been unable to recapitulate a role for MAVS in NLRP3 inflammasome activation in response to ATP or nigericin [60, 63, 64]. Biochemical studies evaluating the interactions of MAVS with NLRP3 during stimulation with NLRP3 agonists will shed light onto how MAVS are specifically required for poly I:C induced NLRP3 inflammasome activation. Moreover, studies elucidating the contribution of the mitochondrial membrane in these interactions will be important to fully understand the specific nature of MAVS in activating NLRP3 inflammasome.
Negative regulators of mitochondrial damage and NLRP3 inflammasome activation
Mitochondria have emerged as central regulators of NLRP3 inflammasome activation and a plethora of studies have elucidated the various mitochondrial components required to trigger inflammasome signaling. Studies examining the mitochondrial pathways and components that negatively regulate NLRP3 inflammasome activation are scarce. Autophagy is a fundamental cellular process that is required for clearance of unwanted or damaged organelles within the cell. Autophagy is also a mechanism for the cell to recycle critical nutrients. Inhibition of autophagy machinery by 3-methyladenine (3-MA) instigated activation of the NLRP3 inflammasome [25]. Nakahira et al. showed that autophagy regulates the clearance of damaged mitochondria from the cytoplasm, a process now known as mitophagy [59]. Genetic ablation of LC3B or Beclin 1, both critical modulators of autophagy, results in the accumulation of damaged mitochondria and enhanced NLRP3 inflammasome activation during ATP stimulation of macrophages [59]. As such, mice deficient in LC3B or Beclin 1 are highly susceptible to sepsis and LPS-induced endotoxemia. Similarly, mice deficient in Atg16L1, a critical component of autophagy machinery, show enhanced NLRP3 inflammasome activation [65].
In a separate study involving influenza virus infections, Lupfer et al. demonstrated a role for the nucleotide-binding oligomerization domain containing 2 (NOD2) protein and its adaptor receptor interacting protein kinase 2 (RIPK2) in promoting mitophagy to clear damaged mitochondria from the cells [66]. NOD2 and RIPK2-deficient mice are highly susceptible to influenza virus infection as a result of uncontrolled inflammation and hyperactive NLRP3 inflammasomes [66]. Consistent with their role in autophagy [67], NOD2 and RIPK2-deficient cells accumulate damaged mitochondria. Phosphorylation of unc-51 like autophagy activating kinase 1 (ULK1) is an important event that promotes mitophagy [68]. Mechanistic studies showed that RIPK2 promotes ULK1 phosphorylation to initiate the formation of the autophagosome and clearance of damaged mitochondria [66]. These studies clearly highlight the need for future work to examine regulatory pathways that purge damaged mitochondria during inflammation, infection and cellular stress. Any defects in these pathways can promote uncontrolled inflammation and immunopathology that can be detrimental to the host. Thus, understanding the molecular mechanisms of how these pathways can be intersected will be helpful in designing potential therapeutics.
Mitochondria dysfunction-induced inflammasome activation drives obesity-associated disease
Mitochondria play essential roles in the maintenance of metabolic health and disruption of mitochondrial activity is believed to be a leading cause of obesity and associated metabolic disorders. Obesity-driven inflammation provokes a perpetual cycle of metabolic dysfunction and contributes to the development of obesity-related disorders including type 2 diabetes mellitus (T2DM), hepatosteatosis, dyslipidemia and atherosclerosis [69]. Under healthy conditions, mitochondria contribute to the regulation of metabolic homeostasis by supplying cells with ATP, controlling energy expenditure, coordinating the disposal of ROS and regulating intrinsic cell death. However, nutrient overload that results from excessive consumption of energy rich foods and lack of physical activity can cause the accumulation of partially oxidized substrates (such as fatty acids) in mitochondria and aberrant mitochondrial ROS production. Unchecked ROS production and the buildup of improperly oxidized substrates ultimately lead to impaired mitochondrial function [70]. Indeed, mitochondria in obese individuals exhibit altered morphology, decreased ATP output, impaired bioenergetics and diminished fatty acid oxidation [71–77].
