

Abstract
Orexins (OXs) are peptides produced by perifornical (PeF) and lateral hypothalamic neurons that exert a prominent role in arousal-related processes, including stress. A critical role for the orexin-1 receptor (OX1R) in complex emotional behavior is emerging, such as overactivation of the OX1R pathway being associated with panic or anxiety states. Here we characterize a brain-penetrant, selective, and high-affinity OX1R antagonist, compound 56 [N-({3-[(3-ethoxy-6-methylpyridin-2-yl)carbonyl]-3-azabicyclo[4.1.0]hept-4-yl}methyl)-5-(trifluoromethyl)pyrimidin-2-amine]. Ex vivo receptor binding studies demonstrated that, after subcutaneous administration, compound 56 crossed the blood-brain barrier and occupied OX1Rs in the rat brain at lower doses than standard OX1R antagonists GSK-1059865 [5-bromo-N-({1-[(3-fluoro-2-methoxyphenyl)carbonyl]-5-methylpiperidin-2-yl}methyl)pyridin-2-amine], SB-334867 [1-(2-methyl-1,3-benzoxazol-6-yl)-3-(1,5-naphthyridin-4-yl)urea], and SB-408124 [1-(6,8-difluoro-2-methylquinolin-4-yl)-3-[4-(dimethylamino)phenyl]urea]. Although compound 56 did not alter spontaneous sleep in rats and in wild-type mice, its administration in orexin-2 receptor knockout mice selectively promoted rapid eye movement sleep, demonstrating target engagement and specific OX1R blockade. In a rat model of psychological stress induced by cage exchange, the OX1R antagonist prevented the prolongation of sleep onset without affecting sleep duration. In a rat model of panic vulnerability (involving disinhibition of the PeF OX region) to threatening internal state changes (i.e., intravenous sodium lactate infusion), compound 56 attenuated sodium lactate–induced panic-like behaviors and cardiovascular responses without altering baseline locomotor or autonomic activity. In conclusion, OX1R antagonism represents a novel therapeutic strategy for the treatment of various psychiatric disorders associated with stress or hyperarousal states.
Introduction
Orexins (OXs), also known as hypocretins, are the common names given to a pair of excitatory neuropeptides called OX-A and OX-B derived from the common precursor prepro-OX that is exclusively produced by perifornical (PeF) and lateral hypothalamic neurons (de Lecea et al., 1998; Sakurai et al., 1998). OX-producing neurons project widely to key areas of the brain (Peyron et al., 1998) and are predominantly involved in the control of wakefulness and the regulation of food intake, reward, addictive behaviors, and stress (Li et al., 2014). The OX neuropeptides mediate their effect by stimulating two distinct G protein–coupled receptors, orexin-1 receptor (OX1R) and orexin-2 receptor (OX2R), that are colocated or selectively located in specific brain areas, suggesting differentiated roles (Trivedi et al., 1998; Marcus et al., 2001). OX1Rs are more selectively expressed in the bed nucleus of the stria terminalis, amygdala, cingulate cortex, and are exclusive to noradrenergic neurons in the locus coeruleus, which play a role in panic and anxiety. Conversely, OX2Rs are exclusive to histaminergic neurons in the tuberomammillary nucleus, which play a critical role in wake promotion and are predominantly expressed in the paraventricular hypothalamic nucleus (PVN), which is involved in the hypothalamic-pituitary-adrenal (HPA) axis regulation. In other brain regions such as the dorsal raphe, the ventral tegmental area, or the prefrontal cortex, OX1R and OX2R are coexpressed.
Although dual OX1/2R antagonists have been shown to promote sleep in various species by inhibiting the output of wake-active neurons in the hypothalamus and brainstem regions (Hoyer and Jacobson, 2013), antagonism of the OX2R alone is sufficient to induce and prolong sleep in rodents (Dugovic et al., 2009; Mang et al., 2012). In parallel, evidence has accumulated that demonstrates an involvement of OX signaling via OX1R in reward pathways associated with drug dependence (Sharf et al., 2010). The role of the OX1R in emotional behavior is emerging (Johnson et al., 2012). Human subjects with panic anxiety symptoms have elevated levels of OX-A in the cerebrospinal fluid compared with subjects without panic anxiety symptoms (Johnson et al., 2010). Sodium lactate (NaLac) infusions or acute hypercapnia induction signal internal body state (interoceptive) threats and reliably cause panic attacks in humans by activating OX neurons in the PeF of rats (Johnson et al., 2010, 2012). This activation correlates with anxiety in the social interaction test. Blocking OX signaling with either small interfering RNA or selective OX1R antagonists attenuates panic-like responses to NaLac. Overall, there is compelling evidence for the overactivation of the OX1R pathway in hyperactive states; thus, conceptually, a selective OX1R antagonist might normalize overexcited networks without inducing sedation. Notably, OX1R antagonism fails to reduce anxiety-like behavior under baseline conditions in both elevated plus maze and social interaction tests (Yeoh et al., 2014). Efficacy of an OX1R antagonist has been observed in fear-conditioned startle and in a resident-intruder paradigm involving social interaction (Steiner et al., 2012, 2013a). OX neurons are activated during arousal or psychological stress but not by passive restraint stress or cold exposure (Furlong et al., 2009). In rats exposed to cage exchange, a model of psychological stress, cFos expression, was elevated in hypothalamic OX neurons and in the PVN (involved in HPA axis and autonomic control) relative to control (Cano et al., 2008).
Here we report the characterization of a novel selective OX1R antagonist recently described in the patent literature, compound 56 [N-({3-[(3-ethoxy-6-methylpyridin-2-yl)carbonyl]-3-azabicyclo[4.1.0]hept-4-yl}methyl)-5-(trifluoromethyl)pyrimidin-2-amine] (Alvaro et al., 2010) (Fig. 1). We compared its properties to the literature standards GSK-1059865 [5-bromo-N-({1-[(3-fluoro-2-methoxyphenyl)carbonyl]-5-methylpiperidin-2-yl}methyl)pyridin-2-amine], SB-334867 [1-(2-methyl-1,3-benzoxazol-6-yl)-3-(1,5-naphthyridin-4-yl)urea], and SB-408124 [1-(6,8-difluoro-2-methylquinolin-4-yl)-3-[4-(dimethylamino)phenyl]urea] (Fig. 1). In vitro affinity and potency for the human and rat OX1R were determined by radioligand binding and in vitro functional assays. Compound 56 was tritiated, tested as a tracer for autoradiography on rat tissue sections, and in vivo target engagement was measured after subcutaneous injection of compound 56 in rat brain using ex vivo receptor occupancy. We have recently shown that, although selective OX1R antagonism did not affect sleep-wake states, additional pharmacologic blockade of OX1R to OX2R antagonism elicited a disinhibition of rapid eye movement (REM) sleep at the expense of non-REM (NREM) sleep in rats (Dugovic et al., 2014). Therefore, the effect of the selective OX1R antagonist on sleep was tested in OX2R knockout (KO) mice to demonstrate in vivo functional target engagement. Finally, the activity of compound 56 was evaluated in two models of stress-induced hyperarousal, a psychological exteroceptive stress (i.e., external threat) elicited by cage exchange and a previously described interoceptive stress (i.e., internal body state threat) elicited by intravenous NaLac provocation in a panic vulnerability model (Johnson et al., 2010) in rats.
Fig. 1.

Chemical structure of OX1R antagonists compound 56, GSK-1059865, SB-408124, and SB-334867.
Materials and Methods
All animal procedures performed in this study were in accordance with the Guide for the Care and Use of Laboratory Animals adopted by the U.S. National Institutes of Health (publication no. 80-23, revised 1996) and the guidelines of the Institutional Animal Care and Use Committee. Animals were housed individually under controlled conditions with a 12-hour light/dark schedule and temperature of 22 ± 2°C. Food and water were provided ad libitum. Experiments were performed after animals had acclimated for at least 1 week unless stated otherwise.
In Vitro Radioligand Binding Assays.
