

Abstract
Sepiapterin reductase (SPR) catalyzes the reduction of sepiapterin to dihydrobiopterin (BH2), the precursor for tetrahydrobiopterin (BH4), a cofactor critical for nitric oxide biosynthesis and alkylglycerol and aromatic amino acid metabolism. SPR also mediates chemical redox cycling, catalyzing one-electron reduction of redox-active chemicals, including quinones and bipyridinium herbicides (e.g., menadione, 9,10-phenanthrenequinone, and diquat); rapid reaction of the reduced radicals with molecular oxygen generates reactive oxygen species (ROS). Using recombinant human SPR, sulfonamide- and sulfonylurea-based sulfa drugs were found to be potent noncompetitive inhibitors of both sepiapterin reduction and redox cycling. The most potent inhibitors of sepiapterin reduction (IC50s = 31–180 nM) were sulfasalazine, sulfathiazole, sulfapyridine, sulfamethoxazole, and chlorpropamide. Higher concentrations of the sulfa drugs (IC50s = 0.37–19.4 μM) were required to inhibit redox cycling, presumably because of distinct mechanisms of sepiapterin reduction and redox cycling. In PC12 cells, which generate catecholamine and monoamine neurotransmitters via BH4-dependent amino acid hydroxylases, sulfa drugs inhibited both BH2/BH4 biosynthesis and redox cycling mediated by SPR. Inhibition of BH2/BH4 resulted in decreased production of dopamine and dopamine metabolites, 3,4-dihydroxyphenylacetic acid and homovanillic acid, and 5-hydroxytryptamine. Sulfathiazole (200 μM) markedly suppressed neurotransmitter production, an effect reversed by BH4. These data suggest that SPR and BH4-dependent enzymes, are “off-targets” of sulfa drugs, which may underlie their untoward effects. The ability of the sulfa drugs to inhibit redox cycling may ameliorate ROS-mediated toxicity generated by redox active drugs and chemicals, contributing to their anti-inflammatory activity.
Introduction
Using NADPH as a source of reducing equivalents, sepiapterin reductase (SPR) catalyzes the reduction of sepiapterin to dihydrobiopterin (BH2), a precursor for tetrahydrobiopterin (BH4), an important cofactor in nitric oxide biosynthesis and alkylglycerol and aromatic amino acid metabolism (Thony et al., 2000; Crabtree and Channon, 2011; Werner et al., 2011; Watschinger and Werner, 2013) (see Fig. 1 showing reactions of sepiapterin reductase). SPR is widely distributed in tissues, including lung, liver, kidney, and brain (Kato, 1971). Several mutations in the enzyme have been identified in humans, and these mutations compromise the enzyme’s ability to generate the biopterin cofactor and are associated with neurologic deficits, including delayed psychomotor development, movement disorders, and early onset Parkinson’s disease (Bonafe et al., 2001; Farrugia et al., 2007; Verbeek et al., 2008). Similarly, in mice, knockout of SPR results in reduced levels of neurotransmitters and movement disorders (Yang et al., 2006; Homma et al., 2011). Deficiencies in BH4 in humans are also associated with dystonia, atypical phenylketonuria, and Parkinson’s and Alzheimer’s diseases (Curtius et al., 1984; Tani et al., 1994; Blau et al., 2011). Loss of BH4 is known to reduce nitric oxide production, compromising nitric oxide–dependent signaling, which can be deleterious to many organs, including the cardiovascular system, leading to alterations in cardiac function, blood pressure, and inflammation (Li and Forstermann, 2013; Bendall et al., 2014).
Fig. 1.

Enzymatic reactions catalyzed by SPR. Reduction of sepiapterin generates 7,8-dihydrobiopterin, which is converted to (6R)-5,6,7,8-tetrahydrobiopterin by additional cellular reductases. The one-electron reduction of redox cycling chemicals, such as menadione, generates radical cations. The rapid reaction of these cations with molecular oxygen regenerates the redox active chemical, and in the process superoxide anion (O2.–) is formed. Sepiapterin reduction is catalyzed by NADPH, whereas chemical redox cycling is catalyzed by NADPH and/or NADH.
In recent studies, we demonstrated that, in addition to reducing sepiapterin and generating BH2, SPR is highly efficient in mediating chemical redox cycling (Yang et al., 2013). In this reaction, SPR catalyzes the pyridine nucleotide cofactor–dependent one-electron reduction of both endogenous and exogenous redox-active chemicals, including quinones and bipyridyl herbicides, a process that generates radical ions. Reacting rapidly with molecular oxygen, these radicals generate superoxide anion and regenerate the parent compounds (Dicker and Cederbaum, 1991; Rashba-Step and Cederbaum, 1994; Bonneh-Barkay et al., 2005). Dismutation of superoxide anion leads to the formation of hydrogen peroxide (H2O2) and, in the presence of trace metals, to highly toxic hydroxyl radicals (Winterbourn, 1995). Preferential utilization of NADPH during redox cycling is at the expense of their supply to sepiapterin, and this effectively blocks the formation of BH2 and BH4 (Yang et al., 2013). In tissues sensitive to chemical redox cyclers, including the lung and brain, this could lead to aberrant physiologic functioning and contribute to disease pathology (Yang et al., 2013).
A number of inhibitors of sepiapterin reduction and chemical redox cycling by SPR have been identified; these include compounds structurally similar to its natural substrates, such as N-acetylserotonin, N-acetyldopamine, and N-acetyl-m-tyramine, as well as the structurally unrelated active drug product and aldo-keto reductase/cyclooxygenase inhibitor indomethacin (Smith et al., 1992; Yang et al., 2013). By inhibiting SPR in vivo, these compounds can compromise the accumulation of BH2/BH4 in tissues and alter aromatic amino acid metabolism, a process required for the formation of catecholamines and indoleamines, and nitric oxide synthases, which are required for nitric oxide biosynthesis. In the case of a nonsteroidal anti-inflammatory agent such as indomethacin, this may contribute to its therapeutic activity; alternatively, this may represent an “off-target” action of the drug, potentially resulting in toxicity. Recently, using a yeast-hybrid system, an anti-inflammatory sulfonamide, sulfasalazine, was identified as an inhibitor of SPR (Chidley et al., 2011). Subsequently, sulfasalazine metabolites, as well as structurally related sulfonamide- and sulfonylurea-based antimicrobial/anti-inflammatory and antidiabetic drugs, were shown to be inhibitors of SPR (Chidley et al., 2011; Haruki et al., 2013). Inhibition of SPR by these drugs could result in reduced levels of BH2 and BH4, causing inhibition of enzymes that require the biopterin cofactor and leading to toxicity. The present studies were aimed at assessing whether sulfa drugs also inhibit chemical redox cycling by SPR. Our findings that sulfa drugs are effective inhibitors of redox cycling suggest an additional off target action of these drugs that may contribute to unexpected side effects.
Materials and Methods
Chemicals and Reagents.