Caloric restriction and exercise can promote improved mitochondrial bioenergetics efficiency and biogenesis [78]. Reductions in ROS production and proliferation of mitochondria are believed to be responsible for the enhanced mitochondrial activity that results from restrictions in caloric intake [79, 80]. Mitochondrial dysfunction has also been reported to contribute to other metabolic syndrome disorders that are associated with obesity. For instance, defective mitochondrial activity is believed to be involved in T2DM pathogenesis [74]. T2DM is associated with impaired mitochondrial respiration and bioenergetics [73, 75], and inherited defects in mitochondrial function are reported to contribute to the increased risk of severe insulin-resistance in offspring from parents with T2DM [76].
Although defective mitochondrial activity has been extensively linked to obesity-associated disease, how aberrant regulation of mitochondria mechanistically contributes to the induction of the inflammatory responses that are responsible for driving obesity and metabolic dysfunction had remained poorly characterized until recently. The NLRP3 inflammasome can sense danger-associated signals that are induced by defective mitochondria. An NLRP3 inflammasome-mediated inflammatory cascade then promotes adipose tissue inflammation and metabolic dysfunction. Specifically, it was discovered that the NLRP3 inflammasome can detect various obesity-associated DAMPs including ROS, damaged mitochondria, saturated fatty acids and ceramides; and that the activation of caspase-1 in the inflammasome complex spurs inflammatory signaling [24, 81–84]. Additionally, the development of adipose tissue inflammation and metabolic impairment in both diet-induced and genetic models of obesity is associated with deregulated caspase-1 activation and hyperinflammatory secretion of the inflammasome-derived cytokines IL-1β and IL-18 in various metabolic organs [81, 83, 85].
Defective regulation of mitochondrial activity has recently been identified as a central instigator of NLRP3 inflammasome-mediated inflammation in obesity-associated disease. Caloric overload causes adipocytes to undergo considerable differentiation and expansion in order to accommodate the storage of energy in the form of lipids [86]. These changes result in the dramatic upregulation of mitochondrial ROS levels. ROS is a potent inducer of NLRP3-inflammasome activation [24, 25, 59] and ROS is believed to be a prominent inducer of proinflammatory responses that are mediated by adipose tissue infiltrating macrophages. The buildup of damaged mitochondria that results from defective autophagy in obese individuals can also spur NLRP3-dependent inflammasome activation and associated cytokine production. In a recent study, it was shown that the excess saturated fatty acid palmitate causes the buildup of defective mitochondria as a result of impaired autophagy [82]. In this model, palmitate-mediated disruption of AMP-activated kinase (AMPK) activity caused impaired autophagy and the resulting accumulation of damaged mitochondria was responsible for inciting downstream NLRP3 inflammasome-induced inflammatory signaling.
NLRP3 inflammasome-induced cytokine production contributes to impaired insulin signaling and glucose homeostasis by inducing apoptosis in insulin-producing β-cells in the pancreas [87] and by promoting insulin resistance in adipocytes [83]. The release of IL-1β and IL-18 also results in potent recruitment of inflammatory macrophages and T cells into adipose tissue. The mobilization of activated immune cells into metabolic organs provokes the release of additional obesity-associated danger signals from damaged tissue and spurs the development of a perpetual state of low-grade inflammation. Importantly, genetic ablation of critical NLRP3 inflammasome components and pharmacological inhibition of caspase-1 in high fat diet-fed mice markedly limited the development of obesity and resulted in improved insulin sensitivity and glucose tolerance [83].
In recent clinical trials, blockade of IL-1 signaling with IL-1R antagonist treatment was found to stabilize blood glucose levels and improve pancreatic β-cell function [88]. The efficacy of IL-1R antagonists in the treatment of T2DM underscores the central importance of inflammasome-derived cytokines in driving metabolic disease, however complete abrogation of IL-1 signaling can pose serious side effects including increased susceptibility to infections and some forms of cancer. Thus, improved strategies are needed to target the specific inflammatory pathways that instigate metabolic disease and obesity. Recent studies in animal models, which identify critical roles for NLRP3 inflammasome-mediated cytokine production in driving obesity-associated inflammation and metabolic dysfunction, suggest that therapeutics that target inflammasome activation may hold great promise in the treatment of obesity and associated metabolic disorders.
