

Abstract
Activation of human ether-a-go-go–related gene 1 (hERG1) K+ channels mediates repolarization of action potentials in cardiomyocytes. RPR-260243 [(3R,4R)-4-[3-(6-methoxy-quinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5-trifluorophenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid] (RPR) slows deactivation and attenuates inactivation of hERG1 channels. A detailed understanding of the molecular mechanism of hERG1 agonists such as RPR may facilitate the design of more selective and potent compounds for prevention of arrhythmia associated with abnormally prolonged ventricular repolarization. RPR binds to a hydrophobic pocket located between two adjacent hERG1 subunits, and, hence, a homotetrameric channel has four identical RPR binding sites. To investigate the stoichiometry of altered channel gating induced by RPR, we constructed and characterized tetrameric hERG1 concatemers containing a variable number of wild-type subunits and subunits containing a point mutation (L553A) that rendered the channel insensitive to RPR, ostensibly by preventing ligand binding. The slowing of deactivation by RPR was proportional to the number of wild-type subunits incorporated into a concatenated tetrameric channel, and four wild-type subunits were required to achieve maximal slowing of deactivation. In contrast, a single wild-type subunit within a concatenated tetramer was sufficient to achieve half of the maximal RPR-induced shift in the voltage dependence of hERG1 inactivation, and maximal effect was achieved in channels containing three or four wild-type subunits. Together our findings suggest that the allosteric modulation of channel gating involves distinct mechanisms of coupling between drug binding and altered deactivation and inactivation.
Introduction
Congenital long QT syndrome (LQTS) increases the risk of life-threatening cardiac arrhythmia and is commonly caused by loss-of-function mutations in KCNH2 (Curran et al., 1995), the gene that encodes human ether-a-go-go–related gene 1 (hERG1) K+ channels. In the heart, hERG1 channels conduct the rapid delayed rectifier K+ current (Sanguinetti et al., 1995; Trudeau et al., 1995). Reduced rapid delayed rectifier K+ current caused either by KCNH2 mutations or unintended block of hERG1 channels by medications (Fenichel et al., 2004) results in delayed ventricular repolarization, prolonged QT interval, and a type of ventricular arrhythmia called torsades de pointes (Keating and Sanguinetti, 2001). First-line therapy for congenital LQTS is administration of β-adrenergic receptor blockers, but these drugs are not always effective. Implantable cardioverter defibrillators can be very effective in preventing lethal arrhythmia, but these devices are costly and unavailable for most patients. Thus, there is an unmet need for additional pharmacotherapies for LQTS. Compounds that activate hERG1 channels were recently discovered, providing a novel molecular mechanism for drug treatment of LQTS.
Based on their differential effects on channel gating, four types of hERG1 agonists have been described (Sanguinetti, 2014) and include RPR-260243 [(3R,4R)-4-[3-(6-methoxy-quinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5 trifluorophenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid] (type 1), PD-118057 [2-[[4-[2-(3,4-dichlorophenyl)ethyl]phenyl]amino]benzoic acid] (type 2), ICA-105574 [3-nitro-N-(4-phenoxyphenyl) benzamide] (type 3), and mallotoxin (type 4). PD-118057 (PD) mainly increases single-channel open probability (Zhou et al., 2005; Perry et al., 2009), ICA-105574 (ICA) attenuates inactivation (Gerlach et al., 2010; Garg et al., 2011), whereas mallotoxin shifts the voltage dependence of activation to more negative potentials than normal (Zeng et al., 2006). The type 1 agonist, RPR-260243 (RPR), profoundly slows the rate of channel deactivation and causes a modest positive shift in the voltage dependence of inactivation (Kang et al., 2005; Perry et al., 2007). Despite their disparate effects on gating, type 1, 2, and 3 hERG1 agonists interact with a similar region of the channel. Site-directed mutagenesis and molecular modeling were used to define the putative agonist binding site as a hydrophobic pocket located between two adjacent hERG1 subunits (Perry et al., 2007, 2009; Garg et al., 2011). Accordingly, a homotetrameric hERG1 channel contains four identical agonist binding sites. We recently reported that ICA and PD must occupy all four binding sites to obtain their maximal effects on the attenuation of inactivation gating and the increase of single-channel open probability, respectively (Wu et al., 2014b). In this study, we characterize the stoichiometric basis of RPR-mediated effects on hERG1 channel gating. Plasmids were constructed to encode concatenated hERG1 tetramers with a variable, but defined number and positioning of wild-type (WT) and mutant subunits. Mutant subunits each contained a single L553A mutation that we previously reported prevents RPR activity (Perry et al., 2007). Our findings indicate that the stoichiometry of RPR action varies: all four binding sites are required to achieve the maximal effect in slowing of hERG1 deactivation, whereas three binding sites were sufficient to acquire the maximal effect on inactivation.
Materials and Methods
Construction of hERG1 Concatemers.