BH2, BH4, and biopterin were from Santa Cruz Biotechnology (Dallas, TX), and Amplex Red reagent was from Invitrogen (Carlsbad, CA). Human recombinant cytochrome P450 reductase was from Life Technologies (Grand Island, NY). Sepiapterin, horseradish peroxidase, NADPH, sulfasalazine, sulfamethoxazole, sulfathiazole, sulfapyridine, tolbutamide, glibenclamide, chlorpropamide, 9,10-phenanthrenequinone, menadione, protease inhibitor mixture 1, and all other chemicals and enzymes were from Sigma-Aldrich (St. Louis, MO), unless otherwise indicated. Protease inhibitor mixture 1 contained 4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatin A, E-64 [trans-epoxysuccinyl-l-leucylamido-(4-guanidino) butane], bestatin, leupeptin, and aprotinin.
Cell Culture.
PC12 cells were obtained from the American Type Culture Collection (Manassas, VA) and grown in Dulbecco’s modified Eagle’s medium supplemented with penicillin (100 IU/ml), streptomycin (100 μg/ml), 5% fetal bovine serum, and 5% horse serum. Cells were maintained at 37°C with 5% CO2 in a humidified incubator.
Assays for Sepiapterin Reductase Activity and Redox Cycling.
Purified recombinant human SPR was prepared and assayed as described elsewhere (Yang et al., 2013). Activity for sepiapterin reduction of the enzyme was measured spectrophotometrically in standard 0.2-ml reaction mixes containing 100 mM potassium phosphate buffer, pH 6.4, 100 μM NADPH, 50 μM sepiapterin, and 1 μg SPR. The reactions were started by the addition of SPR; decreases in absorbance of sepiapterin at 420 nm were monitored at 37°C using a SpectraMax M3 Fluorescence/Absorbance Microplate Reader (Molecular Devices, Sunnyvale, CA). Redox cycling by SPR was quantified by measuring the H2O2, superoxide anion, or hydroxyl radicals in reaction mixes as previously described (Yang et al., 2013). The Amplex-Red/horseradish peroxidase method was used to measure H2O2 (Yang et al., 2013). Briefly, standard 0.1-ml reaction mixes contained 50 mM phosphate buffer, pH 7.8, 25 μM Amplex-Red, 0.1 IU horseradish peroxidase, 0.2 mM NADPH, 0.1–2 μg of SPR, and appropriate concentrations of redox cycling chemicals. Reactions were initiated by addition of the enzyme, and the formation of fluorescent product, resorufin, was quantified by fluorescence using the fluorescence microplate reader with excitation and emission wavelengths set at 530 and 587 nm, respectively. H2O2 formed in the reactions was calculated from a calibration curve prepared using appropriate standards.
The generation of hydroxyl radicals was quantified in Fenton-dependent reactions by monitoring the formation of 2-hydroxyterephthalate from terephthalate (Mishin and Thomas, 2004). In these assays, 4.5 μg of recombinant SPR/100 μl of reaction mix were used. Superoxide anion was measured spectrophotometrically by the reduction of acetylated cytochrome c at 550 nm as described previously (Yang et al., 2013). Reaction mixes contained 100 mM potassium phosphate buffer, pH 7.8, 0.05 mM acetylated cytochrome c, 200 μM NADPH, 200 μM menadione, and 20 μg of SPR in a final volume of 1 ml. SPR activity was also measured by monitoring decreases in absorbance of NADPH or NADH at 340 nm (Yang et al., 2013). Oxygen utilization in enzyme reactions during redox cycling was measured using a traditional Clark-type oxygen electrode (Hansatech Instruments, Norfolk, UK) as described elsewhere (Yang et al., 2013). In these assays, 16 μg of recombinant SPR/0.8 ml of reaction mix were used. Kinetic parameters of the enzyme were calculated from linear portions of reaction curves using Dixon plots.
Assays for Sepiapterin Reductase in PC12 Cells.
SPR activity in PC12 cells was assayed by monitoring the formation of BH2 and BH4 from sepiapterin as previously described (Ferre and Naylor, 1988) with modifications (Yang et al., 2013). Briefly, cells were plated (2 × 105 cells/well) in six-well culture dishes and grown until 80% confluent (∼1 × 106 cells/well). The growth medium was then removed and the cells treated with 100 μM sepiapterin in growth medium. After 2 hours, cells were removed from the plates by scraping in growth medium and centrifuged at 1500g for 5 minutes. Cell pellets were then suspended in 200 μl of lysis buffer (50 mM Tris-HCl, pH 7.4, containing 0.5% NP40 and 0.5 mM EDTA), centrifuged (20,000g for 20 minutes), and 12.5 μl of 1 M NaOH and 12.5 μl of a 1% iodine/2% KI solution added to 100 μl of supernatant. After 1 hour, samples were acidified with 25 μl of 1 M H3PO4. Excess iodine was reduced by addition of 6.3 μl of freshly prepared ascorbic acid (20 mg/ml). After standing for 10 minutes at room temperature, the precipitated proteins were removed by centrifugation at 20,000g (10 minutes). BH2 and BH4 formed in the assay were analyzed isocratically on a Jasco high-performance liquid chromatography system (Easton, MD) fitted with a Maxsil-10 C18 column (250 × 4 mm; Phenomenex, Torrance, CA) using a mobile phase consisting of 5% methanol in water at a flow rate of 1.0 ml/min. Fluorescence was monitored using a Jasco FP-2020 spectrofluorometer with excitation and emission wavelengths set at 362 and 435 nm, respectively. In inhibitor experiments, cells were treated with the sulfa drugs for 2 hours prior to the addition of sepiapterin.
Assays for Redox Cycling in PC12 Cells.
Redox cycling in S9 fractions of PC12 cells was assayed as previously described (Yang et al., 2013). Briefly, PC12 cells were plated (2 × 106 cells) in 10-cm culture dishes and grown until 80% confluent (8 × 106 cells). The cells were removed from the dishes by scraping in growth medium and sedimented at 1500g for 5 minutes. Cell pellets were resuspended in phosphate-buffered saline containing protease inhibitor mixture 1, and sonicated on ice with three 15-second bursts and 1 minute of cooling on ice between each sonication burst. Disrupted cells were centrifuged (9000g, 10 minutes) to remove cellular debris, nucleus, and mitochondria, and the supernatants were assayed immediately for H2O2 generation as described above for recombinant enzyme.
Assay for Neurotransmitter Production by PC12 Cells.
PC12 cells grown in 10-cm culture dishes were treated in the absence or presence of the sulfa drugs and/or BH4 in 10 ml of growth medium. After 48 hours, cells were removed from the plates as described above, washed with phosphate-buffered saline, and then centrifuged at 1500g. Cell pellets, containing approximately 8 × 106 cells, were resuspended in 1 ml of 0.1 N perchloric acid containing 200 μM EDTA and 350 μM sodium metabisulfite and then sonicated on ice. Cell extracts were centrifuged (20,000g, 10 minutes) to remove protein precipitates, and supernatants were filtered using 0.22-μm filters and analyzed for neurotransmitters and their metabolites using a Waters 2695 high-performance liquid chromatography system (Waters Corp., Milford, MA) fitted with a cation exchange column (MD-150 3.2 × 15 mm column; ESA Biosciences, Inc., Chelmsford, MA) using an isocratic mobile phase (MD-TM mobile phase; ESA Biosciences Inc.) containing 2.2 mM NaCl pumped at constant flow rate of 0.5 ml/min. The compounds were quantified by electrochemical detection using a glassy carbon working electrode (2 mm in diameter) with an in situ silver reference electrode (flow cell, 2 mm GC WE, ISAAC; Waters Corp.) (Yochum et al., 2014).