Human or rat OX1R binding was measured in competitive radioligand binding assays using [3H]SB-674042 [1-(5-(2-fluoro-phenyl)-2-methyl-thiazol-4-yl)-1-((S)-2-(5-phenyl-(1,3,4)oxadiazol-2-ylmethyl)-pyrrolidin-l-yl)-methanone] as a tracer (4 nM, specific activity 35 Ci/mmol) (Langmead et al., 2004). Cell membranes were prepared from clonal CHO-K1 cells transfected with the human OX1R or clonal human embryonic kidney 293 (HEK-293) cells transfected with the rat OX1R. Dilutions of test compounds were made in Dulbecco’s phosphate-buffered saline from 10 mM stocks dissolved in dimethylsulfoxide. After a 60-minute incubation at room temperature, binding reactions were filtered. The membranes were counted in a scintillation counter. Nonspecific binding was determined in the presence of 10 μM almorexant. Affinities of compounds for the human OX2R were measured by competitive radioligand binding using tritiated EMPA (N-ethyl-2-[(6-methoxy-pyridin-3-yl)-(toluene-2-sulfonyl)-amino]-N-pyridin-3-ylmethyl-acetamide) (2 nM, specific activity 27 Ci/mmol) (Malherbe et al., 2009). Cell membranes were prepared from a stable pool of HEK-293 cells transfected with the human OX2R. Nonspecific binding was determined with 10 μM almorexant.
The Ki of the test compounds was calculated based on nonlinear regression (one site competition) using GraphPad Prism (GraphPad Software, San Diego, CA).
Compound 56 was tritiated via palladium-mediated hydrogenolysis (27.5 Ci/mmol; Moravek, Brea, CA) of the corresponding bromo precursor. The bromo precursor is available from HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate)-mediated amide-coupling of the corresponding amine and carboxylic acid fragments (Alvaro et al., 2010; Maton et al., 2010; Verdelet et al., 2011), and its binding parameters (Kd and Bmax) for the human and rat OX1R were determined using conditions identical to those described for [3H]SB-674042.
The selectivity of compound 56 was evaluated in a large variety of ion channels, transporters, and receptor-binding assays. These assays were performed by Eurofins (Celles L’Evescault, France).
[3H]Compound 56 In Vitro Receptor Autoradiography on Tissue Section.
Adult male Sprague-Dawley rats (300–350 g; Charles River Laboratories, San Diego, CA) were euthanized using carbon dioxide, and brains were immediately removed and rapidly frozen in dry ice. Twenty-micrometer-thick coronal sections were cut using a cryostat microtome (Leica CM 1950; Leica Mircosystems, Wetzlar, Germany) and thaw-mounted on adhesive microscope slides (Superfrost Plus; VWR, Radnor, PA). The sections were kept at –80°C until use. Brain sections were preincubated for 15 minutes at room temperature in 50 mM Tris, 10 mM MgCl2, 5 mM EDTA buffer at pH 7.4, and then incubated for 60 minutes in a fresh preparation of the same buffer supplemented with 5 nM [3H]compound 56. Nonspecific binding was determined using adjacent sections incubated in the presence of 10 μM GSK-1059865. At the end of the incubation, sections were washed three times (10 minutes each) in ice-cold buffer, dipped in deionized water, and rapidly dried under a stream of cold air. Image acquisition was performed using a β-Imager TRacer (BioSpace, Paris, France).
In Vitro Functional Assays (Calcium Mobilization Assays).
Stably transfected CHO-K1 cells for the human OX1R or HEK-293 cells for the rat OX1R were used for the in vitro functional assays. The human OX2R functional assay used PFSK-1 cells, which are a human neuroectodermal cell line that innately expresses the OX2R. Since the intracellular calcium response is transient and not consistent with equilibrium assumptions, the assays were performed by giving a standard, EC80 dose of the OX agonist and calculating a pKB from inhibition of the agonist response by a dose range of the antagonists. The cells were plated in black 96-well tissue culture plates with clear bottoms at 50,000 cells/well and grown overnight at 37°C in 5% CO2. Dilutions of the antagonist were prepared in Hanks’ balanced salt solution from 10 mM dimethylsulfoxide stocks, whereas dilutions of OX peptides (OX-A for OX1R assays, OX-B for OX2R assays) were prepared in Hanks’ balanced salt solution plus 0.1% bovine serum albumin. On the day of the assay, a 2× dye-loading solution (BD Calcium Assay Kit; BD Biosciences, San Jose, CA) was added to the cells and incubated for 45 minutes at 37°C in 5% CO2. Dilutions of the test compounds were added and the cells were incubated at room temperature for 15 minutes. The cell plate was then transferred to the Molecular Devices (Sunnyvale, CA) Fluorometric Imaging Plate Reader Tetra instrument, which adds the OX agonist and monitors changes in fluorescence which reflect intracellular calcium levels.
Results were calculated using GraphPad Prism software. Raw data from the Fluorometric Imaging Plate Reader Tetra were exported as the difference between the maximum and minimum fluorescence observed for each well. A nonlinear regression was used to determine the agonist EC50 and antagonist IC50 for each plate, then the antagonist KB was calculated according to Cheng and Prusoff (1973).
Ex Vivo Receptor Occupancy Assays.
Experiments were performed as previously described (Dugovic et al., 2009) in male Sprague-Dawley rats (300–400 g; Charles River Laboratories). The animals were euthanized using carbon dioxide and decapitated at different time points after drug administration (n = 3 per time point or dose regimen). Brains were rapidly frozen on powdered dry ice and stored at −80°C before sectioning. Plasma samples were also collected for bioanalysis (liquid chromatography–tandem mass spectrometry). Twenty-micrometer-thick tissue sections at the level of the tenia tecta were prepared for autoradiography. OX1R radioligand binding autoradiography was determined at room temperature with 5 nM [3H]SB-674042. Sections were incubated for 10 minutes to minimize dissociation. Nonspecific binding was determined in the presence of 10 μM GSK-1059865. Sections were allowed to dry before acquisition with β-Imager (BioSpace) for 6 hours. Quantitative analysis was performed using the β-Imager TRacer. Ex vivo receptor labeling was expressed as the percentage of receptor labeling in corresponding brain areas (i.e., tenia tecta) of vehicle-treated animals. The percentage of receptor occupancy was plotted against time or dosage using GraphPad Prism. The percentage of receptor occupancy was also plotted against drug plasma or brain concentration. Pharmacokinetic parameters were analyzed using a noncompartmental model using the software package WinNonlin Version 4.0.1. (Pharsight, Palo Alto, CA).
Sleep Recording and Analysis in Rats and Mice.
Sleep experiments were conducted in male Sprague-Dawley rats (350–450 g; Harlan Laboratories, Livermore, CA) and in male C57Bl6 OX2R KO and corresponding wild-type mice (30–35 g; Charles River Laboratories) as described previously (Dugovic et al., 2009). Animals were chronically implanted with telemetric devices (Data Sciences International, St. Paul, MN) for the recording of electroencephalogram (EEG) and electromyogram signals. Polysomnographic wave forms were analyzed per 10-second epoch and classified as wake, NREM, or REM sleep using the computer software program SleepSign (Kissei Comtec, Nagano, Japan). For each experiment, EEG and electromyogram signals were recorded for up to 6 hours after administration of the tested compounds. Analysis of sleep parameters included latency to NREM sleep (defined as the time interval to the first six consecutive NREM epochs) and REM sleep (the first two consecutive REM epochs post-treatment), and the duration of NREM, REM, and total sleep.
Results were averaged and expressed as the mean ± S.E.M. in defined time intervals. To determine whether differences were significant at a given interval, either paired Student’s t test or one-way analysis of variance (ANOVA) followed by Newman-Keuls multiple comparison post-hoc analysis was performed.
Cage-Exchange Stress Model in Rats and Mice.
The cage-exchange stress procedure was performed in male Sprague-Dawley rats (350–450 g; Harlan Laboratories) and in male C57Bl6 mice (30–35 g; The Jackson Laboratory, Bar Harbor, ME) at 4 hours into the light phase, a period when the animals normally exhibit the maximal amount of physiologic sleep. The animal was removed from its home cage and placed into a dirty cage previously occupied by another animal for at least 1 week during an 8-hour period (until dark onset). As a control procedure, the animal was removed from its home cage and returned to the same cage (brief handling).
Adrenocorticotropic Hormone Measurement in Mice.