WT KCNH2 (HERG1, isoform 1a, National Center for Biotechnology Information Reference Sequence NM_000238) cDNA was cloned into the pSP64 oocyte expression vector, and mutations were introduced using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Methods for constructing concatenated tetramers containing a variable number of WT and/or mutant KCNH2 cDNAs with defined positioning were the same as previously described (Wu et al., 2014b). In the text and figures, concatenated tetramers formed by four WT or L553A (LA) mutant subunits are designated as WT4 and LA4, respectively. Heterotypic tetramers were named to indicate the relative positioning of the WT and LA mutant subunits. For example, the LA1/WT1/LA2 concatemer contains a WT subunit in the second position and subunits harboring the L553A mutation in the first, third, and fourth position of the channel. All constructs were verified by DNA sequence analysis. Tetrameric KCNH2 plasmids were linearized with EcoR1 prior to in vitro transcription using the mMessage mMachine SP6 kit (Ambion, Austin, TX).
Solutions and Drugs.
The extracellular solution used for voltage-clamp experiments contained (in mM) the following: 96 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, 5 HEPES; pH was adjusted to 7.6 with NaOH. RPR-260243 (ChemShuttle, Wuxi, China) was dissolved in dimethylsulfoxide to make a 10 mM stock solution, and final drug concentrations (1–50 µM) were obtained by dilution of the stock solution with extracellular solution.
Isolation and Voltage Clamp of Xenopus Oocytes.
Procedures used for the surgical removal of ovarian lobes from Xenopus laevis and isolation of oocytes were approved by the University of Utah Institutional Animal Care and Use Committee and performed as described previously (Abbruzzese et al., 2010). Single oocytes were injected with 10 ng cRNA encoding single hERG1 subunits and studied 1–3 days later. HERG1 tandem dimers and concatenated tetramers expressed poorly and, therefore, oocytes were injected with 40 ng cRNA and studied 3–8 days later. Ionic currents were recorded using agarose-cushion microelectrodes (Schreibmayer et al., 1994) and standard two-electrode voltage-clamp techniques (Goldin, 1991; Stühmer, 1992). A GeneClamp 500 amplifier, Digidata 1322A data acquisition system, and pCLAMP 8.2 software (Molecular Devices, Sunnyvale, CA) were used to produce command voltages and to record current and voltage signals.
Voltage Pulse Protocols and Data Analysis.
Digitized data were analyzed with pCLAMP 8.2 and ORIGIN 8.5 (OriginLab, Northhampton, MA) software. To determine the voltage dependence of activation of hERG1 channel currents, 4-second pulses were applied in 10-mV increments to test potentials (Vt) that ranged from −70 to +40 mV. The amplitude of tail currents (Itail) measured at −70 mV was normalized to their maximum value (Itail-max) and plotted as a function of Vt. The resulting normalized, isochronal conductance-voltage (g/gmax-Vt) relationship was fitted with a Boltzmann function:
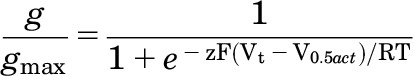 |
(1) |
where V0.5act is the half-point for activation, z is the equivalent charge, F is Faraday’s constant, R is the universal gas constant, and T is the absolute temperature.
The voltage dependence of the recovery from C-type inactivation was estimated using a different two-pulse protocol. Currents were first activated by a 2-second pulse to +40 mV, and then tail currents were elicited with a 1.5-second pulse to a variable return potential (Vret), applied in 10-mV increments that ranged from +30 to −140 mV. Normalized values of Itail/(Vret − Erev) were plotted as a function of Vret, and the resulting relationship was fitted with a Boltzmann function to determine the half-point (V0.5inact) and z for inactivation:
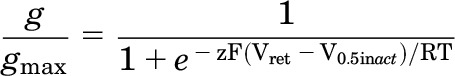 |
(2) |
A two-state model (O↔I) was assumed for the analysis of free energy change associated with inactivation gating (ΔGinact). The calculated V0.5inact and z values were used to estimate free energy change in the absence and presence of 50 µM RPR:
| (3) |
The energy change caused by RPR was defined as the difference of ΔGinact before and after RPR application:
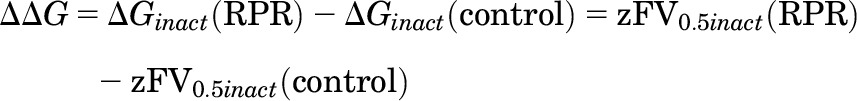 |
(4) |
The effects of RPR on the kinetics of hERG1 deactivation were quantified by calculating the integral of leak-subtracted tail currents (∫Itail). Channels were activated with a 4-second pulse to +20 mV, and tail currents were elicited at a Vret of −70 or −120 mV. The pulse to −70 was always 30 seconds in duration, and ∫Itail was determined using the full trace. At −120 mV, ∫Itail was determined at the longest time required (6–20 seconds) for full deactivation of currents in the presence of RPR.
Data are presented as mean ± S.E.M. (n = number of oocytes). Data were analyzed by analysis of variance (one- or two-way), followed by a Tukey multiple comparison test where appropriate, or with a paired Student’s t test. A P value of <0.05 was considered significant. For linear regression analysis, the goodness of fit was evaluated by the coefficient of determination (adjusted R2).
Results
Comparison of the Effects of RPR on Gating of WT4 and WTmonomer hERG1 Channels.