Molecular Modeling and Docking.
The 3D model of mouse and human SPR were generated using PyMOL v1.7.2.2 (The PyMOL Molecular Graphics System; Schrödinger LLC, Cambridge, MA). The crystal structures of SPR used (PDB IDs 1SEP, 1NAS, 4HWK, 4J7U, and 4J7X) and the structure of menadione (PDB ID 2QR2) were obtained from the Protein Data Bank. For docking modeling, ligands and water were removed from the protein structure (4J7X) using Discovery Studio v4.0 (Accelrys Inc., San Diego, CA). The addition of polar hydrogen to the modified protein and setting of rotatable bonds on menadione were performed using AutoDock Tools v1.5.6 (The Scripps Research Institute, La Jolla, CA) and then converted to PDBQT format for docking screening. The grid size was set to 44 × 34 × 12 xyz points with grid spacing of 1 Å and grid center was designated at dimensions (x, y, and z): –28.903, –48.162 and 22.510. Exhaustiveness was set to 8 with the remaining parameters set on default as described previously (Trott and Olson, 2010). Molecular docking was performed using AutoDock Vina v1.1.2 (The Scripps Research Institute). The best conformation with the lowest docked energy was extracted and visualized using PyMol software.
Results
Effects of Sulfa Drugs on Sepiapterin Reduction and Redox Cycling by Human Sepiapterin Reductase.
In initial studies, we examined the effects of the sulfonamide sulfasalazine on sepiapterin reduction by recombinant human SPR (see Table 1 for the structure of sulfasalazine and related sulfa drugs used in this study). Sulfasalazine was found to cause a concentration-dependent inhibition of the formation of BH2 from sepiapterin (Figs. 2A and 3A). Three additional amide derivatives of the sulfa drugs tested were also found to inhibit sepiapterin reduction (Fig. 3A). Sulfasalazine was the most potent (IC50 = 31 nM) followed by sulfathiazole (IC50 = 62 nM), sulfapyridine (IC50 = 141 nM), and sulfamethoxazole (IC50 = 180 nM) (Fig. 3A; Table 2). Three antidiabetic sulfonylureas, chlorpropamide, glibenclamide, and tolbutamide, also caused a concentration-dependent inhibition of sepiapterin reduction, with IC50s of 120, 340, and 510 nM, respectively (Fig. 3B; Table 2). All of the sulfa drugs were noncompetitive inhibitors of the enzyme with respect to sepiapterin (inset to Fig. 2A; Table 2). The relative IC50s for inhibition of sepiapterin reduction were generally similar to those reported previously (Haruki et al., 2013), although in our studies 2- to 9-fold higher concentrations of sulfa drugs were needed to inhibit enzyme activity. This is probably attributable to different enzyme preparations, including the use of different expression vectors to produce SPR.
TABLE 1.
Sulfa drugs tested as inhibitors of sepiapterin reduction and redox cycling of SPR
| Drug Name | Core Structure | Structure |
|---|---|---|
| Sulfasalazine | Sulfonamide | 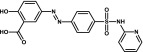 |
| Sulfathiazole | Sulfonamide | 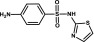 |
| Sulfapyridine | Sulfonamide | 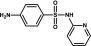 |
| Sulfamethoxazole | Sulfonamide | 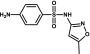 |
| Chlorpropamide | Sulfonylurea | 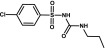 |
| Glibenclamide | Sulfonylurea | 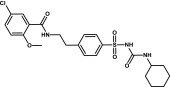 |
| Tolbutamide | Sulfonylurea | 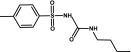 |
Fig. 2.

Effects of sulfasalazine on human recombinant SPR activity. (A) Inhibition of sepiapterin reduction by sulfasalazine. Sepiapterin reduction was measured by decreases in sepiapterin absorbance at 420 nm. Inset: Dixon plot showing noncompetitive type of inhibition of sepiapterin reduction by sulfasalazine. Reactions were run with increasing concentrations of sulfasalazine in the presence of 25 μM (a), 50 μM (b), or 75 μM (c) sepiapterin. (B) Inhibition of redox cycling of SPR by sulfasalazine. Redox cycling was measured in the presence of 200 μM menadione. In the absence of sulfasalazine, menadione redox cycling induced 23.6 nmol of H2O2/min per milligram of protein. In the absence of menadione, the rate of formation of H2O2 was 0.3 nmol/min per milligram of protein. Inset: Dixon plot showing noncompetitive type of inhibition of redox cycling by sulfasalazine inhibition. Reactions were run with increasing concentrations of sulfasalazine in the presence of 100 μM (a), 200 μM (b), or 300 μM (c) menadione. (C) Effects of 100 μM sulfasalazine on superoxide anion production by SPR. Menadione (MD; 200 μM) was used to induce superoxide anion production; 50 IU/ml superoxide dismutase (SOD) inhibited accumulation of superoxide anion. Superoxide anion was measured spectrophotometrically by monitoring changes in absorbance of acetylated cytochrome c at 550 nm. Reactions were run in the absence and presence of SPR. (D) Effects of 100 μM sulfasalazine on H2O2 production by SPR. Menadione was used to stimulate H2O2 production; catalase (1000 IU/ml) inhibited accumulation of H2O2. H2O2 was measured using the Amplex Red assay. (E) Effects of 100 μM sulfasalazine on the formation of hydroxyl radicals by SPR. Menadione (200 μM) was used to stimulate hydroxyl radical production. Dimethylsulfoxide (1%) was used as a control to trap hydroxyl radicals. Hydroxyl radical production was measured using the terephthalate assay; the product of the reaction, 2-hydroxyterephthalate (2-OH TPT), was used to monitor the formation of hydroxyl radicals. Reactions were run for 10 minutes before analysis.
Fig. 3.

Effects of sulfa drugs on sepiapterin reduction and redox cycling by human recombinant SPR. Effects of sulfonamides (A) and sulfonylureas (B) on sepiapterin reduction by SPR. Effects of sulfonamides (C) and sulfonylureas (D) on chemical redox cycling by SPR. SPR activity in enzyme assays was measured by decreases in sepiapterin absorbance at 420 nm. Redox cycling was measured by the formation of H2O2 in enzyme assays in the presence of 200 μM menadione. Data are presented as percentage of activity of control enzyme reactions run in the absence of inhibitors (100% = 3.2 μmol/min per milligram of protein.
TABLE 2.