Male C57Bl6 mice (30–35 g; The Jackson Laboratory, Sacramento, CA) were used to assess adrenocorticotropic hormone (ACTH) serum levels. On the day of the experiment, a blood sample was collected in a serum separator tube (Becton Dickinson, Piscataway, NJ) via the submandibular punch of each mouse. Each tube was then allowed to sit on ice for 1 hour and then spun at 10,000 rpm (Eppendorf Microfuge; Hamburg, Germany) at 4°C for 10 minutes to allow for separation of the serum from the remaining whole-blood constituents. Serum was then collected into a sterile Eppendorf tube and frozen at −80°C until the assay was performed. The Mouse Bone Magnetic Bead Luminex Panel (EMD Millipore, Billerica, MA) was used to measure serum levels of ACTH. Results were averaged and expressed as mean ± S.E.M. A one-way ANOVA followed by a Neuman-Keuls post-hoc analysis was used to determine if any of the treatment groups were significant from each other.
Sodium Lactate Panic Provocation Model.
Experiments were performed in male Sprague-Dawley rats (300–350 g; Harlan Laboratory, Indianapolis, IN). Prior to and during surgeries, rats were anesthetized with a nose cone connected to an isoflurane system (MGX Research Machine; Vetamac, Rossville, IN). Radiotelemetry probes (Data Science International St. Paul, MN) were surgically implanted into the peritoneal cavity and sutured to the muscle wall to assess general motor activity and core body temperature (CBT). A pressure transducer was implanted into the femoral artery to assess cardiovascular responses [i.e., mean arterial blood pressure (MAP) and heart rate (HR)]. Rats were also fitted with femoral venous catheters for 0.5 M i.v. NaLac infusions, as previously described (Shekhar et al., 1996). After 3–5 days of recovery, rats were anesthetized, and 26-gauge T-shaped cannulae (Plastics One Inc., Roanoake, VA) were directed at cardioexcitatory PeF regions (Shekhar and Keim, 1997) (bregma: 1.2 mm posterior, +2.1 mm lateral, +9.1 mm ventral and adjusted for approaching at a 10° angle toward the midline with the stereotaxic incisor bar elevated 5 mm above the interaural line) and cemented into place. The 22-gauge side arm was then attached, via PE-60 tubing, to an osmotic minipump [prefilled with 1-allyglycine (l-AG) solution chronically infused at 3.5 nmol/0.5μl per hour] and sutured into place subcutaneously at the nape of the neck (model no. 2002; DURECT Corporation, Cupertino, CA). Previous studies have determined that the dose of l-AG used here reduces local GABA concentrations by approximately 60% following unilateral infusions (Shekhar and DiMicco, 1987; Abshire et al., 1988; Shekhar et al., 1996, 2006; Shekhar and Keim, 1997).
Five days following l-AG infusion onset, in a counterbalanced design and with 48 hours between crossover, rats received a subcutaneous injection of either compound 56 at 3, 10, or 30 mg/kg or vehicle as a control group 60 minutes prior to the 15-minute 0.5 M NaLac challenge. Data Science International Dataquest software was used to monitor and record MAP, HR, CBT, and motor activity, which were recorded continuously in freely moving conscious rats and are expressed as a 20-minute time course. The data reported are changes in MAP, HR, CBT, and activity from the average of the baseline (t − 5 to t − 1) from each rat.
Baseline social interaction (SI) testing was performed 7–8 days following radiotelemetry surgery recovery, and repeated again 2–3 days later during drug treatment crossover. On experimental drug testing days, the SI test was performed 5 minutes after the offset of the NaLac challenge, with different partners each time. The SI box dimensions were 0.9 m (length) × 0.9 m (width) × 0.3 m (height). The SI test is a validated test of experimental anxiety-like behavior in rats that is sensitive to Food and Drug Administration–approved treatments for anxiety disorder symptom management that includes benzodiazepines and selective serotonin reuptake inhibitors (Sanders and Shekhar, 1995; Shekhar and Katner, 1995). All behavioral tests are digitally video recorded with a camera above the box. The “experimental” rat and an unfamiliar “partner” rat are both allowed to habituate individually to the box for a 5-minute period 24 hours prior to each SI test. During the SI test, the two rats are placed together in the center of the box, and the total duration (seconds) of nonaggressive physical contact (grooming, sniffing, crawling over and under, etc.) initiated by the experimental rat is quantified over a 5-minute duration. A baseline SI test was performed 72+ hours after intravenous catheterization, but prior to osmotic minipump implantation. Another SI test was performed 5 days following minipump infusions and immediately following saline or NaLac infusions. Video-recorded sessions were scored at a later time by S.D.F., who was blind to any drug treatment.
Following experiments, all rats were anesthetized and decapitated, and their brains were removed, frozen, sectioned (30 μm), and stained with neutral red for determination of injection cannulae placements.
Each dependent variable for assessment of behavior and radiotelemetry data were analyzed using a one-way ANOVA, or a one-way ANOVA with repeated measures with drug treatment as the main factor and time as repeated measures, respectfully. In the presence of significant main effects, post-hoc tests were conducted using a parametric Fisher’s least significant difference test. Within-subjects time effects were also assessed using a Dunnett’s one-way analysis with the minute prior to the intravenous infusion used as the control. Statistical significance was accepted with P < 0.05. All statistical analyses were carried out using SPSS 13.0 (SPSS Inc., Chicago, IL), and all graphs were generated using SigmaPlot 2001 for Windows (SPSS Inc.).
Chemicals.
SB-334867 and SB-408124 were purchased from Tocris Cookson (Ellisville, MO). Almorexant, GSK-1059865, compound 56, EMPA, and SB-674042 were synthesized at Janssen Research & Development LLC (San Diego, CA). Peptides were obtained from Bachem (Torrance, CA). For in vitro assays, compounds were dissolved in dimethylsulfoxide (stock solution at 10 mM) and further diluted in assay buffer. For in vivo studies, SB-334867, SB-408124, and compound 56 were formulated in 30% sulfobutyl ether-β-cyclodextrin. GSK-1059865 was formulated in 5% N-methyl-2-pyrrolidone, 20% Solutol (BASF Canada, Mississauga, ON, Canada), and 75% 2-hydroxypropyl-β-cyclodextrin (20%). All compounds were administered orally or subcutaneously in a volume of 5 ml/kg (receptor occupancy studies) or 1 ml/kg (sleep and NaLac experiments) in rats and in a volume of 10 ml/kg in mice.
Results
Compound 56 Is a Selective High-Affinity OX1R Antagonist
The affinity of compound 56 for the human and rat OX1R was determined by competitive radioligand binding using [3H]SB-674042. GSK-1059865, SB-334867, and SB-408124, OX1R antagonist literature standards, were also included for comparison purposes. To determine the selectivity ratio versus the OX2R, the affinity of the compounds for the human OX2R was determined by competitive radioligand binding using [3H]EMPA as the radioligand.
Compound 56 showed high-affinity binding to the human and rat OX1R, with Ki values of 3.6 and 3.2 nM, respectively (Table 1). GSK-1059865 also displayed high affinity for human and rat OX1R. Compound 56 showed greater affinity for the human OX1R compared with SB-334867 and SB-408124 (13- and 31-fold, respectively). The binding selectivity of compound 56 at the human OX1R compared with the human OX2R is substantial (44-fold).
TABLE 1.
Comparative binding parameters (Ki values [in nM]) of compound 56, GSK-1059865, SB-334867, and SB-408124 at human OX1R, rat OX1R, and human OX2R
The selectivity ratio versus the human OX2R is also listed. Data represent the mean ± S.E.M. of at least six determinations, each performed in duplicate.
|
Ki |
||||
|---|---|---|---|---|
| hOX1R CHO [3H]SB-674042 | rOX1R HEK-293 [3H]SB-674042 | hOX2R PFSK-1 [3H]EMPA | hOX2R/hOX1R Ratio | |
| Compound 56 | 3.6 ± 0.9 | 3.2 ± 0.4 | 160 ± 5 | 44 |
| GSK-1059865 | 5.0 ± 0.3 | 6.4 ± 0.2 | 409 ± 10 | 82 |
| SB-334867 | 47.6 ± 6.4 | 144 ± 7 | 3781 ± 280 | 79 |
| SB-408124 | 112 ± 8 | 375 ± 56 | >10,000 | >100 |
hOX1R, human OX1R; hOX2R, human OX2R; rOX1R, rat OX1R.