RPR profoundly slows the rate of deactivation (Kang et al., 2005; Perry et al., 2007) and causes a modest positive shift in the voltage dependence of inactivation (Perry et al., 2007) of hERG1 channels. We previously reported that the biophysical properties and response to PD and ICA were similar for concatenated WT hERG1 tetramers (WT4) and WTmonomer channels formed by coassembly of single WT hERG subunits (Wu et al., 2014b). In this study, we compare RPR-altered deactivation and inactivation between these two types of WT channels. Following a 4-second pulse to +20 mV to activate channels, tail currents were elicited at −70 and −120 mV. At a concentration of 50 µM RPR, the rate of tail current decay (deactivation) was profoundly slowed at −70 mV and −120 mV for both channel types (Fig. 1A). The fold increase in tail current integral (∫Itail) by 50 µM RPR was greater for WTmonomer than WT4 channels (Fig. 1B). The attenuation of inward rectification of the fully activated Itail-Vret relationship (Fig. 1C) and positive shift in the voltage dependence of recovery from inactivation (Fig. 1D) by 50 µM RPR were also greater for WTmonomer than WT4 channels. Thus, although WT4 channels were less sensitive than WTmonomer to the effects of RPR on channel deactivation and inactivation, the qualitative effects of the compound on gating were similar for both channel types.
Fig. 1.

RPR exerts less potent effects on inactivation and deactivation of homotetrameric hERG1 concatemers (WT4) than WT hERG1 channels formed from monomers. (A) Effect of 50 µM RPR on WT4 and WTmonomer hERG1 channel currents elicited by a 30-second pulse to −70 mV (left panels) or an 18-second pulse to −120 mV (right panels). (B) Fold increase in ∫Itail for WT4 and WTmonomer hERG1 channels at indicated Vret; *P = 0.015. (C) Fully-activated Itail-Vret relationships in the absence and presence of 50 µM RPR for WT4 (n = 6) and WTmonomer (n = 7) hERG1 channels, respectively. Currents were normalized to Itail at −140 mV measured under control conditions. (D) Voltage dependence of recovery from inactivation for WT4 and WTmonomer channels in the absence and presence of 50 µM RPR. For WTmonomer channels, control: V0.5inact = −50.2 ± 1.4 mV; z = 0.91 ± 0.01; 50 µM RPR: V0.5inact = −18.4 ± 2.8 mV; z = 1.52 ± 0.08 (n = 7). For WT4 channels, control: V0.5inact = −51.6 ± 1.6 mV; z = 0.92 ± 0.02; 50 µM RPR: V0.5inact = −27.6 ± 0.8 mV; z = 1.48 ± 0.05 (n = 6). For (C and D), relationships for WT4 and WTmonomer channels in the presence of 50 µM RPR were significantly different from one another (P < 0.0001, two-way analysis of variance).
Concatenated hERG1 Tetramers Containing a Variable Number of WT and LA Mutant Subunits Have Similar, but Not Identical Biophysical Properties.
RPR binds to a hydrophobic pocket between S5 and S6 segments of adjacent hERG1 subunits (Perry et al., 2007). Accordingly, a homotetrameric WT hERG1 channel should contain four identical RPR binding sites. A point mutation, L553A, located within the hydrophobic pocket prevents RPR-induced slowing of deactivation and shift in the voltage dependence of inactivation of hERG1 channel (Perry et al., 2007). However, in the absence of a ligand-binding assay for RPR, it is unclear whether the L553A mutation disrupts RPR binding to the hERG1 channel or prevents the compound from exerting its multiple effects by an allosteric mechanism. We constructed eight LAn/WT4-n concatenated hERG1 tetramers containing 0–4 WT subunits to determine the number of intact ligand binding sites required for maximal RPR effect and to characterize potential subunit interactions. Figure 2 compares the voltage-dependent biophysical properties of the eight hERG1 concatemers, including Itest-Vt relationships for 4-second pulses (Fig. 2A), voltage dependence of activation (Fig. 2B), fully activated Itail-Vret relationships (Fig. 2C), and voltage dependence of inactivation (Fig. 2D). The V0.5 and z values for activation and inactivation of channels are summarized in Tables 1 and 2. Incorporation of 1-4 LA subunits into a concatenated tetramer had only relatively minor effects on the voltage dependence of activation or inactivation gating. The maximum difference in V0.5act was 4.8 mV (WT3/LA1 versus LA1/WT1/LA2 channels). The maximum difference in V0.5inact was 11.4 mV (LA4 versus WT3/LA1 channels). The rate of current deactivation was estimated from single exponential fitting of tail current traces measured at −70 mV and −120 mV. Concatenated tetramers containing one or more LA mutant subunits deactivated faster (reduced τdeact) at −70 mV compared with WT4 channels (Fig. 2E). At −120 mV (Fig. 2F), τdeact was similar for channels containing two or more WT subunits in adjacent positions, but deactivation was accelerated for LA4 channels and concatemers with only one WT subunit or having two WT subunits arranged diagonal to one another (P < 0.01). In summary, the voltage dependence of activation and recovery from inactivation of concatenated channels containing a variable number of WT and LA subunits were similar, but the rate of channel deactivation was often faster for channels containing LA subunits.
Fig. 2.