Ability of sulfa drugs to inhibit sepiapterin reduction and redox cycling by SPR
| Inhibitor | Sepiapterin Reductiona | Redox Cycling | ||
|---|---|---|---|---|
| Inhibition Type | IC50 | Inhibition Type | IC50 | |
| nM | μM | |||
| Antibacterial | ||||
| Sulfasalazine | Noncompetitive | 31 | Noncompetitive | 0.53 |
| Sulfathiazole | Noncompetitive | 62 | Noncompetitive | 8.02 |
| Sulfapyridine | Noncompetitive | 141 | Noncompetitive | 19.41 |
| Sulfamethoxazole | Noncompetitive | 180 | Noncompetitive | 10.97 |
| Antidiabetic | ||||
| Chlorpropamide | Noncompetitive | 120 | Noncompetitive | 0.37 |
| Glibenclamide | Noncompetitive | 340 | Noncompetitive | 1.11 |
| Tolbutamide | Noncompetitive | 510 | Noncompetitive | 4.58 |
Sepiapterin reduction and redox cycling were measured as described in Materials and Methods. For redox cycling assays, 200 μM menadione was used.
In earlier studies we showed that SPR mediates redox cycling of a variety of quinones, including menadione (2-methylnaphthalene-1,4-dione) and 9,10-phenanthrenequinone (Yang et al., 2013). The redox cycling process with menadione and 9,10-phenanthrenequinone generates superoxide anion, which dismutates into H2O2, and in the presence of transition metals, hydroxyl radicals are formed (Fig. 2, C–E, and not shown). Accumulation of superoxide anion and H2O2 are inhibited by superoxide dismutase and catalase, respectively (Fig. 2, C and D), and dimethylsulfoxide, a trap for hydroxyl radicals, inhibited the formation of hydroxyl radicals (Fig. 2E). Sulfasalazine was found to readily block the formation of superoxide anion, H2O2, and hydroxyl radicals (Figs. 2, C–E, and 4). Redox cycling by SPR, an oxygen-dependent reaction, is NADPH- and, to a lesser extent, NADH-dependent; sulfasalazine inhibited both NAD(P)H utilization and oxygen consumption (Fig. 5 and not shown). Similarly, sulfasalazine inhibited NADPH utilization during sepiapterin reduction (Fig. 5E). The effects of sulfasalazine on redox cycling were found to be concentration-dependent (IC50 = 0.53 μM) (Figs. 2B and 3C) as measured by H2O2 formation. Additional sulfonamides, including sulfathiazole (IC50 = 8.02 μM), sulfamethoxazole (IC50 = 10.97 μM), and sulfapyridine (IC50 = 19.41 μM), also inhibited redox cycling (Fig. 3C; Table 2). Sulfonylureas also effectively inhibited redox cycling by SPR (Fig. 3D and not shown); chlorpropamide was the most active (IC50 = 0.37 μM) followed by glibenclamide (IC50 = 1.11 μM) and tolbutamide (IC50 = 4.58 μM) (Fig. 3D; Table 2). The sulfa drugs were also found to be noncompetitive inhibitors of menadione-mediated chemical redox cycling by SPR (Fig. 2B, inset; Table 2).
Fig. 4.

Effects of sulfa drugs on enzymes known to mediate chemical redox cycling. Human sepiapterin reductase (hSPR; 60 nM), human cytochrome P450 reductase (hOR; 1 nM), rat cytosolic TrxR (rTrxR1; 30 nM), or human glutathione reductase (hGR; 70 nM) was incubated in 50 mM potassium phosphate buffer (pH 7.8) with menadione (MD; 200 μM) in the absence or presence of sulfasalazine (SSZ; 100 μM) or sulfamethoxazole (SMX; 100 μM). The reactions were initiated by the addition of NADPH (200 μM) and H2O2 production was measured by the Amplex Red assay. Data are presented as percentage of activity of control enzyme reactions run in the absence of inhibitors. Data are the mean ± S.E. (n = 3). *Significantly different (P < 0.05) from menadione-treated samples in the absence of inhibitors.
Fig. 5.

Effects of sulfa drugs on oxygen consumption and NADPH utilization by SPR during chemical redox cycling. A Clark-type oxygen electrode was used to detect oxygen consumption during redox cycling. Reactions, run in the presence of NADPH (200 μM) and 9,10-phenanthrenequinone (10 μM), were initiated by the addition of 16 μg/ml SPR at the indicated times in 0.8 ml of 50 mM phosphate buffer, pH 7.8. Sulfasalazine (20 μM) or chlorpropamide (50 μM) were added to the assays as indicated. Sodium dithionite was added to deplete oxygen and served as a zero oxygen control. (A and B) Effects sulfasalazine on oxygen utilization during chemical redox cycling. (C and D) Effects of chlorpropamide on oxygen utilization during chemical redox cycling. Note that the addition of the sulfa drugs either after (A and C) or before (B and D) the start of the redox cycling reactions inhibited oxygen consumption. 9,10-Phenanthrenequinone (10 μM) was used as the redox cycling chemical. (E and F) Effects of sulfa drugs on NADPH utilization by SPR during sepiapterin reduction and chemical redox cycling. Standard reaction mixes contained 200 μM NADPH and 20 μM sulfasalazine during sepiapterin reduction by SPR. Menadione (200 μM) was used as the redox cycling chemical. Control reactions contain NADPH, but no SPR. Inset: inhibition of NADH utilization by sulfasalazine during menadione-supported redox cycling by SPR. Control reactions contained NADH, but no SPR.
The effects of the sulfa drugs on chemical redox cycling were specific for SPR. As shown in Fig. 4, whereas sulfasalazine and sulfamethoxazole readily inhibited menadione-mediated redox cycling by SPR, they had no significant effects on other enzymes able to mediate redox cycling, including NADPH cytochrome P450 reductase, thioredoxin reductase, or glutathione reductase (Grover and Piette, 1981; Jan et al., 2014; unpublished data).
Effects of Sulfa Drugs on Sepiapterin Reduction and Redox Cycling by PC12 Cells.
We next determined if the sulfa drugs could inhibit SPR in intact cells and cell lysates. For these studies we used PC12 cells, a neuroectodermal-derived rat cell line known to require BH4 for the activity of aromatic amino acid hydroxylases needed to generate catecholamines and monoamine neurotransmitters (Greene and Tischler, 1976; Kittner et al., 1987). Very low or undetectable levels of BH2 and BH4 were noted in intact PC12 cells and cell lysates, respectively (Fig. 6, C and D). Treatment of the cells with 100 μM sepiapterin resulted in a marked increase in levels of BH2 and BH4 in intact cells and lysates (Fig. 6, C and D); greater levels of BH4 were detected in intact cells. Sulfathiazole (100 μM) inhibited sepiapterin-induced BH2 and BH4 production (Fig. 6, C and D). BH4 is a cofactor required for aromatic amino acid hydroxylases that mediate catecholamine and monoamine neurotransmitter biosynthesis (Carlsson and Lindqvist, 1978; Hufton et al., 1995; Werner et al., 2011). We next determined if inhibition of BH4 production by sulfa drugs inhibited catecholamine and monoamine neurotransmitter production in PC12 cells. Figure 7 shows that PC12 cells readily generate dopamine and the dopamine metabolites, 3,4-dihydroxyphenylacetic acid and homovanillic acid, as well as 5-hydroxytryptamine. Treatment of the cells with sulfathiazole (200 μM) suppressed neurotransmitter production (Fig. 7); this was completely restored when PC12 cells were treated with BH4 (Fig. 7).