The four compounds were assayed by binding in a panel of 50 receptors, ion channels, and transporter assays including adenosine (A1, A2A, A3), adrenergic (α1, α2, α1), angiotensin (AT1), dopamine (D1, D2), bradykinin (B2), cholecystokinin (CCKA), galanin (GAL2), melatonin (ML1), muscarinic (M1, M2, M3), neurotensin (NT1), neurokinin (NK2, NK3), opiate (μ, κ, δ), serotonin (5-HT1A, 5-HT1B, 5-HT2A, 5-HT3, 5-HT5A, 5-HT6, 5-HT7), somatostatin, vasopressin (V1a), norepinephrine transporter, dopamine transporter, and ion channels (sodium, calcium, potassium, and chloride). Compound 56 at concentrations up to 1 μM had no significant affinity for any receptor/transporter/ion channel (<50% inhibition at 1 μM) other than the OX1R. GSK-1059865 had some weak affinity for hNK2 (Ki = 920 nM) and hκ (Ki = 620 nM). SB-334867 had weak affinity for the h5-HT2B receptor (750 nM).
Compound 56 was radiolabeled and tested as a radioligand for the human and rat OX1R. Characterization of the binding of [3H]compound 56 to the human and rat OX1R was performed on membranes. Specific binding of [3H]compound 56 was saturable with Kd = 0.8 ± 0.1 nM and Bmax = 879 ± 26 fmol⋅mg for the human OX1R (Fig. 2A) and Kd = 0.3 ± 0.1 nM and Bmax = 280 ± 8 fmol⋅mg for the rat OX1R (Fig. 2B).
Fig. 2.
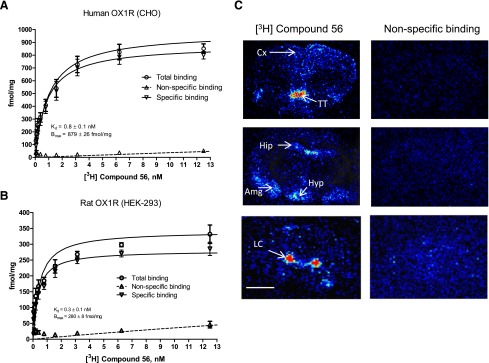
Characterization of [3H]compound 56. (A and B) Saturation study: total, specific, and nonspecific binding of [3H]compound 56 to CHO or HEK-293 cells stably expressing the human (A) or rat (B) OX1R receptor with increasing radioligand concentration. Nonspecific binding was defined as that remaining in the presence of 10 μM almorexant. Vertical lines show the S.E.M. Data are the mean of three (rat OX1R) or four (human OX1R) experiments. (C) Digitized computer images of the distribution of [3H]compound 56 binding sites in coronal rat brain sections at three different levels; nonspecific binding was determined in the presence of 10 μM GSK-1059865. Color represents relative levels of optical density ranging red > yellow > green > blue > black. Scale bar, 0.25 cm. Amg, amygdala; Cx, cortex; Hip, hippocampus; Hyp, hypothalamus; LC, locus coeruleus TT, tenia tecta.
[3H]Compound 56 was then tested on rat brain tissue sections using autoradiography (Fig. 2C). Specific labeling was observed in brain regions known to contain the OX1R, with the highest binding densities observed in the locus coeruleus and tenia tecta. Specific binding densities were also observed in the cortex, hippocampus, hypothalamus, and amygdala. Very low nonspecific binding was observed.
The functional antagonism of compound 56 for the human or rat OX1R was determined by measuring changes in intracellular calcium in cell culture assays in response to an EC80 dose of OX-A. Functional antagonism data, summarized in Table 2, show that the high-affinity OX1R binding of compound 56 is reflected in potent functional activity. The KB values correlated well with the Ki values for the human and rat OX1R. The binding selectivity of compound 56 at the OX1R compared with the OX2R was confirmed at the functional level (Table 2).
TABLE 2.
Comparative in vitro functional potencies (KB values [in nM]) of compound 56, GSK-1059865, SB-334867, and SB-408124 at human OX1R, rat OX1R, and human OX2R
Data represent the mean ± S.E.M. of at least three determinations, each performed in duplicate.
|
KB |
|||
|---|---|---|---|
| hOX1R CHO | rOX1R HEK-293 | hOX2R PFSK-1 | |
| Compound 56 | 2.9 ± 0.5 | 5.2 ± 1.1 | 92 ± 22 |
| GSK-1059865 | 13.3 ± 1.5 | 20.7 ± 3.8 | 304 ± 31 |
| SB-334867 | 163 ± 29 | 232 ± 53 | <5.0 |
| SB-408124 | 127 ± 16 | 210 ± 55 | <5.0 |
hOX1R, human OX1R; hOX2R, human OX2R; rOX1R, rat OX1R.
Compound 56 Crosses the Blood-Brain Barrier and Occupies the OX1R in Rat Brain after Systemic Administration
In vivo occupancy of the OX1R was assessed by ex vivo receptor binding autoradiography of [3H]SB-674042 in rat brain tissue sections at the level of the tenia tecta. Time dependency and dose dependency were assessed after subcutaneous dosing (Fig. 3).
Fig. 3.

Ex vivo brain OX1R binding autoradiography by compound 56, GSK-1059865, SB-334867, and SB-408124. (A) Duration of occupancy for OX1R antagonists postsubcutaneous administration in rats (10 mg/kg). (B) Occupancy versus dose measured at 0.5 hour. Results are expressed as average percentage receptor occupancy versus vehicle-treated rats ± S.E.M. (n = 3). OX1R occupancy was measured in the tenia tecta.
After subcutaneous administration of a 10-mg/kg dose, compound 56 quickly occupied the OX1R in the rat brain (Fig. 3A). The plateau for maximal receptor occupancy (above 90%) was achieved within 15 minutes and lasted for 4 hours. At the 6-hour time point, occupancy levels dropped to 60–70%. GSK-1059865 had a very similar profile. SB-334867 achieved slightly lower levels of OX1R occupancy (80–90%) and started to significantly decrease (∼50%) at the 4-hour time point. Lower levels of OX1R occupancy (∼70%) were observed for SB-408124 with a fast clearance (below 50% after 1 hour).
Dose-response studies were performed at the 0.5-hour time point to compare the four compounds (Fig. 3B). The lowest ED50 value was obtained for compound 56 (0.01 mg/kg) followed by GSK-1059865 (0.9 mg/kg). Much higher ED50 values were measured for SB-334867 (7.0 mg/kg) and SB-408124 (7.6 mg/kg).
Blood-brain barrier and pharmacokinetic parameters are listed in Table 3. Overall, there was a good correlation between receptor occupancy levels and plasma/brain concentrations, i.e., receptor occupancy levels dropped when plasma/brain levels dropped. Compound 56 had the highest brain-to-plasma ratio. Although the highest plasma exposure values were reached after administration of SB-408124, this compound poorly crossed the blood-brain barrier (brain-to-plasma ratio = 0.02).
TABLE 3.
Pharmacokinetic and blood-brain barrier parameters of subcutaneous administration of a 10-mg/kg dose of compound 56, GSK-1059865, SB-334867, and SB-408124 in rats
| Compound 56 | GSK-1059865 | SB-334867 | SB-408124 | |
|---|---|---|---|---|
| Plasma | ||||
| Tmax (h) | 0.25 | 2.00 | 0.50 | 0.50 |
| T1/2 (h) | 0.62 | 2.95 | 1.11 | 2.31 |
| Cmax (ng/ml) | 1871 | 756 | 5407 | 37,028 |
| AUCinf (h*ng/ml) | 3350 | 9142 | 18,068 | 153,418 |
| Brain | ||||
| Tmax (h) | 0.25 | 1.00 | 0.50 | 0.50 |
| Cmax (ng/ml) | 903 | 254 | 1563 | 896 |
| AUCinf (h*ng/ml) | 1515 | 1520 | 5586 | 4081 |
| Brain/plasma ratio | 0.48 | 0.34 | 0.29 | 0.02 |
AUCinf, area under the curve extrapolated to infinity.
Compound 56 was also found to cross the blood-brain barrier after systemic administration in mice (10 mg/kg s.c., Cmax in plasma: 1970 ng/ml, in brain: 929 ng/ml).
The OX1R Antagonist Compound 56 Does Not Affect Spontaneous Sleep in Rats
Rats were orally dosed with the OX1R antagonist compound 56 (10 mg/kg) at the onset of the dark/active phase, the optimal phase to detect any hypnotic activity for OX1/2R antagonists. There were no significant differences between compound 56 and vehicle conditions (paired Student’s t test) in any of the sleep measures examined over the 6-hour period following the treatment, specifically NREM sleep latency or duration (Fig. 4, A and B) and REM sleep latency or duration (Fig. 4, C and D).