Biophysical properties of concatenated hERG1 channels. (A–D) Normalized Itest-Vt relationships (A), voltage dependence of activation (B), fully activated Itail-Vret relationships (C), and voltage dependence of inactivation (D) for LAn/WT4-n tetrameric hERG1 channels. To adjust for variable channel expression in oocytes, currents were normalized to peak currents measured at −20 mV (A), peak Itail (B), Itail at −140 mV (C), and to extrapolated peak (D). Symbol legend in (D) refers to (A–D) (n = 4–7). In (B and D), smooth curves represent best fits of data to a Boltzmann function. Values of V0.5 and z for activation and recovery from inactivation are presented in Tables 1 and 2, respectively. (E) Plot of τdeact measured at −70 mV as a function of the number of WT subunits contained in a concatenated tetramer (n = 4–11). *P < 0.05 compared with all other tetramers (one-way analysis of variance). Diagram located above each bar in the graph indicates positioning of WT and L553A hERG1 subunits in a concatenated tetramer. (F) Plot of τdeact measured at −120 mV as a function of the number of WT subunits contained in a concatenated tetramer (n = 4–10). *P < 0.05; **P < 0.01; †P < 0.001 (one-way analysis of variance) compared with WT4 channels.
TABLE 1.
Summary of the voltage dependence of activation for hERG1 tetramers containing 0–4 L553A (LA) subunits
Data are expressed as mean ± S.E.M. (n = number of oocytes).
| Channel Type | V0.5act (mV) | Z | n |
|---|---|---|---|
| WT4 | −28.7 ± 0.7 | 3.76 ± 0.18 | 10 |
| LA1/WT3 | −31.0 ± 0.7 | 4.07 ± 0.11 | 5 |
| WT3/LA1 | −27.8 ± 0.6 | 3.61 ± 0.09 | 5 |
| LA2/WT2 | −29.7 ± 0.8 | 3.78 ± 0.12 | 5 |
| LA1/WT1/LA1/WT1 | −31.1 ± 0.1 | 3.87 ± 0.10 | 5 |
| LA1/WT1/LA2 | −32.6 ± 0.4 | 3.26 ± 0.16 | 4 |
| LA3/WT1 | −30.3 ± 1.0 | 3.54 ± 0.17 | 4 |
| LA4 | −30.6 ± 1.3 | 3.16 ± 0.31 | 4 |
TABLE 2.
Summary of effects of RPR on the voltage dependence of recovery from inactivation for WT hERG1 monomers and tetramers containing 0–4 L553A (LA) subunits
| Channel Type | Control |
50 µM RPR |
n | ||
|---|---|---|---|---|---|
| V0.5inact (mV) | z | V0.5inact (mV) | z | ||
| WTmonomer | −50.2 ± 1.4 | 0.91 ± 0.01 | −18.4 ± 2.8 | 1.52 ± 0.08 | 7 |
| WT4 | −51.6 ± 1.6 | 0.92 ± 0.02 | −27.6 ± 0.8 | 1.48 ± 0.05 | 6 |
| LA1/WT3 | −55.9 ± 1.9 | 0.92 ± 0.03 | −36.6 ± 1.0 | 1.27 ± 0.08 | 6 |
| WT3/LA1 | −57.1 ± 1.3 | 0.88 ± 0.01 | −33.6 ± 0.8 | 1.38 ± 0.04 | 5 |
| LA2/WT2 | −56.2 ± 1.3 | 0.93 ± 0.04 | −37.2 ± 1.2 | 1.38 ± 0.04 | 4 |
| LA1/WT1/LA1/WT1 | −53.8 ± 1.8 | 0.84 ± 0.01 | −34.8 ± 0.9 | 1.10 ± 0.03 | 5 |
| LA1/WT1/LA2 | −54.1 ± 3.1 | 0.91 ± 0.03 | −42.3 ± 3.8 | 1.07 ± 0.03 | 5 |
| LA3/WT1 | −46.5 ± 3.6 | 0.87 ± 0.03 | −34.6 ± 1.9 | 1.04 ± 0.04 | 4 |
| LA4 | −45.7 ± 5.7 | 0.93 ± 0.01 | −44.7 ± 5.7 | 1.12 ± 0.05 | 4 |
Stoichiometry of Slowed hERG1 Channel Deactivation by RPR.
We next examined the concentration-dependent effects of RPR on deactivation gating of LAn/WT4-n tetrameric channels. Under control conditions, deactivation of hERG1 channel current is a biexponential process, but at some potentials the process is dominated by a single exponential component. In the presence of RPR, the deactivation of hERG1 channel current is characterized by complex kinetics, requiring the sum of three or more exponential components to fully describe the time-dependent decay of current amplitude; however, a very slow component dominates the deactivation process altered by RPR. Therefore, to simplify the kinetic analysis of gating, tail currents recorded at −70 and −120 mV were fitted with a single exponential function (Fig. 3A). The fold increase in the single time constants for deactivation (τdeact) induced by 50 µM RPR is summarized in Fig. 3B. Deactivation was slowed in rough proportion to the number of WT subunit present in a concatenated tetrameric channel, whereas the rate of LA4 channel deactivation was unaffected by RPR.
Fig. 3.