Fig. 6.

Effects of sulfa drugs on chemical redox cycling and the formation of BH2 and BH4 in PC12 cells. (A) Effects of sulfasalazine on menadione-generated H2O2 in cell lysates of PC12 cells. (B) Effects of different sulfa drugs on menadione-generated H2O2 in lysates of PC12 cells. H2O2 formation during chemical redox cycling was assayed using 100 μg/ml of a cell lysate prepared from PC12 cells. Assays contained 100 μM NADPH and 200 μM menadione. Control assays were run in the absence of cell lysates and sulfa drugs. (C) Effects of sulfathiazole on the formation of BH2 and BH4 in PC12 cell lysates. Lysates were incubated for 2 hours in the presence [upper and middle panels; high-performance liquid chromatography (HPLC) tracings] and absence (lower HPLC tracing) of 100 μM sepiapterin plus 200 μM NADPH. The middle HPLC tracing was from cell lysates that also contained 100 μM sulfathiazole. (D) Effects of sulfathiazole on BH2 and BH4 formation in intact PC12 cells. Cells were treated for 2 hours in the presence (upper and middle HPLC tracings) and absence (lower HPLC tracing) of 100 μM sepiapterin. In the middle HPLC tracing, cells were treated with 100 μM sulfathiazole 2 hours before the addition of sepiapterin and when sepiapterin was present. BH2 and BH4 formation were measured using HPLC with fluorescence detection.
Fig. 7.

Effects of sulfathiazole on the production of catecholamine and monoamine neurotransmitters by PC12 cells. Cells (8 × 106/10-cm dish) were pretreated with control medium [upper high-performance liquid chromatography (HPLC) tracing], medium containing 200 μM sulfathiazole (center HPLC tracing), or medium containing sulfathiazole plus 100 μM BH4 (lower HPLC tracing). After 8 hours, catecholamine/monoamine neurotransmitters or metabolites (5-HT, 5-hydroxytryptamine; DA, dopamine; DOPAC, 3,4-dihydroxyphenylacetic acid; HVA, homovanillic acid) were extracted from the cells and analyzed by HPLC with electrochemical detection as described in Materials and Methods.
We also found that menadione stimulates production of reactive oxygen species (ROS) in S9 fractions from PC12 cells. (Fig. 6A and not shown). In this preparation, each of the sulfa drugs was found to cause a concentration-dependent inhibition of menadione redox cycling (Fig. 6, A and B). Redox cycling was most sensitive to inhibition by sulfasalazine (IC50 = 29 μM) followed by sulfathiazole (IC50 = 1.4 mM), sulfamethoxazole (IC50 = 2.0 mM), and sulfapyridine (IC50 = 2.2 mM).
Discussion
Previously, we demonstrated that, in addition to reducing sepiapterin, SPR mediates chemical redox cycling (Yang et al., 2013). Moreover, these activities are mediated by distinct mechanisms, on the basis of our findings that: 1) The substrate sepiapterin had no inhibitory effect on redox cycling, whereas redox cycling quinones (e.g., menadione and 9,10-phenanthrenequinone) inhibited sepiapterin reduction; 2) inhibitors of sepiapterin reduction, including dicoumarol, N-acetylserotonin, indomethacin, ethacrynic acid, and rutin, had no effects on redox cycling; 3) nonredox cycling quinones inhibited sepiapterin reduction and redox cycling by distinct mechanisms; and 4) site-directed mutagenesis of the C-terminal substrate-binding site (D257H) of SPR completely inhibited sepiapterin reduction but had minimal effects on redox cycling. The present studies demonstrate that sulfa drugs are also effective inhibitors of the two enzymatic reactions of SPR. However, unlike other inhibitors, sulfa drugs are noncompetitive inhibitors of both sepiapterin reduction and redox cycling. Importantly, much lower concentrations of sulfa drugs are needed to inhibit sepiapterin reduction when compared with redox cycling. Thus, the IC50s for inhibition of sepiapterin reduction and redox cycling for the sulfonamides is 31–180 and 530–10,970 nM, respectively, and for the sulfonylureas, 120–510 and 370–4458 nM, respectively. These data suggest that, despite similarities in the type of inhibition by sulfa drugs, they probably bind to different sites in the enzyme that lead to differences in electron transfer for sepiapterin reduction and redox cycling.
Crystallographic studies with SPR complexed with sulfasalazine, sulfapyridine, or sulfathiazole and other inhibitors have provided insight into the mechanism underlying inhibition of sepiapterin reduction and redox cycling (Auerbach et al., 1997; Haruki et al., 2013). A highly conserved sequence motif in the catalytic site of the human enzyme is highlighted by a critical triad pocket composed of Ser157, Tyr170, and Lys174 (Ser158, Tyr171 and Lys175 in the mouse enzyme), which is important in stabilizing protein structure, maintaining substrate/cofactor proximity, and proton transfer (Auerbach et al., 1997). Sulfa drugs, which form a ternary complex with the enzyme and NADP+, appear to anchor by forming hydrogen bonds with Tyr170 and Ser157 in the triad pocket (Haruki et al., 2013). Tyr170 forms a hydrogen bond with the aromatic nitrogen in the R-group pyridine or thiazole ring in the sulfonamides, whereas Ser157 forms hydrogen bonds with nitrogen and/or oxygen in the sulfonamide groups (see Figs. 8 and 9 for structures of sulfa drugs bound to the active site in SPR). The pyridine or thiazole rings in the sulfa drugs also stack in close proximity to nicotinamide in NADP+, resulting in hydrophobic and/or π-electron interactions. Thus, sulfa drugs have the capacity to block binding of sepiapterin, and potentially its access to the active site in the enzyme. In the case of the redox cycling chemicals, proximity of the sulfa drugs to NADP+ may block binding and interactions required for one-electron reduction reactions. It should be noted that in contrast to the sulfa drugs, N-acetylserotonin, a competitive inhibitor of sepiapterin, binds in a different orientation in the active site triad. By anchoring its 5-hydroxyl substituent to Asp258 and modifying the substrate binding site, N-acetylserotonin can block binding of sepiapterin via its pyrimidine center (see Fig. 8). The fact that N-acetylserotonin binding is not in proximity to NADP+ may explain the fact that it does not interfere with chemical redox cycling reactions (Auerbach et al., 1997; Yang et al., 2013).
Fig. 8.