Fig. 4.

Effects of the OX1R antagonist compound 56 on sleep parameters in rats. NREM latency (A), NREM duration (B), REM latency (C), and REM duration (D) are determined for the 6-hour period after oral dosing (10 mg/kg) at the onset of the dark phase. Results are expressed in minutes and are represented as means ± S.E.M. of eight animals.
Transient OX1R Blockade Selectively Promotes REM Sleep in OX2R Knockout Mice
Since the functional activity of an OX1R antagonist is not evident and cannot be revealed under baseline conditions, the challenge was to identify an in vivo pharmacodynamic model of efficacy. Previous studies have demonstrated that OX1R antagonists minimally affect spontaneous sleep-wake states in rodents, but produce a disinhibition of REM sleep in the presence of OX2R antagonism (Dugovic et al., 2014). In the present study, the in vivo functional target engagement was investigated in a model of permanent inhibition of OX2R, and the effects of compound 56 on sleep-wake states were examined in mice lacking the OX2R relative to their corresponding wild-type (WT) mice. As compared with vehicle, oral dosing of compound 56 (30 mg/kg) to OX2R KO mice at 2 hours into the light phase significantly reduced the latency for REM sleep and prolonged the time spent in REM sleep during the first 6 hours post-treatment (Fig. 5A), but did not alter REM sleep in WT mice (Fig. 5B). Moreover, OX2R KO mice dosed with the OX1R antagonist exhibited a dysregulation in REM sleep onset as evidenced by the average of REM sleep latency occurring less than 10 minutes before the average of NREM sleep latency, indicative of sleep-onset REM sleep episodes that were observed in 5 out of 7 mice. As opposed to REM sleep parameters, NREM sleep latency and NREM sleep duration were not significantly affected by the OX1R antagonist in either genotype (Fig. 5, C and D).
Fig. 5.

REM sleep-promoting effects of the OX1R antagonist compound 56 in OX2R KO. REM sleep latency and duration in OX2R KO (A), OX2R WT (B), and NREM sleep latency and duration in OX2R KO (C) and OX2R WT (D) for the 6-hour period after oral dosing (30 mg/kg) during the light phase are expressed in minutes. Values are means ± S.E.M. of seven OX2R KO and five OX2R WT mice. *P < 0.05 and **P < 0.01 versus vehicle as determined by paired Student’s t test for each genotype.
Selective OX1R Antagonism Attenuates Cage-Exchange Stress-Induced Hyperarousal without Affecting ACTH Release
The effect of the OX1R antagonist compound 56 was investigated in a rat model of acute psychological stress–induced hyperarousal elicited by cage exchange during the light/rest phase (Cano et al., 2008). Rats were dosed with compound 56 (10 mg/kg s.c.) or vehicle, and 30 minutes later (4 hours into the light phase) were submitted to the cage exchange or control (brief handling) procedure. For each condition (cage exchange and control), animals were injected with the opposite treatment (vehicle or compound 56) in a counterbalanced manner and a crossover design. Analysis of EEG sleep parameters revealed a significant main effect (four treatment conditions, crossover design) for NREM sleep latency [F(3,27) = 8.67, P < 0.001], REM sleep latency [F(3,27) = 6.47, P < 0.01], and total sleep in the first 2 hours [F(3,27) = 5.18, P < 0.01]. In vehicle-treated rats, cage-exchange stress induced a significant delay in both latencies for NREM sleep (P < 0.05; Fig. 6A) and REM sleep (P < 0.05; Fig. 6B) associated with a decrease in total sleep duration (P < 0.05; Fig. 6C) as compared with the control condition. Administration of compound 56 did not affect any EEG sleep parameters relative to vehicle in control conditions, but significantly attenuated the increase in both NREM sleep latency (P < 0.05; Fig. 6A) and REM sleep latency (P < 0.05; Fig. 6B) induced by cage exchange. In contrast, the OX1R antagonist did not prevent the decrease in total sleep duration (Fig. 6C). Thus, compound 56 could attenuate the sleep-onset insomnia (i.e., prolongation of NREM and REM sleep latencies) elicited by cage exchange without impacting sleep duration.
Fig. 6.

Effects of the OX1R antagonist compound 56 on cage-exchange stress-induced hyperarousal in rats and ACTH release in mice. Vehicle or compound 56 was administered subcutaneously (10 mg/kg in rats and 30 mg/kg in mice) 30 minutes prior to cage exchange or control condition. NREM latency (A), REM latency (B), and total sleep (C) in rats for the first 2 hours are expressed in minutes and are represented as means ± S.E.M. of seven animals (four treatment conditions, crossover design). (D) ACTH release in mice is expressed in picograms per milliliter of serum and is represented as the mean ± S.E.M. of 11–13 animals per condition (four treatment conditions). Statistical significance (*P < 0.05, **P < 0.01, and ***P < 0.00) was based on one-way ANOVA followed by Newman-Keuls multiple comparison test.
To assess whether the cage-exchange procedure would activate the HPA axis, ACTH levels were evaluated as a biomarker for the stress response. We examined the effects of the selective OX1R antagonist to address the involvement of OX1R in modulating the potential ACTH response to this psychological stress. ACTH serum levels were measured 20 minutes after the cage exchange or control conditions in mice treated at 4 hours into the light phase with compound 56 (30 mg/kg s.c.) or vehicle 30 minutes before the procedure. The analysis of ACTH levels revealed a significant main effect (four treatment conditions in separate groups) [F(3,44) = 6.77, P < 0.001]. Following cage-exchange stress, vehicle-treated mice exhibited a 3.4-fold increase of ACTH levels as compared with the control conditions (P < 0.05; Fig. 6D), consistent with an activation of the HPA axis. Administration of compound 56, which had no effect by itself in control conditions, did not alter the cage exchange–induced ACTH release.
Compound 56 Attenuated NaLac-Induced Panic/Anxiety-Associated Behavioral and Physiologic Responses in Panic-Prone Rats.
In a final experiment, the effect of the OX1R antagonist compound 56 was investigated in a panic vulnerability to a well known interoceptive panicogenic challenge (i.e., NaLac) following chronic disinhibition of the PeF OX region.
Analysis of Autonomic and Locomotor-Associated Behavioral Responses.
An overall ANOVA with time as a repeated measure detected a drug treatment × time interaction for NaLac-induced changes in MAP [F(57,608) = 1.8, P < 0.001; Fig. 7A], HR [F(57,608) = 1.6, P = 0.005; Fig. 7B], CBT [F(57,608) = 1.8, P < 0.001; Fig. 7D], and general motor activity [F(57,608) = 2.1, P = 0.024; Fig. 7E]. A within-subjects analysis using a repeated-measures ANOVA plus Dunnett’s post-hoc test revealed that, compared with baseline, NaLac 1) increased MAP in the vehicle group [F(8,179) = 7.1, P < 0.001], 3 mg/kg group [F(8,179) = 5.3, P < 0.001], and 10 mg/kg group [F(8,179) = 4.7, P < 0.001], but not the 30 mg/kg group [F(8,179) = 1.6, P = 0.067]; 2) increased HR in the vehicle group [F(8,179) = 9.5, P < 0.001], 3 mg/kg group [F(8,179) = 4.6, P < 0.001], and 10 mg/kg group [F(8,179) = 5.6, P < 0.001], but not the 30 mg/kg group [F(8,179) = 1.6, P = 0.072]; 3) increased CBT in the vehicle group [F(8,179) = 2.2, P = 0.004], 10 mg/kg group [F(8,179) = 8.7, P < 0.001], and 30 mg/kg group [F(8,179) = 2.1, P = 0.008; Dunnett’s failed to reveal post-hoc significance], but not the 3 mg/kg group [F(8,179) = 0.5, P = 0.992]; and 4) increased general motor activity in the vehicle group [F(3,35) = 12.0, P < 0.001], 3 mg/kg group [F(3,35) = 3.7, P = 0.026], and 10 mg/kg group [F(3,35) = 4.5, P = 0.012], but not the 30 mg/kg group [F(3,35) = 2.2, P = 0.114]. Final n/group was 9/group.
Fig. 7.