Slowing of deactivation by RPR is dependent on the number of WT subunits in a concatenated hERG1 tetramer. (A) Representative current traces of LA2/WT2 tetrameric channels in an oocyte before and after treatment with 50 µM RPR. Channels were activated with 4-second pulse to +20 mV, and tail currents were elicited at a Vret of −70 mV (left panels) or −120 mV (right panels). Upper panels depict voltage pulse protocol; middle panels show control tail currents; lower panels show tail currents after treatment of oocyte with 50 µM RPR. Tail currents were fitted to a single exponential function (dotted red traces) to estimate τdeact for the slow component of deactivation. (B) Bar graphs indicating that the fold increase in τdeact by 50 µM RPR at −70 mV (left panel) or −120 mV (right panel) increases as a function of the number of WT subunits contained in a concatenated tetramer (P < 0.0001, one-way analysis of variance; n = 4–6). *P < 0.05; **P < 0.01; †P < 0.001 compared with WT4 channels.
Another way to analyze the effects of RPR on deactivation that avoids the complications associated with complex kinetics is to simply calculate the time integral of the tail current before and after treatment of an oocyte with RPR. This approach was used to measure the effects of 10, 30, and 50 µM RPR on tail currents elicited at a Vret of −70 and −120 mV. Representative current traces elicited with a 2-second test pulse to +20 mV, followed by repolarization to −70 mV for 30 seconds in the absence and presence of 30 µM RPR, are illustrated in Fig. 4A for all eight concatemers. LA4 mutant channels had almost no response to 30 µM RPR, similar to channels formed by coassembly of L553A monomers (Perry et al., 2007). The ability of RPR at concentrations of 10, 30, and 50 µM to slow hERG1 deactivation was enhanced in direct proportion to the number of WT subunits contained in a concatenated tetramer (Fig. 4A). This relationship is quantified in Fig. 4B, in which the fold increase in ∫Itail determined by integration of outward tail currents measured at −70 mV for 30 seconds is plotted as a function of the number of WT subunits contained in a concatenated tetramer. RPR exerted its effect on concatemers regardless of the positioning of the WT subunits (e.g., LA1/WT1/LA2 versus LA3/WT1). The data for 50 µM RPR are replotted in Fig. 4C and analyzed by linear regression. The fold increase in ∫Itail induced by RPR was strongly correlated to the number of WT subunits present in a concatenated tetramer (R2 = 0.97). We also evaluated the concentration-dependent effects of RPR on ∫Itail for tail currents measured at −120 mV. Compared with WT4 channels, deactivation of LA4 channels was unaffected by RPR, and increasing the number of WT subunit contained within a concatenated tetramer resulted in a graded increase in response to the compound (Fig. 5A). Figure 5B summarizes the fold increase in ∫Itail at −120 mV in response to 10 µM RPR, 30 µM RPR, and 50 µM RPR, and Fig. 5C illustrates again the strong correlation between the number of WT subunits present in a concatenated tetramer and the fold increase in ∫Itail induced by RPR (R2 = 0.96). Together these findings indicate that increasing the number of WT subunits contained within a tetramer from 0 to 4 is associated with an additive enhancement of the RPR effect and that all four WT subunits are required to achieve maximal slowing of hERG1 channel deactivation.
Fig. 4.

RPR-induced enhancement of tail current integral at −70 mV is directly proportional to the number of WT subunits contained within a concatenated hERG1 tetramer. (A) Representative current traces for indicated LAn/WT4-n tetrameric hERG1 channels, recorded before and after treatment of oocyte with 30 µM RPR. Channels were activated with 4-second pulse to +20 mV, and tail currents were elicited at a Vret of −70 mV. (B) Bar graphs showing that the fold increase in ∫Itail induced by indicated concentration of RPR is increased (P < 0.0001, one-way analysis of variance) as a function of the number of WT subunits contained in a concatenated tetramer (n = 4–9). (C) Data for 50 µM RPR replotted and analyzed by linear regression (red line; y = 9.87x − 1.93; R2 = 0.97).
Fig. 5.

RPR-induced enhancement of tail current integral at −120 mV is directly proportional to the number of WT subunits contained within a concatenated hERG1 tetramer. (A) Representative current traces for indicated LAn/WT4-n tetrameric hERG1 channels, recorded before and after treatment of oocyte with 30 µM RPR. Channels were activated with 4-second pulse to +20 mV, and tail currents were elicited at a Vret of −120 mV. (B) Bar graphs showing that the fold increase in ∫Itail induced by indicated concentration of RPR is increased (P < 0.0001, one-way analysis of variance) as a function of the number of WT subunits contained in a concatenated tetramer (n = 4–9). (C) Data for 50 µM RPR replotted and analyzed by linear regression (red line; y = 4.74x − 0.3; R2 = 0.96).
Stoichiometry of Altered hERG1 Channel Inactivation by RPR.