Molecular models of human SPR complexed with sulfa drugs. (A) Superposition of human and mouse SPR. Crystal structure of human SPR is shown in cyan (PDB ID 4J7X) and crystal structures of mouse SPR are shown in yellow (PDB ID 1SEP) and pink (PDB ID 1NAS), respectively. NADP+ and sulfasalazine complexed with human SPR are shown in green and blue, respectively. Sepiapterin and N-acetylserotonin bound with mouse SPR are shown in yellow and magenta, respectively. Superimposed structures of the human and mouse SPR complexes reveal high similarities in the active site and cofactor binding regions; it also allows us to compare the binding site of the sulfa drugs with sepiapterin and N-acetylserotonin. (B) Close-up view of sepiapterin and inhibitors in SPR complexes. The stereo view of the SPR complexes shows that sepiapterin, sulfasalazine, and N-acetyl serotonin interact at distinct sites relative to the cofactor. Both sepiapterin and sulfasalazine bind in proximity to the nicotinamide group of NADP+, whereas N-acetyl serotonin binds in a region distal to the cofactor. The distances between the nicotinamide of NADP and pyridine ring of sulfasalazine are around 3.2–4.0 Å. This may contribute to the fact that the sulfa drugs, but not N-acetyl serotonin, inhibit chemical redox cycling. (C) Binding interactions of sulfasalazine with key amino acid residues in the SPR crystal structure. The nitrogen atom in the pyridine ring of sulfasalazine forms hydrogen bonds with catalytic residues Ser157 and Tyr170. The pyridine ring of sulfasalazine also forms hydrophobic and/or π-stacking interactions with residues Leu104, Leu158, Cys159, Trp167, Tyr170, Met205, and Ala209 (not shown). Hydrogen bonds with Ser157 and Tyr170 are shown by broken lines, and the corresponding distances (Å) are indicated. (D) Superposition of sulfa drugs in the active site of human SPR. The binding modes of sulfasalazine, sulfapyridine (PDB ID 4HWK), and sulfathiazole (PDB ID 4J7U) to human SPR show that the positioning of the sulfa drugs are highly similar to the heteroaromatic ring stacked on top of the nicotinamide ring of NADP+. (E) The docking model of menadione bound to human SPR (PDB ID 4J7X). Docking was performed using AutoDock Vina and the docking results were analyzed using PyMOL. The docked menadione (shown in orange) locates in close proximity to the nicotinamide group of NADP+ cofactor (∼2.4–4.0 Å) forming π-stacking interactions and develops hydrogen bond interactions (labeled by broken lines) with catalytic residues Tyr170 and/or Ser157. (F) Superimposition of the docked menadione with sulfasalazine in the active site of human SPR. Note the overlap of the quinone ring of menadione and the pyridine ring of sulfasalazine. This overlaps with the C1′ side chain of the pterin moiety in sepiapterin, which interacts with Tyr170 and NADP+.
Fig. 9.

Interaction of the sulfa drugs with active site residues and NADP+ in human SPR. The ternary complexes of human SPR with NADP+ (nicotinamide moiety) and sulfasalazine (A), sulfapyridine (B), or sulfathiazole (C) are derived from their reported crystal structures in the Protein Data Bank. The sulfa drugs are anchored by hydrogen bonding to catalytic residues tyrosine 170 and serine 157 and hydrophobic and/or π-electron interactions of the pyridine or thiazole rings in the sulfa drugs with the nicotinamide group of the NADP+ cofactor. The distances between the nicotinamide of NADP and pyridine or thiazole ring of sulfa drugs are around 3.2–4.0 Å. Hydrogen bonds are shown by broken lines, and the corresponding distances (Å) are indicated.
The structure of menadione bound to SPR has not been reported. To better understand the mechanism of inhibition of menadione redox cycling by the sulfa drugs, we performed molecular docking screening of menadione to the SPR active site (Fig. 8, E and F). Menadione was found to fit in the hydrophobic cleft in the active site of SPR forming hydrophobic interactions with Leu104, Leu158, Cys159, Trp167, Tyr170, Met205, and Ala209 (Auerbach et al., 1997; Haruki et al., 2013). Menadione is in close proximity (∼2.4–4 Å) to the nicotinamide group of the NADP+ cofactor forming π-stacking interactions and forms hydrogen bonds with catalytic residues Tyr170 and/or Ser157. This is superimposed on the binding site for sulfasalazine as well as the carbonyl side chain of the pterin moiety in sepiapterin. This provides a mechanism by which the sulfa drug inhibits menadione redox cycling. The close proximity of menadione with NADP+ is likely important in electron transfer during redox cycling. Thus, during redox cycling, one electron is transferred from the four position in the pyridine ring to an oxygen atom on the quinone, forming a semiquinone radical. Addition of new NADPH or its regeneration during the redox cycling process allows for the continuous formation of the semiquinone and subsequent formation of ROS. This mechanism is distinct from redox cycling mediated by flavin-containing enzymes, such as NADPH cytochrome P450 reductase, in which a one-electron reduction of the redox active quinone is mediated by an electron transfer from flavin adenine dinucleotide and flavin mononucleotide (Murataliev et al., 1997).
Differences in the chemical properties of the core structure of the sulfonamides and sulfonylureas and their interaction with the active site in SPR probably contributes to differences in their activity as inhibitors of sepiapterin reduction. In general, sulfonylureas are more acidic than sulfonamides. This is owing to the –N–H group on sulfonylureas, which is most ionizable, and the additional acidification effect of being adjacent to the urea carbonyl. The pKa of these drugs can also vary depending on R-group substituents (Newton and Kluza, 1978). Although one might expect that a stronger acid would bind more tightly to the hydroxyl group on Ser157, if the serine becomes a hydrogen bond acceptor, i.e., the N–H of the sulfa drug is the donor and is bonded to the lone pair of electrons on the serine OH, the greater acidity of the sulfonylureas would tend to antagonize sulfa drug binding. This is because the sulfonylureas will be substantially ionized at physiologic pH with a negative charge on the nitrogen and virtually no –N–H in the medium.
BH4 is a critical cofactor for aromatic amino acid hydroxylases, including phenylalanine hydroxylase, tyrosine hydroxylase, and tryptophan hydroxylase; it is thus essential for catecholamine neurotransmitter biosynthesis (Carlsson and Lindqvist, 1978; Hufton et al., 1995; Werner et al., 2011). Using PC12 cells, a catecholamine neurotransmitter producing cell line, we demonstrated that BH2 and BH4 were generated in intact cells and S9 fractions of cells treated with sepiapterin. The formation of BH4 from BH2 requires additional enzymes, such as dihydropteridine reductase (Werner et al., 2011). Thus, limited formation of BH4 in S9 fractions when compared with intact cells is probably attributable to the fact that conditions for the enzymatic conversion of BH2 to BH4 were not optimized in our assays. In sulfathiazole-treated PC12 cells, the formation of BH2 and BH4 was essentially eliminated. This resulted in inhibition of cellular production of catecholamine, catecholamine metabolite, and monoamine neurotransmitter. The fact that BH4 was able to reverse the inhibitory effects of sulfathiazole on catecholamine/monoamine production in PC12 cells indicated that the sulfa drug was targeting SPR. Our data are consistent with earlier studies showing that various sulfa drugs are effective inhibitors of BH4 biosynthesis in cytokine-stimulated primary human dermal fibroblasts, PC12 cells, and human neuroblastoma cells SK-N-BE(2), as well as the formation of l-3,4-dihydroxyphenylalanine and 4-methoxytyramine in SK-N-BE(2) cells (Chidley et al., 2011; Haruki et al., 2013).