Concluding remarks
Mitochondria have diverse functions that range from generating and storing energy for the cell, controlling cell death, inflammation and regulating NLRP3 inflammasome activation. Thus it is not surprising that defects in either mitochondrial function or NLRP3 inflammasome drive similar metabolic disorders such as obesity-associated diseases. While recent studies have provided an improved insight into how mitochondria regulate NLRP3 inflammasome activation, there are still multiple important questions that remain (Box 1). One of the limitations in understanding the role of mitochondrial function in vivo has been the generation of mice with deficiencies in mitochondrial proteins. This is mostly due to the critical roles played by these molecules in development and cell death. With the development of newer technologies such as the Crispr/Cas9 gene targeting system and conditional deletion of genes, several of these questions will hopefully be answered in coming years. Such future studies will result in a more complete understanding of how mitochondria are involved in regulating these diverse functions within the cell.
Box 1. Outstanding Questions.
How do mitochondria decide the fate of the cell between apoptotic and pyroptotic cell death?
Are mitochondria required for NLRP3 inflamamsome activation?
Several mitochondrial components promote NLRP3 inflammasome activation following cellular stress, injury or infection. Is there a common pathway where these mitochondrial components converge to activate the NLRP3 inflammasome?
Why is mitochondrial-dependent regulation of inflammasome activation specific to the NLRP3 inflammasome and not to other types of inflammasomes such as the NLRC4, AIM2 and NLRP1b inflammasomes?
What comes first: inflammasome activation or mitochondrial dysfunction?
What are the common metabolic pathways triggered by mitochondrial and NLRP3 dysfunction that could be targeted to generate novel therapeutics?
Highlights.
Mitochondrial dysfunction induces specific activation of NLRP3 inflammasome.
Mitochondria recruit NLRP3 and aid the assembly of NLRP3 inflammasome.
Autophagy/mitophagy clear damaged mitochondria and inflammasome activation.
Mitochondria and NLRP3 dysfunction promote metabolic diseases.
Acknowledgments
We thank Christopher Lupfer and Deepika Sharma for reviewing and editing the manuscript. Although we tried our best to be comprehensive and include most of the literature, we sincerely apologize to authors whose work was not cited in this review. PG is a postdoctoral fellow supported by Paul Barrett Endowed Fellowship from St. Jude Children’s Research Hospital. This work was supported in part by grants from the National Institute of Health (Grants AR056296, CA163507 and AI101935) and the American Lebanese Syrian Associated Charities (ALSAC) to T-D.K.
Glossary
- Apoptosis
Programmed cell death that is executed by caspases. Apoptosis is commonly known as a silent cell death as this process does not induce inflammation
- Apoptosome
During mitochondrial apoptosis (intrinsic cell death), cytochrome c release from the mitochondria prompts the formation of a multimeric protein complex containing APAF-1. The APAF-1 complex, known as the apoptosome then recruits and activates executioner caspases to initiate apoptosis
- Adenosine triphosphate (ATP)
ATP is the cellular source of energy. Cells store energy in the form of ATP in the mitochondria. Energy is stored in the phosphate bonds of ATP, such that when ATP is broken down to ADP, energy is released
- Autophagy
A highly conserved cellular mechanism that is responsible for recycling of damaged cellular organelles within the cytoplasm of the cell
- Cardiolipin
Lipids present in the inner membrane of mitochondria. While cardiolipin is known for its role in mitochondrial oxidative phosphorylation and induction of apoptosis, recent work has shown that cardiolipin recruits and activates the NLRP3 inflammasome