To investigate the stoichiometry of attenuated hERG1 inactivation by RPR, we compared the shifts in voltage-dependent inactivation for each LAn/WT4-n tetrameric channel. Figure 6A illustrates the effect of 50 µM RPR on fully activated Itail-Vret relationships for all eight tetrameric concatemers. RPR reduced the extent of inward rectification of tetramers containing two or more WT subunits, but slightly inhibited currents conducted by LA4 and channels containing only one WT subunit. This antagonist effect of RPR in the absence of WT subunits was previously reported for the type 3 hERG1 agonist ICA (Garg et al., 2011). The effects of 50 µM RPR on the voltage dependence of inactivation are summarized in Fig. 6B and Table 2. RPR shifted V0.5inact of WT4 channels by 24.0 ± 1.1 mV (n = 6) and significantly increased the slope (z) of the g/gmax-Vret relationship. In contrast, V0.5inact and z values for LA4 channel were not changed by RPR. The effect of RPR on V0.5inact for voltage-dependent inactivation for all the concatenated tetramers is summarized in Fig. 7A. The shift in V0.5inact produced by RPR was half maximal for concatemers containing a single WT subunit, and WT3/LA1 channels exhibited a similar shift to that measured for WT4 channels (Fig. 7B). RPR also increased z, indicating enhanced sensitivity of the inactivation process to membrane voltage (Fig. 7C). The free energy associated with inactivation gating was calculated as ∆G = zFV0.5inact, which assumes a two-state model for channel gating. This is a reasonable assumption for these experiments in which essentially all channels were either in an open or inactivated state by the end of the prepulse to +40 mV. RPR (50 µM) induced a slight change in the free energy of inactivation (∆∆G = ∆GRPR − ∆Gcontrol) in some channels; however, ∆∆G was not increased in proportion to the number of WT subunits/tetramer (Fig. 7D) because the RPR-induced positive shift in V0.5inact was counterbalanced by an increase in z.
Fig. 6.

Effect of 50 µM RPR on inactivation of concatenated LAn/WT4-n hERG1 tetramers. (A) Fully activated Itail-Vret relationships in the absence and presence of 50 µM RPR for concatenated LAn/WT4-n hERG1 tetramers (n = 4–6). (B) The voltage dependence of inactivation in the absence and presence of 50 µM RPR for concatenated LAn/WT4-n hERG1 tetramers. Smooth curves represent best fits of data to a Boltzmann function (n = 4–6). Values of V0.5-inact and z for inactivation are presented in Table 2. Diagram shown in each panel indicates positioning of WT and L553A hERG1 subunits in the concatenated tetramer. Control versus 50 µM RPR: †P < 0.0001 (two-way analysis of variance).
Fig. 7.

Effects of 50 µM RPR on parameters of inactivation of concatenated hERG1 tetramers. (A) Plot of V0.5inact versus the number of WT subunits contained within a concatenated tetramer. Control versus 50 µM RPR: **P < 0.01; †P < 0.001. (B) Shift in V0.5inact induced by RPR plotted as a function of the number of WT subunits contained in a concatenated tetramer. †P < 0.001 compared with WT4 channels (one-way analysis of variance). (C) Plot of equivalent charge (z) before and after 50 µM RPR versus the number of WT subunits contained within a concatenated tetramer. Control versus 50 µM RPR: *P < 0.05; **P < 0.01; †P < 0.001. (D) Plot of ΔΔG [= zFV0.5inact (50 µM RPR) − zFV0.5inact (control)], versus number of WT subunits contained in a concatenated tetramer. *P < 0.05; **P < 0.01 (paired t test). n = 4–6 for all panels.
Discussion
In this study, we constructed concatenated hERG1 tetramers containing a variable number of WT subunits and mutant subunits harboring a point mutation (L553A) that abolishes the effects of RPR on channel gating. Theoretically, self-assembly of the concatenated subunits into a tetramer with the intended stoichiometry should be highly favored over more complex structures formed by coassembly of multiple tetramers. However, unusual stoichiometry with overrepresentation of WT subunits or underrepresentation of mutant subunits has been reported (McCormack et al., 1992; Hurst et al., 1995; Sack et al., 2008). For several reasons, we suggest that formation of multimerized hERG1 tetramers is unlikely. First, based on current magnitudes, functional expression was similar for the various LAn/WT4-n tetrameric channels. Lower levels of expression would be expected if tetramers formed complex assemblies. Second, the biophysical properties of the various tetrameric channels were similar, with the exception of faster rates of deactivation for channels containing one or more LA subunits compared with WT4 channels. Third, the response to RPR was graded in proportion to the number of WT subunits contained within a tetramer. Finally, concatemers that contained the same number of WT subunits (i.e., 1, 2, or 3) responded similarly to RPR irrespective of their position within a tetramer. Therefore, we conclude that for the most part LAn/WT4-n tetramers self-assembled to form channels with the intended stoichiometry.
Slow deactivation of hERG1 channels is dependent on an interaction between the N- and C-termini of adjacent subunits (Gianulis et al., 2013). In a WT4 channel, three of the four C- and N-termini are linked together in the tetramer. Thus, it was unexpected to find that the deactivation rates for WTmonomer channels, formed by coassembly of four single subunits, and WT4 channels were indistinguishable (Thomson et al., 2014; Wu et al., 2014b). In a previous study, we found that concatenation of four WT hERG1 subunits did not alter the pharmacological response to PD or ICA (Wu et al., 2014b). However, with regard to the alterations in channel gating induced by RPR, concatenated WT4 hERG1 channels were less sensitive than WTmonomer hERG1 channels formed naturally by coassembly of single WT subunits. Presumably, slow deactivation induced by RPR is facilitated by the greater flexibility of the N- and C-terminal cytoplasmic domains present in naturally formed channels as compared with the tethered cytoplasmic domains present in WT4 channels. Interaction between the N-terminal eag domain and the C-terminus of an adjacent subunit has also been reported to affect inactivation gating (Gustina and Trudeau, 2013). However, we also found that concatenation of four WT hERG1 subunits caused no significant change in the steady-state voltage dependence of inactivation (Wu et al., 2014b) and that RPR disrupted inactivation of WTmonomer channels more than WT4 channels. Together our findings indicate that whereas linking four subunits together does not affect the gating of WT channels, unrestrained cytoplasmic N- and C-termini are required to achieve maximal effects of RPR on hERG1 channel gating.