Sulfa drugs are indicated for a variety of medical conditions, including microbial infections, inflammation, and diabetes. Earlier studies suggested that by interfering with BH2/BH4 biosynthesis, and in turn, enzymes that require biopterin as a cofactor, sulfa drugs could cause “off-target” actions with resulting deleterious side effects (Haruki et al., 2013). This is supported by findings that average steady-state serum or blood concentrations of sulfa drugs at therapeutic doses exceed their IC50s for inhibition of sepiapterin reduction by SPR (Table 2; Haruki et al., 2013), a condition that could result in localized tissue injury or systemic toxicity. Relatively high concentrations of sulfa drugs in the cerebral spinal fluid could also result in off-target actions of these drugs in the brain (Haruki et al., 2013). On the basis of our data and those of Haruki et al. (2013), these off-target actions of the sulfa drugs, which include central nervous system– and gastrointestinal-related side effects, may be caused by inhibition of BH4-dependent aromatic amino acid hydroxylases important in catecholamine production. However, it should be noted that additional enzymes also require BH4 as cofactor, including the three isoforms of nitric oxide synthase responsible for nitric oxide production, and alkylglycerol monooxygenase, an enzyme that degrades ether lipids (Tejero and Stuehr, 2013; Watschinger and Werner, 2013) Nitric oxide is known to mediate many physiologic processes, including inflammation, vascular tone, and neurotransmission, and its production has been shown to be suppressed by inhibitors of BH2/BH4 biosynthesis (Gross and Levi, 1992; Sakai et al., 1993). Less is known about the role of BH2/BH4 in regulation of alkylglycerol monooxygenase and the metabolism of ether lipids. Ether lipid derivatives, notably platelet-activating factor, control many functions, including inflammation and anaphylaxis, and these processes are also likely to be modulated as a consequence of altering BH2/BH4 formation via inhibition of SPR (Watschinger and Werner, 2013). Thus, a variety of mechanisms, in addition to limiting catecholamine production, may be responsible for the off-target actions of sulfa drugs.
The present studies show that sulfa drugs were also effective inhibitors of chemical redox cycling by recombinant SPR, as measured by inhibition of quinone-mediated production of superoxide anion, hydrogen peroxide, and hydroxyl radicals. This appeared to be a result of inhibition of NAD(P)H and oxygen utilization, which are required for redox cycling. Of note, higher concentrations of sulfa drugs were required to inhibit redox cycling, when compared with sepiapterin reduction. This is probably attributable to the fact that there are different sites in SPR that mediate sepiapterin reduction and redox cycling; sulfa drugs appear to be significantly less efficient in affecting the site-mediating redox cycling, which may be related to how well the sulfa drugs block access to NAD(P)H and/or the active site of the enzyme. Sulfa drugs also inhibited redox cycling in PC12 cells; sulfasalazine was the most active, followed by sulfathiazole, sulfapyridine, and sulfamethoxazole. We used S9 cell fractions to measure the effects of sulfa drugs on redox cycling and this and/or the fact that it is a rat cell line may explain differences in activity when compared with purified human recombinant SPR. Alternatively, additional enzymes in the cell lysates that are not inhibited by the sulfa drugs may mediate chemical redox cycling. This is supported by our findings that the sulfa drugs do not inhibit chemical redox cycling by NADPH cytochrome P450 reductase, thioredoxin reductase, or glutathione reductase. Further studies are needed to better understand mechanisms underlying differences in the activities of sulfa drugs in PC12 cells.
A question arises as to the functional significance of sulfa drug–induced inhibition of chemical redox cycling by SPR. Although higher concentrations of the sulfa drugs are required to inhibit chemical redox cycling by the enzyme, when compared with sepiapterin reduction, for almost all the sulfa drugs, concentrations in the blood and serum are significantly higher than the IC50 for inhibiting redox cycling (Chidley et al., 2011; Haruki et al., 2013). SPR mediates redox cycling of both endogenous and exogenous redox active compounds. It is generally thought that redox cycling is deleterious as it generates highly toxic ROS, which promote cell damage and cause inflammation. ROS can also modulate the activity of the immune system (Klotz et al., 2014). By inhibiting redox cycling, sulfa drugs may protect against ROS, and this may account, at least in part, for their anti-inflammatory and/or immunomodulatory activity.
In summary, the present studies demonstrate that sulfonamides and sulfonylureas are effective inhibitors of the two enzymatic reactions of SPR: reduction of sepiapterin and chemical redox cycling. Inhibition of chemical redox cycling was specific for SPR, as other enzymes known to mediate this process, including NADPH cytochrome P450 reductase, thioredoxin reductase and glutathione reductase, were not inhibited by sulfa drugs. Inhibition of SPR suppresses BH2/BH4 formation and in turn, amino acid hydroxylases important in catecholamine and monoamine neurotransmitter biosynthesis; these could be important off-targets of sulfa drugs. Sulfa drugs also have the potential to inhibit other enzymes that require BH4 as a cofactor, including nitric oxide synthases and alkylglycerol monooxygenase. If inhibition of SPR is an underlying mechanism causing adverse reactions to the sulfa drugs, then it might be expected that administration of BH2/BH4 could overcome toxicity and this remains to be evaluated. However, as indicated above, inhibition of redox cycling by the sulfa drugs may protect against ROS-mediated toxicity. This will depend on the concentrations of the sulfa drugs; at relatively low concentrations, inhibition of BH2/BH4 production prevails. In contrast, at relatively high concentrations, both BH2/BH4 production and ROS formation are inhibited. Further studies are needed to determine which mechanism is likely to explain the toxicity versus the therapeutic activity of the sulfa drugs.
Abbreviations
- BH2
dihydrobiopterin
- BH4
tetrahydrobiopterin
- E-64
trans-epoxysuccinyl-l-leucylamido-(4-guanidino) butane
- ROS
reactive oxygen species
- SPR
sepiapterin reductase
Authorship Contributions
Participated in research design: Yang, Jan, Mishin, Richardson, Heindel, Heck, D. L. Laskin, J. D. Laskin.
Conducted experiments: Yang, Jan, Mishin, Hossain.
Contributed new reagents or analytic tools: Richardson, Mishin.
Performed data analysis: Yang, Jan, D. L. Laskin, J. D. Laskin, Heindel.
Wrote or contributed to the writing of the manuscript: Yang, Jan, Mishin, Richardson, Heindel, Heck, D. L. Laskin, J. D. Laskin.
Footnotes
This work was supported by the National Institutes of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases [Grant U54-AR055073]; the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant U01-NS079249]; the National Institutes of Health National Cancer Institute [Grant R01-CA132624]; and the National Institutes of Health National Institute of Environmental Health Sciences [Grants R01-ES004738, R01-ES021800, and P30-ES005022].