- Crispr/Cas9
A novel gene editing system that allows very efficient and targeted gene editing in both cells and mice in vivo
- Danger associated molecular patterns (DAMPs)
DAMPs are cues that inform a cell about a possible invasion or danger. Some common examples of DAMPs include DNA in the cytosol, ATP, heat shock proteins and HMGB1
- Electron transport chain
The electron transport chain present in the inner-membrane of the mitochondria is the major site for oxidative phosphorylation, where electrons from NADH and FADH2 generated in glycolysis and the TCA cycle are used to generate ATP
- Glycolysis
Glucose is converted into pyruvate by a metabolic pathway known as glycolysis. This is the first critical step for conversion of glucose molecules into ATP, as products generated in the glycolysis step are fed into the TCA cycle. Glycolysis also generates ATP, although it is not as efficient
- Inflammasome
The inflammasome is a multimeric protein complex that consists of a Nod-like receptor sensor, bipartite adaptor ASC and caspase-1. Assembly of the inflammasome complex results in the activation of caspase-1. Bioactive caspase-1 cleaves pro-IL-1β and pro-IL-18 into their active forms and also provokes an inflammatory form of cell death that is referred to as pyroptosis
- Mitochondrial antiviral signaling protein (MAVS)
MAVs is an adaptor protein localized in the outer-membrane of the mitochondria. MAVS is required for interferon signaling downstream of nucleic acid sensors such as RIG-I and MDA-5 and thus MAVS is a critical initiator of antiviral immune responses
- Metabolic disorders
Disorders that arise as a result of disruption in the process of metabolism which is required to generate energy for the cell
- Mitophagy
A process whereby damaged mitochondria are targeted for degradation and recycling via autophagy
- Mitofusins
Mitofusins are GTPases present in the outer-membrane of mitochondria and mediate mitochondrial fusion. Mitofusins can interact with NLRP3 and regulate inflammasome activation
- Mitochondrial outer membrane permeabilization (MOMP)
MOMP is a result of series of events that leads to pore formation in the mitochondrial membrane. MOMP results in the release of mitochondrial contents into the cytoplasm, which activates caspases to induce apoptosis
- Nod-like receptors (NLRs)
NLRs are cytoplasmic pattern recognition receptors that are characterized by the presence of a C-terminal leucine rich repeat, a mid nucleotide oligomerization domain (NOD) [32] and an N-terminal activation domain. NLRs detect PAMPs and DAMPs in the cytoplasm and initiate immune responses by instructing inflammatory signaling and inflammasome formation
- Pathogen associated molecular patterns (PAMPs)
PAMPs can be recognized by cells to initiate immune responses against pathogens. Some common pathogen associated molecular patterns that are recognized by our immune cells include lipopolysaccharides (LPS), peptidoglycans and double stranded RNA
- Pyroptosis
Programmed cell death that is mediated specifically by activation of inflammasomes. Caspase-1 and caspase-11 have been shown to induce pyroptotic cell death, which results in the release of cellular contents and is inflammatory
- Reactive oxygen species (ROS)
ROS are chemically reactive compounds that consist of oxygen ions and peroxidases. ROS are generated as byproducts of several metabolic pathways and are used by cells to combat infection
- TCA cycle
The tricarobxylic acid cycle (also known as the citric acid cycle, or Krebs cycle; named after Hans Krebs) takes place in the mitochondrial matrix and is the second major metabolic pathway involved in the generation of ATP from glucose. TCA cycle accepts pyruvate generated from glycolysis to generate ATP and reducing agents NADH and FADH2.