RPR binds to a hydrophobic pocket located between adjacent hERG1 subunits and formed by specific residues in the S5 and S6 segments. Similar to the subunit stoichiometry associated with altered channel gating induced by PD or ICA (Wu et al., 2014b), four WT subunits were required to achieve maximal slowing of hERG1 deactivation by RPR. Although RPR slowed the rate of deactivation of hERG1 channels in direct proportion to the number of WT subunits present in a concatenated tetramer, the presence of a single WT subunit was sufficient to achieve half of the maximum shift in the voltage dependence of hERG1 inactivation, and full effects were achieved with only three WT subunits.
These findings suggest that distinct molecular pathways mediate the allosteric coupling between drug binding and altered properties of deactivation and inactivation. Concatemers containing an equal number of WT subunits responded similarly to RPR irrespective of their position within a tetramer. For example, concatemers with two WT subunits arranged in an adjacent or diagonal configuration responded similarly to RPR. This finding is unlike what we previously reported for the effects of PD or ICA on hERG1 concatemers. Channels with adjacent positioning of two WT subunits were more sensitive to ICA than channels with diagonally oriented WT subunits, and the opposite was observed for PD (Wu et al., 2014b). Together these findings suggest that agonist-induced subunit interactions differ for compounds that primarily affect open probability (PD), or the gating associated with inactivation (ICA) or deactivation (RPR) of hERG1 channels.
Both RPR and ICA disrupt hERG1 inactivation, but do so by apparently different mechanisms. First, RPR at 50 µM shifted V0.5inact by about +25 mV, whereas ICA at 30 µM shifted V0.5inact by >100 mV. Second, the ∆∆Ginact for RPR was minimal (<0.15 kcal/mol) and not enhanced when the number of WT subunits/tetramer was increased from 1 to 4. In contrast, ∆∆Ginact for ICA was increased from ∼0.25 kcal/mol to ∼3 kcal/mol when the number of WT subunits/tetramer was increased from 1 to 4. Third, the full effect of ICA on inactivation required all four ligand binding sites, whereas three binding sites were sufficient to achieve maximal effects of RPR. An extreme example of agonist-dependent binding site stoichiometry is exemplified by PNU-120596 [N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea] and acetylcholine. PNU-120596 is a potentiator that slows desensitization of pentameric α7 nicotinic receptors only when at least four and perhaps all five available binding sites are occupied by the compound (daCosta and Sine, 2013). In contrast, occupancy of only a single binding site by the native ligand (acetylcholine) is sufficient to fully activate α7 nicotinic receptors (Andersen et al., 2013). Thus, the stoichiometric basis of allosteric modification of multimeric channels can be ligand specific (i.e., RPR versus ICA; PNU-120596 versus acetylcholine).
Our study has several limitations. First, it is unclear whether the L553A mutation disrupts RPR binding to the hERG1 channel or prevents the compound from exerting its multiple effects by an allosteric mechanism. Second, because the time course for deactivation of hERG1 currents in the presence of RPR was highly complex, we were unable to perform a quantitative analysis of the energetics associated with subunit interactions. Energetic analysis requires that channel gating can be reasonably simplified to a two-state (open↔closed) model (Ogielska et al., 1995; Wu et al., 2014a). Nonetheless, the slowing of deactivation by RPR, characterized by additive contributions from each WT subunit, is clearly distinct from the natural deactivation process. Slow deactivation of WT hERG1 channels is obligate, requiring the presence of four intact N-terminal domains (Thomson et al., 2014). Third, the effects of RPR on deactivation and inactivation parameters were not saturated at its maximal soluble concentration (50 µM), preventing determination of a complete concentration-response relationship. In the absence of an accurate estimate of EC50 or Hill coefficient, we were unable to determine whether RPR binding was cooperative. We previously reported that the binding of PD and ICA to hERG1 channels was not cooperative (Wu et al., 2014b) and that RPR binds to a similar hydrophobic region of the channel as these other agonists (Perry et al., 2007, 2009; Garg, et al., 2011). Thus, we speculate that the effects of RPR are not mediated by cooperative ligand binding.
In summary, the effects of RPR on deactivation and inactivation gating are characterized by distinct subunit stoichiometry. RPR slowed channel deactivation in direct proportion to the number of WT subunits present in a concatenated tetramer, and all four WT subunits were required to achieve maximum efficacy. In contrast, inhibition of inactivation by a high concentration of RPR was half maximal in channels containing a single WT subunit, and maximum effects required that only three of the four subunits be WT. A detailed understanding of how gating modifiers allosterically enhance hERG1 channel function may facilitate discovery of novel compounds for prevention of arrhythmias associated with LQTS.
Abbreviations
- ICA
ICA-105574, 3-nitro-N-(4-phenoxyphenyl) benzamide
- LA
L553A mutation
- LQTS
long QT syndrome
- PD
PD-118057, 2-[[4-[2-(3,4 dichlorophenyl)ethyl]phenyl]amino]benzoic acid
- PNU-120596
N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea
- RPR
RPR-260243, (3R,4R)-4-[3-(6-methoxy-quinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5 trifluorophenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid
- WT
wild-type
Authorship Contributions
Participated in research design: Wu, Gardner, Sanguinetti.