The effects of systemic subcutaneous injections of 3-,10-, and 30-mg/kg doses of compound 56 on NaLac-induced panic-associated autonomic responses [MAP (A), HR (B), and CBT (D)] and behavioral responses [social interaction duration (C) and general motor activity (E)] in rats made panic-prone with chronic reductions of GABA synthesis in the PeF region (i.e., local l-AG infusions). Line graphs are expressed as average percentage ± S.E.M. (n = 9/group). +Between-subjects differences using a Fisher’s least significant difference post-hoc test that was protected with an ANOVA at each time point and significance with a one-way ANOVA with repeated measures was determined as P < 0.05. *Within-subjects differences using a Dunnett’s post-hoc test against baseline with a repeated measures P < 0.05. Bar graph in (C) is expressed as the average percentage ± S.E.M. (n = 9, 9, 8, 9, and 9); + and * indicate between-subjects differences using a Fisher’s least significant difference post-hoc test that was protected with an ANOVA P < 0.05. There was no significant baseline HR, MAP, CBT, or general motor activity between groups (see legends on figures). (F) Coronal brain section from a standard stereotaxic atlas of the rat brain (Paxinos and Watson, 2005) illustrating cannula placements (black circles) in the PeF region and medial nuclei. BPM, beats per minute; bsln, baseline; DA, dorsal hypothalamic area; DMN, dorsomedial hypothalamic nucleus; f, fornix; LH, lateral hypothalamus; mt, mammillothalamic tracts; veh, vehicle.
Analysis of Anxiety-Associated Behavioral Responses Following the NaLac Infusion
An ANOVA with a Fisher’s least significant difference post-hoc test revealed that rats treated with vehicle had a decreased social interaction duration following the NaLac challenge [F(4,39) = 6.0, P = 0.047; Fig. 7C], which was attenuated with the 30-mg/kg dose of compound 56, and approached significance with the 3-mg/kg dose (P = 0.076). Final n values/group were, respectively, 9, 9, 8, 9, and 9, with one video capture malfunction.
Histology revealed that all minipump cannulae placements resided in the PeF or just medial to the PeF in the dorsomedial hypothalamic nuclei or dorsal hypothalamic area, where acute disinhibition or chronic disinhibition plus NaLac produces anxiety-associated behaviors and robust cardioexcitatory responses (Shekhar and DiMicco, 1987; Samuels et al., 2004; Johnson et al., 2008) (Fig. 7F).
Discussion
In the present study, we characterized a novel selective OX1R antagonist that showed efficacy in two models of stress, a psychological stress elicited by cage exchange model and a NaLac provocation panic model in rats. We also demonstrated that the compound did not alter sleep-wake states in baseline conditions, and therefore was devoid of any hypnotic effect.
Over the last 15 years, most of the drug discovery efforts in the OX field have been directed toward developing nonselective OX receptor antagonists. At least four dual OX receptor antagonists have entered human trials for the treatment of sleep-related disorders (Hoyer and Jacobson, 2013). Recently, the U.S. Food and Drug Administration approved suvorexant (Belsomra; Merck, White House Station, NJ) for the treatment of insomnia (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm409950.htm). It is now well established that selective OX2R antagonists have robust hypnotic activity in preclinical species, and transient pharmacological inhibition of the two receptors, either by a dual OX1/2R antagonist or by simultaneous blockade of OX1R to OX2R antagonism, disrupts sleep architecture by shifting the balance in favor of REM sleep at the expense of NREM sleep (Dugovic et al., 2014). The role of the OX1R in emotional behavior is starting to emerge (Johnson et al., 2012). Most of these preclinical studies used the OX1R antagonist SB-334867 (Smart et al., 2001), which has been reported to be hydrolytically unstable, and therefore, possible confounding effects on in vivo and in vitro studies cannot be ruled out (McElhinny et al., 2012). We confirmed that SB-334867 has moderate in vitro affinity for the OX1R and a decent selectivity profile. Our data showed that SB-334867 occupied the OX1R for several hours after systemic administration, indicating that some of the effects of SB-334867 can be attributed to OX1R blockade. The related compound SB-408124 is more stable but has lower in vitro affinity/potency for the OX1R (Langmead et al., 2004). Despite poor brain penetration, sufficient exposure was achieved to sustain a significant level of OX1R occupancy. We also confirmed and extended the excellent selectivity profile of GSK-1059865 (Gozzi et al., 2011). Compound 56 had a similar in vitro profile to GSK-1059865 but slightly better brain penetration. Compound 56 was therefore selected for thorough in vivo characterization.
Previous studies have demonstrated that OX1R antagonists minimally affect sleep-wake states in basal conditions (Smith et al., 2003; Dugovic et al., 2009, 2014; Gozzi et al., 2011; Steiner et al., 2013a) but produce a disinhibition of REM sleep in the presence of OX2R antagonism (Dugovic et al., 2014). In rats, the coadministration of the OX2R antagonist JNJ-10397049 [N-(2,4-dibromophenyl)-N′-[(4S,5S)-2,2-dimethyl-4-phenyl-1,3-dioxan-5-yl]-urea] and the OX1R antagonist SB-408124 or GSK-1059865 promoted REM sleep, whereas REM sleep was not affected by either OX2R or OX1R blockade alone (Dugovic et al., 2009, 2014). Consistent with these data, we found that compound 56 was devoid of sleep-promoting effects under basal conditions in rats and WT mice. In line with our previous results on simultaneous pharmacological OX1R and OX2R blockade, compound 56 did promote REM sleep in OX2R KO mice, but not WT mice. These experiments showed that compound 56 fully engaged the OX1R and elicited specific OX1R blockade.
There is literature supporting the view that OX receptor antagonists do not alter basal anxiety levels in rodents but show efficacy when anxiety levels have been exacerbated (Steiner et al., 2012, 2013a). Negative data with OX receptor antagonists in classic anxiety models such as elevated plus-maze or social interaction (under baseline conditions) have been reported (Yeoh et al., 2014). Therefore, we tested compound 56 in two models of psychological exteroceptive (i.e., external threat) and interoceptive (i.e., internal body state threat) stress-induced hyperarousal. In future experiments, it would be interesting to test the fear potentiated startle paradigm, as another selective OX1R antagonist showed anxiolytic effects in this model (Steiner et al., 2013a). OXs exert a prominent role in the arousal-related process including stress by activating OX1R and OX2R. Stress and corticotrophin-releasing factor activate OX neurons, and OX administration stimulates hyperarousal, ACTH, and corticosterone release (for review, see Berridge et al., 2010). We first examined whether a selective OX1R antagonist could attenuate the arousal effects of a psychological stress elicited by cage exchange on EEG sleep in rats and on potential HPA axis activation in mice. Previous studies have shown that psychological stress induced by cage exchange produced an increase in body temperature, activity, arterial pressure, and heart rate in mice (Oka et al., 2003; Lee et al., 2004) and acute insomnia in rats (Cano et al., 2008). In accordance with these results, we found that rats exposed to this psychological stressor exhibited sleep disturbances including increased NREM and REM sleep latencies and decreased total sleep time for at least 2 hours. When the animals were pretreated with an OX1R antagonist before the cage-exchange stress, the effect on sleep latency, an index of the level of arousal, was attenuated, whereas the compound had little impact on sleep duration. The normalization of both NREM and REM sleep latencies by compound 56 administration was not the result of a soporific effect since the associated decrease in total sleep time was not altered between vehicle and compound-treated animals submitted to cage exchange. Together with our finding that compound 56 was inactive on spontaneous sleep, these data suggest that OX1R antagonism may inhibit the cage-exchange stress-induced phasic orexin activation without affecting the physiologic tonic excitability of orexin neurons regulating the basal sleep-wake cycle. In a fear-potentiated startle paradigm, another selective OX1R antagonist, ACT-335827 [(aR,1S)-1-[(3,4-dimethoxyphenyl)methyl]-3,4-dihydro-6,7-dimethoxy-N-(1-methylethyl)-a-phenyl-2(1H)-isoquinolineacetamide], has been shown to elicit anxiolytic-like effects without promoting sleep in rats (Steiner et al., 2013a). In addition to a number of alterations in the autonomic system previously reported following cage exchange in mice (Oka et al., 2003; Lee et al., 2004), our present data showed an increase in ACTH levels, revealing an activation of the HPA axis. In contrast to the hyperarousal response, OX1R blockade did not affect the cage-exchange stress-induced ACTH release in mice, suggesting that OX1Rs are not directly involved in HPA axis activation elicited by this psychological stress model. A putative OX2R-mediated effect might be rather expected since increase in ACTH levels by swimming stress was attenuated with prior central infusion of an OX2R antagonist (Chang et al., 2007), a result consistent with the predominance of OX2R in the PVN (Marcus et al., 2001). However, in a later study, administration of the dual OX1/2R antagonist almorexant in rats failed to alter the release of corticosterone in basal and various stress conditions (novelty, social stress, and restraint stress) or the release of ACTH upon pharmacological challenge with corticotrophin-releasing factor (Steiner et al., 2013b). It remains uncertain whether testing a selective OX2R antagonist in this particular model is relevant since the intrinsic sleep-promoting properties of the antagonist would be a confounding factor.