Footnotes
Conflict of Interest:
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Margulis L. Symbiosis and evolution. Scientific American. 1971;225(2):48–57. doi: 10.1038/scientificamerican0871-48. [DOI] [PubMed] [Google Scholar]
- 2.Zimorski V, et al. Endosymbiotic theory for organelle origins. Current opinion in microbiology. 2014;22C:38–48. doi: 10.1016/j.mib.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Delhumeau G, Cruz-Mendoza AM, Gomez Lojero C. Protection of cytochrome c oxidase against cyanide inhibition by pyruvate and alpha-ketoglutarate: effect of aeration in vitro. Toxicology and applied pharmacology. 1994;126(2):345–51. doi: 10.1006/taap.1994.1125. [DOI] [PubMed] [Google Scholar]
- 4.Webster KA. Evolution of the coordinate regulation of glycolytic enzyme genes by hypoxia. The Journal of experimental biology. 2003;206(Pt 17):2911–22. doi: 10.1242/jeb.00516. [DOI] [PubMed] [Google Scholar]
- 5.Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell biochemistry and biophysics. 2014;68(3):475–8. doi: 10.1007/s12013-013-9750-1. [DOI] [PubMed] [Google Scholar]
- 6.Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochimica et biophysica acta. 2008;1778(10):1978–2021. doi: 10.1016/j.bbamem.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Ke F, et al. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell death & disease. 2013;4:e650. doi: 10.1038/cddis.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindsten T, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Molecular cell. 2000;6(6):1389–99. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei MC, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei MC, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes & development. 2000;14(16):2060–71. [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy KM, et al. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell death and differentiation. 2000;7(1):102–11. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- 12.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature reviews. Molecular cell biology. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 13.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112(4):481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 14.Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annual review of genetics. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harbor perspectives in biology. 2013;5(9) doi: 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tait SW, Green DR. Caspase-independent cell death: leaving the set without the final cut. Oncogene. 2008;27(50):6452–61. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature immunology. 2011;12(3):222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurung P, Kanneganti TD. Novel Roles for Caspase-8 in Interleukin-1β and Inflammasome Regulation. The American Journal of Pathology. 2014 doi: 10.1016/j.ajpath.2014.08.025. http://dx.doi.org/10.1016/j.ajpath.2014.08.025. [DOI] [PMC free article] [PubMed]
- 19.Lupfer CR, Kanneganti TD. The role of inflammasome modulation in virulence. Virulence. 2012;3(3):262–70. doi: 10.4161/viru.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khare S, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36(3):464–76. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61(7):1113–21. doi: 10.1002/glia.22499. [DOI] [PubMed] [Google Scholar]
- 22.Gurung P, et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. Journal of immunology. 2014;192(4):1835–46. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anand PK, Malireddi RK, Kanneganti TD. Role of the nlrp3 inflammasome in microbial infection. Frontiers in microbiology. 2011;2:12. doi: 10.3389/fmicb.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou R, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2009;11(2):136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 25.Zhou R, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 26.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nature immunology. 2005;6(10):981–8. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 27.Seth RB, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–82. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Xu LG, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Molecular cell. 2005;19(6):727–40. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Cruz CM, et al. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. The Journal of biological chemistry. 2007;282(5):2871–9. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrilli V, et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell death and differentiation. 2007;14 (9):1583–9. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 31.Dostert C, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang LS, et al. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. Journal of molecular biology. 2005;351(3):573–97. doi: 10.1016/j.jmb.2005.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li N, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. The Journal of biological chemistry. 2003;278(10):8516–25. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 35.Iyer SS, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39(2):311–23. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ichinohe T, et al. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(44):17963–8. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chinopoulos C, Adam-Vizi V. Mitochondrial Ca2+ sequestration and precipitation revisited. The FEBS journal. 2010;277(18):3637–51. doi: 10.1111/j.1742-4658.2010.07755.x. [DOI] [PubMed] [Google Scholar]
- 38.Brough D, et al. Ca2+ stores and Ca2+ entry differentially contribute to the release of IL-1 beta and IL-1 alpha from murine macrophages. Journal of immunology. 2003;170(6):3029–36. doi: 10.4049/jimmunol.170.6.3029. [DOI] [PubMed] [Google Scholar]