References
- Auerbach G, Herrmann A, Gütlich M, Fischer M, Jacob U, Bacher A, Huber R. (1997) The 1.25 A crystal structure of sepiapterin reductase reveals its binding mode to pterins and brain neurotransmitters. EMBO J 16:7219–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. (2014) Tetrahydrobiopterin in cardiovascular health and disease. Antioxid Redox Signal 20:3040–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U. (2011) Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab 104 (Suppl):S2–S9. [DOI] [PubMed] [Google Scholar]
- Bonafé L, Thöny B, Penzien JM, Czarnecki B, Blau N. (2001) Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet 69:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneh-Barkay D, Reaney SH, Langston WJ, Di Monte DA. (2005) Redox cycling of the herbicide paraquat in microglial cultures. Brain Res Mol Brain Res 134:52–56. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindqvist M. (1978) Dependence of 5-HT and catecholamine synthesis on concentrations of precursor amino-acids in rat brain. Naunyn Schmiedebergs Arch Pharmacol 303:157–164. [DOI] [PubMed] [Google Scholar]
- Chidley C, Haruki H, Pedersen MG, Muller E, Johnsson K. (2011) A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat Chem Biol 7:375–383. [DOI] [PubMed] [Google Scholar]
- Crabtree MJ, Channon KM. (2011) Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 25:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtius HC, Niederwieser A, Levine R, Müldner H. (1984) Therapeutic efficacy of tetrahydrobiopterin in Parkinson’s disease. Adv Neurol 40:463–466. [PubMed] [Google Scholar]
- Dicker E, Cederbaum AI. (1991) NADH-dependent generation of reactive oxygen species by microsomes in the presence of iron and redox cycling agents. Biochem Pharmacol 42:529–535. [DOI] [PubMed] [Google Scholar]
- Farrugia R, Scerri CA, Montalto SA, Parascandolo R, Neville BG, Felice AE. (2007) Molecular genetics of tetrahydrobiopterin (BH4) deficiency in the Maltese population. Mol Genet Metab 90:277–283. [DOI] [PubMed] [Google Scholar]
- Ferre J, Naylor EW. (1988) Sepiapterin reductase in human amniotic and skin fibroblasts, chorionic villi, and various blood fractions. Clin Chim Acta 174:271–282. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73:2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SS, Levi R. (1992) Tetrahydrobiopterin synthesis. An absolute requirement for cytokine-induced nitric oxide generation by vascular smooth muscle. J Biol Chem 267:25722–25729. [PubMed] [Google Scholar]
- Grover TA, Piette LH. (1981) Influence of flavin addition and removal on the formation of superoxide by NADPH-Cytochrome P-450 reductase: a spin-trap study. Arch Biochem Biophys 212:105–114. [DOI] [PubMed] [Google Scholar]
- Haruki H, Pedersen MG, Gorska KI, Pojer F, Johnsson K. (2013) Tetrahydrobiopterin biosynthesis as an off-target of sulfa drugs. Science 340:987–991. [DOI] [PubMed] [Google Scholar]
- Homma D, Sumi-Ichinose C, Tokuoka H, Ikemoto K, Nomura T, Kondo K, Katoh S, Ichinose H. (2011) Partial biopterin deficiency disturbs postnatal development of the dopaminergic system in the brain. J Biol Chem 286:1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufton SE, Jennings IG, Cotton RG. (1995) Structure and function of the aromatic amino acid hydroxylases. Biochem J 311:353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YH, Heck DE, Malaviya R, Casillas RP, Laskin DL, Laskin JD. (2014) Cross-linking of thioredoxin reductase by the sulfur mustard analogue mechlorethamine (methylbis(2-chloroethyl)amine) in human lung epithelial cells and rat lung: selective inhibition of disulfide reduction but not redox cycling. Chem Res Toxicol 27:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S. (1971) Sepiapterin reductase from horse liver: purification and properties of the enzyme. Arch Biochem Biophys 146:202–214. [DOI] [PubMed] [Google Scholar]
- Kittner B, Bräutigam M, Herken H. (1987) PC12 cells: a model system for studying drug effects on dopamine synthesis and release. Arch Int Pharmacodyn Ther 286:181–194. [PubMed] [Google Scholar]
- Klotz LO, Hou X, Jacob C. (2014) 1,4-naphthoquinones: from oxidative damage to cellular and inter-cellular signaling. Molecules 19:14902–14918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Förstermann U. (2013) Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr Opin Pharmacol 13:161–167. [DOI] [PubMed] [Google Scholar]
- Mishin VM, Thomas PE. (2004) Characterization of hydroxyl radical formation by microsomal enzymes using a water-soluble trap, terephthalate. Biochem Pharmacol 68:747–752. [DOI] [PubMed] [Google Scholar]
- Murataliev MB, Klein M, Fulco A, Feyereisen R. (1997) Functional interactions in cytochrome P450BM3: flavin semiquinone intermediates, role of NADP(H), and mechanism of electron transfer by the flavoprotein domain. Biochemistry 36:8401–8412. [DOI] [PubMed] [Google Scholar]
- Newton DW, Kluza RB. (1978) pKa values of medical compounds in pharmacy practice. Drug Intell Clin Pharm 12:547–554. [Google Scholar]
- Rashba-Step J, Cederbaum AI. (1994) Generation of reactive oxygen intermediates by human liver microsomes in the presence of NADPH or NADH. Mol Pharmacol 45:150–157. [PubMed] [Google Scholar]
- Sakai M, Kitamura H, Nagata N. (1993) Lung function in patients with collagen vascular diseases: a study of non-smokers with normal chest roentgenograms. Intern Med 32:365–369. [DOI] [PubMed] [Google Scholar]
- Smith GK, Duch DS, Edelstein MP, Bigham EC. (1992) New inhibitors of sepiapterin reductase. Lack of an effect of intracellular tetrahydrobiopterin depletion upon in vitro proliferation of two human cell lines. J Biol Chem 267:5599–5607. [PubMed] [Google Scholar]
- Tani Y, Fernell E, Watanabe Y, Kanai T, Långström B. (1994) Decrease in 6R-5,6,7,8-tetrahydrobiopterin content in cerebrospinal fluid of autistic patients. Neurosci Lett 181:169–172. [DOI] [PubMed] [Google Scholar]
- Tejero J, Stuehr D. (2013) Tetrahydrobiopterin in nitric oxide synthase. IUBMB Life 65:358–365. [DOI] [PubMed] [Google Scholar]
- Thöny B, Auerbach G, Blau N. (2000) Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J 347:1–16. [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek MM, Blom AM, Wevers RA, Lagerwerf AJ, van de Geer J, Willemsen MA. (2008) Technical and biochemical factors affecting cerebrospinal fluid 5-MTHF, biopterin and neopterin concentrations. Mol Genet Metab 95:127–132. [DOI] [PubMed] [Google Scholar]
- Watschinger K, Werner ER. (2013) Alkylglycerol monooxygenase. IUBMB Life 65:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ER, Blau N, Thöny B. (2011) Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem J 438:397–414. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC. (1995) Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82-83:969–974. [DOI] [PubMed] [Google Scholar]
- Yang S, Lee YJ, Kim JM, Park S, Peris J, Laipis P, Park YS, Chung JH, Oh SP. (2006) A murine model for human sepiapterin-reductase deficiency. Am J Hum Genet 78:575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Jan YH, Gray JP, Mishin V, Heck DE, Laskin DL, Laskin JD. (2013) Sepiapterin reductase mediates chemical redox cycling in lung epithelial cells. J Biol Chem 288:19221–19237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yochum C, Doherty-Lyon S, Hoffman C, Hossain MM, Zelikoff JT, Richardson JR. (2014) Prenatal cigarette smoke exposure causes hyperactivity and aggressive behavior: role of altered catecholamines and BDNF. Exp Neurol 254:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]