As a second model, we assessed the effect of the OX1R antagonist compound 56 in a panic vulnerability model involving chronic disinhibition of the PeF OX regions that has been used for over 15 years (see review in Johnson and Shekhar, 2012). Previously, Johnson et al. (2010) determined that chronically disinhibiting the PeF OX region was critical for stress (NaLac)-induced panic responses in a rat model of panic vulnerability that has robust postdictive, face, predictive, and construct validity. Here we showed that the highest dose of compound 56 (30 mg/kg) attenuated the anxiety-like behavior (reduced social interaction), locomotor, and cardioexcitatory responses induced by the NaLac challenge. The lowest dose tested (3 mg/kg) was also efficacious on locomotor and cardiovascular parameters but not on the behavioral readout. Surprisingly, the intermediate dose was not efficacious on any parameters except the suppression of NaLac-induced locomotor activity. Compound 56 had no significant sedative side effects at any of the doses tested as assessed by monitoring baseline locomotion or autonomic activity. Compound 56 was also tested in a rat model of CO2-induced panic (Johnson et al., manuscript in preparation). As observed in the NaLac model, compound 56 had no effect on baseline cardiovascular or locomotor activity, and a consistent attenuation/blockade of CO2-induced anxiety behavior in the social interaction test was observed at 30 mg/kg. An attenuation of the pressor and bradycardia response was also observed at 10 and 30 mg/kg with a more clear dose-response relationship. Thus, overall the efficacy of compound 56 in these two models of panic was consistent. In the cage-exchange paradigm and the NaLac-provoked panic model, efficacy was observed at doses that achieved maximal levels of OX1R occupancy. However, the lowest doses of compound 56 which were ineffective on the anxiety response in the NaLac model were also found to be near maximal level of receptor occupancy. These data indicate that the receptor occupancy assay does not predict the level of OX1R needed for efficacy in this behavioral model. The reason for this discrepancy is not entirely clear. OX1R occupancy levels were measured in the tenia tecta, a small region where high densities were detected. For practical reasons, we were not able to measure OX1R occupancy in the hypothalamus, bed nucleus of the stria terminalis, or amygdala, which are more closely related to the site of action for the antipanic response. Binding densities in these regions are relatively low, and therefore only a weak signal can be detected in these regions.
Although OX appears to play an important role in the aforementioned stress paradigms (e.g., exteroceptive cage exchange and interoceptive NaLac or CO2), not all stressors engage the OX system. Restraint and cold stress, for example, are not effective stimuli for OX activation, and OX1R and OX2R blockade does not alter the pressor and tachycardia responses to these stressors (Furlong et al., 2009). As active wake/hyperactivity is a prerequisite component to activate OX neurons, only nonstandard preclinical stress models could reveal the potential anxiolytic effects of selective OX1R antagonists. Clinical translations of these findings in the context of complex nervous system disorders remain to be established.
In conclusion, our data indicate that, under aversive conditions, hyperarousal and anxiety-associated behavioral and physiologic responses can be reduced by a selective OX1R antagonist that could represent a new therapeutic strategy without affecting sleep per se.
Acknowledgments
The authors thank Drs. Kevin Sharp and Tatiana Koudriakova and their staff at Janssen Research & Development LLC for assistance.
Abbreviations
- ACT-335827
(aR,1S)-1-[(3,4-dimethoxyphenyl)methyl]-3,4-dihydro-6,7-dimethoxy-N-(1-methylethyl)-a-phenyl-2(1H)-isoquinolineacetamide
- ACTH
adrenocorticotropic hormone
- 1-AG
1-allyglycine
- ANOVA
analysis of variance
- CBT
core body temperature
- compound 56
N-({3-[(3-ethoxy-6-methylpyridin-2-yl)carbonyl]-3-azabicyclo[4.1.0]hept-4-yl}methyl)-5-(trifluoromethyl)pyrimidin-2-amine
- EEG
electroencephalogram
- EMPA
N-ethyl-2-[(6-methoxy-pyridin-3-yl)-(toluene-2-sulfonyl)-amino]-N-pyridin-3-ylmethyl-acetamide
- GSK-1059865
5-bromo-N-({1-[(3-fluoro-2-methoxyphenyl)carbonyl]-5-methylpiperidin-2-yl}methyl)pyridin-2-amine
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HEK-293
human embryonic kidney 293
- HPA
hypothalamic-pituitary-adrenal
- HR
heart rate
- JNJ-10397049
N-(2,4-dibromophenyl)-N′-[(4S,5S)-2,2-dimethyl-4-phenyl-1,3-dioxan-5-yl]-urea
- KO
knockout
- MAP
mean arterial blood pressure
- NaLac
sodium lactate
- NREM
non-REM
- OX
orexin
- OX1R
orexin-1 receptor
- OX2R
orexin-2 receptor
- PeF
perifornical hypothalamus
- PVN
paraventricular hypothalamic nucleus
- REM
rapid eye movement
- SB-334867
1-(2-methyl-1,3-benzoxazol-6-yl)-3-(1,5-naphthyridin-4-yl)urea
- SB-408124
1-(6,8-difluoro-2-methylquinolin-4-yl)-3-[4-(dimethylamino)phenyl]urea
- SB-674042
1-(5-(2-fluoro-phenyl)-2-methyl-thiazol-4-yl)-1-((S)-2-(5-phenyl-(1,3,4)oxadiazol-2-ylmethyl)-pyrrolidin-l-yl)-methanone
- SI
social interaction
- WT
wild-type
Authorship Contributions
Participated in research design: Bonaventure, Johnson, Shekhar, Lovenberg, Dugovic.
Conducted experiments: Yun, Fitz, Nepomuceno, Lord, Wennerholm, Shelton.
Contributed new reagents or analytic tools: Shireman, Lebold, Carruthers.
Performed data analysis: Bonaventure, Johnson, Nepomuceno, Lord, Shelton, Dugovic.
Wrote or contributed to the writing of the manuscript: Bonaventure, Johnson, Dugovic.
Footnotes
This work was supported by Janssen Research and Development, LLC; the National Institutes of Health National Institute on Aging [Grant 1K01-AG044466-01A1]; National Institutes of Health National Center for Research Resources [Grant UL1-RR025761] (to P.L.J.); and the National Institutes of Health National Institute of Mental Health [Grant R01-MH52619 and R01-MH65702] (to A.S.).