- 39.Lee HM, et al. Mycobacterium abscessus activates the NLRP3 inflammasome via Dectin-1-Syk and p62/SQSTM1. Immunology and cell biology. 2012;90(6):601–10. doi: 10.1038/icb.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrari D, et al. The P2X7 receptor: a key player in IL-1 processing and release. Journal of immunology. 2006;176(7):3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 41.Chu J, et al. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. Journal of leukocyte biology. 2009;86(5):1227–38. doi: 10.1189/jlb.0309164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feldmeyer L, et al. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Current biology: CB. 2007;17 (13):1140–5. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 43.Rossol M, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nature communications. 2012;3:1329. doi: 10.1038/ncomms2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee GS, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–7. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murakami T, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(28):11282–7. doi: 10.1073/pnas.1117765109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munoz-Planillo R, et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38(6):1142–53. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Misawa T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature immunology. 2013;14 (5):454–60. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 48.Arias-Cartin R, et al. Cardiolipin binding in bacterial respiratory complexes: structural and functional implications. Biochimica et biophysica acta. 2012;1817(10):1937–49. doi: 10.1016/j.bbabio.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 49.Huang Z, et al. Cardiolipin deficiency leads to decreased cardiolipin peroxidation and increased resistance of cells to apoptosis. Free radical biology & medicine. 2008;44(11):1935–44. doi: 10.1016/j.freeradbiomed.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu CT, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature cell biology. 2013;15(10):1197–205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. The Journal of biological chemistry. 2005;280(28):26185–92. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 52.Chen H, et al. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. The Journal of cell biology. 2003;160(2):189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanneganti TD, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–6. doi: 10.1038/nature04517. [DOI] [PubMed] [Google Scholar]
- 54.Fernandes-Alnemri T, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nature immunology. 2010;11(5):385–93. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rathinam VA, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nature immunology. 2010;11(5):395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patrushev M, et al. Release of mitochondrial DNA fragments from brain mitochondria of irradiated mice. Mitochondrion. 2006;6(1):43–7. doi: 10.1016/j.mito.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464(7285):104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–14. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allam R, et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO reports. 2014;15(9):982–90. doi: 10.15252/embr.201438463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hou F, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–61. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subramanian N, et al. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–61. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franchi L, et al. Cytosolic Double-Stranded RNA Activates the NLRP3 Inflammasome via MAVS-Induced Membrane Permeabilization and K+ Efflux. Journal of immunology. 2014;193(8):4214–22. doi: 10.4049/jimmunol.1400582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park S, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. Journal of immunology. 2013;191(8):4358–66. doi: 10.4049/jimmunol.1301170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 66.Lupfer C, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nature immunology. 2013;14(5):480–8. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature medicine. 2010;16(1):90–7. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 68.Joo JH, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Molecular cell. 2011;43(4):572–85. doi: 10.1016/j.molcel.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lukens JR, Dixit VD, Kanneganti TD. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov Med. 2011;12 (62):65–74. [PMC free article] [PubMed] [Google Scholar]
- 70.Yin X, et al. Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J Clin Endocrinol Metab. 2014;99(2):E209–16. doi: 10.1210/jc.2013-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hahn WS, et al. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am J Physiol Endocrinol Metab. 2014;306(9):E1033–45. doi: 10.1152/ajpendo.00422.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holloway GP, Bonen A, Spriet LL. Regulation of skeletal muscle mitochondrial fatty acid metabolism in lean and obese individuals. Am J Clin Nutr. 2009;89(1):455S–62S. doi: 10.3945/ajcn.2008.26717B. [DOI] [PubMed] [Google Scholar]
- 73.Kelley DE, et al. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51(10):2944–50. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 74.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–7. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 75.Mogensen M, et al. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56(6):1592–9. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- 76.Petersen KF, et al. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350(7):664–71. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Slattery MJ, et al. Insulin resistance and impaired mitochondrial function in obese adolescent girls. Metab Syndr Relat Disord. 2014;12(1):56–61. doi: 10.1089/met.2013.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Toledo FG, Goodpaster BH. The role of weight loss and exercise in correcting skeletal muscle mitochondrial abnormalities in obesity, diabetes and aging. Mol Cell Endocrinol. 2013;379(1–2):30–4. doi: 10.1016/j.mce.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 79.Lopez-Lluch G, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103(6):1768–73. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lanza IR, et al. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab. 2012;16 (6):777–88. doi: 10.1016/j.cmet.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vandanmagsar B, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wen H, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–15. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stienstra R, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12(6):593–605. doi: 10.1016/j.cmet.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lamkanfi M, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stienstra R, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108(37):15324–9. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116(7):1793–801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lagathu C, et al. Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia. 2006;49(9):2162–73. doi: 10.1007/s00125-006-0335-z. [DOI] [PubMed] [Google Scholar]
- 88.Larsen CM, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356(15):1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]