Conducted experiments: Wu, Gardner.
Performed data analysis: Wu, Sanguinetti.
Wrote or contributed to the writing of the manuscript: Wu, Sanguinetti.
Footnotes
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01-HL055236].
References
- Abbruzzese J, Sachse FB, Tristani-Firouzi M, Sanguinetti MC. (2010) Modification of hERG1 channel gating by Cd2+. J Gen Physiol 136:203–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen N, Corradi J, Sine SM, Bouzat C. (2013) Stoichiometry for activation of neuronal α7 nicotinic receptors. Proc Natl Acad Sci USA 110:20819–20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80:795–803. [DOI] [PubMed] [Google Scholar]
- daCosta CJ, Sine SM. (2013) Stoichiometry for drug potentiation of a pentameric ion channel. Proc Natl Acad Sci USA 110:6595–6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenichel RR, Malik M, Antzelevitch C, Sanguinetti M, Roden DM, Priori SG, Ruskin JN, Lipicky RJ, Cantilena LR, Independent Academic Task Force (2004) Drug-induced torsades de pointes and implications for drug development. J Cardiovasc Electrophysiol 15:475–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V, Stary-Weinzinger A, Sachse F, Sanguinetti MC. (2011) Molecular determinants for activation of human ether-à-go-go-related gene 1 potassium channels by 3-nitro-n-(4-phenoxyphenyl) benzamide. Mol Pharmacol 80:630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach AC, Stoehr SJ, Castle NA. (2010) Pharmacological removal of human ether-à-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574). Mol Pharmacol 77:58–68. [DOI] [PubMed] [Google Scholar]
- Gianulis EC, Liu Q, Trudeau MC. (2013) Direct interaction of eag domains and cyclic nucleotide-binding homology domains regulate deactivation gating in hERG channels. J Gen Physiol 142:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. (1991) Expression of ion channels by injection of mRNA into Xenopus oocytes. Methods Cell Biol 36:487–509. [DOI] [PubMed] [Google Scholar]
- Gustina AS, Trudeau MC. (2013) The eag domain regulates hERG channel inactivation gating via a direct interaction. J Gen Physiol 141:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst RS, North RA, Adelman JP. (1995) Potassium channel assembly from concatenated subunits: effects of proline substitutions in S4 segments. Receptors Channels 3:263–272. [PubMed] [Google Scholar]
- Kang J, Chen XL, Wang H, Ji J, Cheng H, Incardona J, Reynolds W, Viviani F, Tabart M, Rampe D. (2005) Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K+ channel. Mol Pharmacol 67:827–836. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. (2001) Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104:569–580. [DOI] [PubMed] [Google Scholar]
- McCormack K, Lin L, Iverson LE, Tanouye MA, Sigworth FJ. (1992) Tandem linkage of Shaker K+ channel subunits does not ensure the stoichiometry of expressed channels. Biophys J 63:1406–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. (1995) Cooperative subunit interactions in C-type inactivation of K channels. Biophys J 69:2449–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Abbruzzese J, Sanguinetti MC. (2009) PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc Natl Acad Sci USA 106:20075–20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Sanguinetti MC. (2007) Structural basis of action for a human ether-a-go-go-related gene 1 potassium channel activator. Proc Natl Acad Sci USA 104:13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack JT, Shamotienko O, Dolly JO. (2008) How to validate a heteromeric ion channel drug target: assessing proper expression of concatenated subunits. J Gen Physiol 131:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC. (2014) HERG1 channel agonists and cardiac arrhythmia. Curr Opin Pharmacol 15:22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307. [DOI] [PubMed] [Google Scholar]
- Schreibmayer W, Lester HA, Dascal N. (1994) Voltage clamping of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflugers Arch 426:453–458. [DOI] [PubMed] [Google Scholar]
- Stühmer W. (1992) Electrophysiological recording from Xenopus oocytes. Methods Enzymol 207:319–339. [DOI] [PubMed] [Google Scholar]
- Thomson SJ, Hansen A, Sanguinetti MC. (2014) Concerted all-or-none subunit interactions mediate slow deactivation of human ether-à-go-go-related gene K+ channels. J Biol Chem 289:23428–23436. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269:92–95. [DOI] [PubMed] [Google Scholar]
- Wu W, Gardner A, Sanguinetti MC. (2014a) Cooperative subunit interactions mediate fast C-type inactivation of hERG1 K+ channels. J Physiol 592:4465–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Sachse FB, Gardner A, Sanguinetti MC. (2014b) Stoichiometry of altered hERG1 channel gating by small molecule activators. J Gen Physiol 143:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Lozinskaya IM, Lin Z, Willette RN, Brooks DP, Xu X. (2006) Mallotoxin is a novel human ether-a-go-go-related gene (hERG) potassium channel activator. J Pharmacol Exp Ther 319:957–962. [DOI] [PubMed] [Google Scholar]
- Zhou J, Augelli-Szafran CE, Bradley JA, Chen X, Koci BJ, Volberg WA, Sun Z, Cordes JS. (2005) Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol 68:876–884. [DOI] [PubMed] [Google Scholar]