References
- Abshire VM, Hankins KD, Roehr KE, DiMicco JA. (1988) Injection of L-allylglycine into the posterior hypothalamus in rats causes decreases in local GABA which correlate with increases in heart rate. Neuropharmacology 27:1171–1177. [DOI] [PubMed] [Google Scholar]
- Alvaro G, Amantini D, Castiglioni E, Di Fabio R and Pavone F (2010) inventors, Glaxo Group Limited UK, assignee. N-{[(1S,4S,6S)-3-(2-Pyridinylcarbonyl)-3-azabicyclo[4.1.0]hept-4-yl]methyl}-2-heteroarylamine derivatives as orexin receptor antagonists and their preparation and use for the treatment of diseases. WO2010063662A1
- Berridge CW, España RA, Vittoz NM. (2010) Hypocretin/orexin in arousal and stress. Brain Res 1314:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Mochizuki T, Saper CB. (2008) Neural circuitry of stress-induced insomnia in rats. J Neurosci 28:10167–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Saito T, Ohiwa N, Tateoka M, Deocaris CC, Fujikawa T, Soya H. (2007) Inhibitory effects of an orexin-2 receptor antagonist on orexin A- and stress-induced ACTH responses in conscious rats. Neurosci Res 57:462–466. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, et al. (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95:322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugovic C, Shelton JE, Aluisio LE, Fraser IC, Jiang X, Sutton SW, Bonaventure P, Yun S, Li X, Lord B, et al. (2009) Blockade of orexin-1 receptors attenuates orexin-2 receptor antagonism-induced sleep promotion in the rat. J Pharmacol Exp Ther 330:142–151. [DOI] [PubMed] [Google Scholar]
- Dugovic C, Shelton JE, Yun S, Bonaventure P, Shireman BT, Lovenberg TW. (2014) Orexin-1 receptor blockade dysregulates REM sleep in the presence of orexin-2 receptor antagonism. Front Neurosci 8:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong TM, Vianna DM, Liu L, Carrive P. (2009) Hypocretin/orexin contributes to the expression of some but not all forms of stress and arousal. Eur J Neurosci 30:1603–1614. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Turrini G, Piccoli L, Massagrande M, Amantini D, Antolini M, Martinelli P, Cesari N, Montanari D, Tessari M, et al. (2011) Functional magnetic resonance imaging reveals different neural substrates for the effects of orexin-1 and orexin-2 receptor antagonists. PLoS ONE 6:e16406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer D, Jacobson LH. (2013) Orexin in sleep, addiction and more: is the perfect insomnia drug at hand? Neuropeptides 47:477–488. [DOI] [PubMed] [Google Scholar]
- Johnson PL, Samuels BC, Fitz SD, Federici LM, Hammes N, Early MC, Truitt W, Lowry CA, Shekhar A. (2012) Orexin 1 receptors are a novel target to modulate panic responses and the panic brain network. Physiol Behav 107:733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PL, Shekhar A. (2012) An animal model of panic vulnerability with chronic disinhibition of the dorsomedial/perifornical hypothalamus. Physiol Behav 107:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PL, Truitt WA, Fitz SD, Lowry CA, Shekhar A. (2008) Neural pathways underlying lactate-induced panic. Neuropsychopharmacology 33:2093–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PL, Truitt W, Fitz SD, Minick PE, Dietrich A, Sanghani S, Träskman-Bendz L, Goddard AW, Brundin L, Shekhar A. (2010) A key role for orexin in panic anxiety. Nat Med 16:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Jerman JC, Brough SJ, Scott C, Porter RA, Herdon HJ. (2004) Characterisation of the binding of [3H]-SB-674042, a novel nonpeptide antagonist, to the human orexin-1 receptor. Br J Pharmacol 141:340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DL, Webb RC, Brands MW. (2004) Sympathetic and angiotensin-dependent hypertension during cage-switch stress in mice. Am J Physiol Regul Integr Comp Physiol 287:R1394–R1398. [DOI] [PubMed] [Google Scholar]
- Li J, Hu Z, de Lecea L. (2014) The hypocretins/orexins: integrators of multiple physiological functions. Br J Pharmacol 171:332–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malherbe P, Borroni E, Gobbi L, Knust H, Nettekoven M, Pinard E, Roche O, Rogers-Evans M, Wettstein JG, Moreau JL. (2009) Biochemical and behavioural characterization of EMPA, a novel high-affinity, selective antagonist for the OX(2) receptor. Br J Pharmacol 156:1326–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mang GM, Dürst T, Bürki H, Imobersteg S, Abramowski D, Schuepbach E, Hoyer D, Fendt M, Gee CE. (2012) The dual orexin receptor antagonist almorexant induces sleep and decreases orexin-induced locomotion by blocking orexin 2 receptors. Sleep 35:1625–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. (2001) Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol 435:6–25. [DOI] [PubMed] [Google Scholar]
- Maton W, Stazi F, Manzo A, Pachera R, Ribecai A, Stabile P, Perboni A, Giubellina N, Bravo F, Castoldi D, et al. (2010) An efficient scalable route for the synthesis of enantiomerically pure tert-Butyl-(1R,4S,6R)-4-(hydroxymethyl)-3-azabicyclo[4.1.0]heptane-3-carboxylate. Org Process Res Dev 14:1239–1247. [Google Scholar]
- McElhinny CJ, Jr, Lewin AH, Mascarella SW, Runyon S, Brieaddy L, Carroll FI. (2012) Hydrolytic instability of the important orexin 1 receptor antagonist SB-334867: possible confounding effects on in vivo and in vitro studies. Bioorg Med Chem Lett 22:6661–6664. [DOI] [PubMed] [Google Scholar]
- Oka T, Oka K, Kobayashi T, Sugimoto Y, Ichikawa A, Ushikubi F, Narumiya S, Saper CB. (2003) Characteristics of thermoregulatory and febrile responses in mice deficient in prostaglandin EP1 and EP3 receptors. J Physiol 551:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. (2005) The Rat Brain in Stereotaxic Coordinates, Elsevier Academic Press, San Diego, CA. [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. (1998) Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18:9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richarson JA, Kozlowski GP, Wilson S, et al. (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585. [DOI] [PubMed] [Google Scholar]
- Samuels BC, Zaretsky DV, DiMicco JA. (2004) Dorsomedial hypothalamic sites where disinhibition evokes tachycardia correlate with location of raphe-projecting neurons. Am J Physiol Regul Integr Comp Physiol 287:R472–R478. [DOI] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A. (1995) Regulation of anxiety by GABAA receptors in the rat amygdala. Pharmacol Biochem Behav 52:701–706. [DOI] [PubMed] [Google Scholar]
- Sharf R, Sarhan M, Dileone RJ. (2010) Role of orexin/hypocretin in dependence and addiction. Brain Res 1314:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A, DiMicco JA. (1987) Defense reaction elicited by injection of GABA antagonists and synthesis inhibitors into the posterior hypothalamus in rats. Neuropharmacology 26:407–417. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Johnson PL, Sajdyk TJ, Fitz SD, Keim SR, Kelley PE, Gehlert DR, DiMicco JA. (2006) Angiotensin-II is a putative neurotransmitter in lactate-induced panic-like responses in rats with disruption of GABAergic inhibition in the dorsomedial hypothalamus. J Neurosci 26:9205–9215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A, Katner JS. (1995) Dorsomedial hypothalamic GABA regulates anxiety in the social interaction test. Pharmacol Biochem Behav 50:253–258. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Keim SR. (1997) The circumventricular organs form a potential neural pathway for lactate sensitivity: implications for panic disorder. J Neurosci 17:9726–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar A, Keim SR, Simon JR, McBride WJ. (1996) Dorsomedial hypothalamic GABA dysfunction produces physiological arousal following sodium lactate infusions. Pharmacol Biochem Behav 55:249–256. [DOI] [PubMed] [Google Scholar]
- Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA, Jerman JC. (2001) SB-334867-A: the first selective orexin-1 receptor antagonist. Br J Pharmacol 132:1179–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MI, Piper DC, Duxon MS, Upton N. (2003) Evidence implicating a role for orexin-1 receptor modulation of paradoxical sleep in the rat. Neurosci Lett 341:256–258. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Gatfield J, Brisbare-Roch C, Dietrich H, Treiber A, Jenck F, Boss C. (2013a) Discovery and characterization of ACT-335827, an orally available, brain penetrant orexin receptor type 1 selective antagonist. ChemMedChem 8:898–903. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Lecourt H, Jenck F. (2012) The brain orexin system and almorexant in fear-conditioned startle reactions in the rat. Psychopharmacology (Berl) 223:465–475. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Sciarretta C, Brisbare-Roch C, Strasser DS, Studer R, Jenck F. (2013b) Examining the role of endogenous orexins in hypothalamus-pituitary-adrenal axis endocrine function using transient dual orexin receptor antagonism in the rat. Psychoneuroendocrinology 38:560–571. [DOI] [PubMed] [Google Scholar]
- Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LHT, Guan X-M. (1998) Distribution of orexin receptor mRNA in the rat brain. FEBS Lett 438:71–75. [DOI] [PubMed] [Google Scholar]
- Verdelet T, Mercey G, Correa N, Jean L, Renard P-Y. (2011) Straightforward and efficient synthesis of 3-benzyloxy-4-bromopicolinate ester and 3-benzyloxy-5-bromopicolinate ester, common building blocks for pharmaceuticals and agrochemicals. Tetrahedron 67:8757–8762. [Google Scholar]
- Yeoh JW, Campbell EJ, James MH, Graham BA, Dayas CV. (2014) Orexin antagonists for neuropsychiatric disease: progress and potential pitfalls. Front Neurosci 8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]