

Abstract
G protein–coupled receptors (GPCRs) often activate multiple signaling pathways, and ligands may evoke functional responses through individual pathways. These unique responses provide opportunities for biased or functionally selective ligands to preferentially modulate one signaling pathway over another. Studies with several GPCRs have suggested that selective activation of signaling pathways downstream of a GPCR may lead to safer and more effective drug therapies. The dopamine D2 receptor (D2R) is one of the main drug targets in the therapies for Parkinson’s disease and schizophrenia. Recent studies suggest that selective modulation of individual signaling pathways downstream of the D2R may lead to safer antipsychotic drugs. In the present study, immediate effectors of the D2R (i.e., Gαi/o, Gβγ, β-arrestin recruitment) and more complex signaling pathways (i.e., extracellular signal-regulated kinase phosphorylation, heterologous sensitization, and dynamic mass redistribution) were examined in response to a series of D2R ligands. This was accomplished using Chinese hamster ovary cells stably expressing the human D2L dopamine receptor in the PathHunter β-Arrestin GPCR Assay Platform. The use of a uniform cellular background was designed to eliminate potential confounds associated with cell-to-cell variability, including expression levels of receptor as well as other components of signal transduction, including G protein subunits. Several well characterized and clinically relevant D2R ligands were evaluated across each signaling pathway in this cellular model. The most commonly used methods to measure ligand bias were compared. Functional selectivity analyses were also used as tools to explore the relative contribution of immediate D2R effectors for the activation of more complex signaling pathways.
Introduction
G protein–coupled receptors (GPCRs) are coupled to multiple signaling pathways (Lefkowitz and Shenoy, 2005), and different ligands can show unique profiles for modulation of these individual pathways. Moreover, some ligands demonstrate functional selectivity (or biased agonism), behaving as agonists for one signaling pathway while acting as antagonists for another (Urban et al., 2007; Kenakin and Christopoulos, 2013b). Functional selectivity has been proposed as a strategy to improve the safety and specificity of drug therapies targeting GPCRs (Whalen et al., 2011). For example, several studies suggest that G protein–biased ligands, which selectively activate G proteins over β-arrestin for the μ-opioid receptor, can lead to enhanced analgesic effects and decreased tolerance (Bohn et al., 1999, 2000, 2004). It has also been suggested that G protein–biased ligands for the β2-adrenergic receptor may lead to reduced receptor tachyphylaxis in bronchodilation therapies for obstructive lung diseases (Deshpande et al., 2008; Wang et al., 2009). Additionally, β-arrestin–biased ligands for the β1-adrenergic receptor may provide the beneficial effects of β-blockers along with increased cell survival, a desired outcome in patients with arrhythmia and hypertension after myocardial infarctions (DeWire and Violin, 2011).
The dopamine D2 receptor (D2R) is the primary target in therapies for treating schizophrenia and Parkinson’s disease, but modulation of D2R activity is also associated with a number of side effects including dysregulation of motor and pituitary function. D2Rs couple to Gαi/o subunits, leading to several signaling events through the release/rearrangement of G proteins, such as inhibition/sensitization of adenylyl cyclase, Gβγ potentiation of adenylyl cyclase 2 (AC2), and extracellular signal-regulated kinase (ERK) activation as well as β-arrestin recruitment (Watts and Neve, 2005; Beaulieu and Gainetdinov, 2011). These diverse signaling pathways make D2Rs of great interest in studies of functional selectivity. Several studies have demonstrated that agonists differ in their ability to activate various pathways. For example, RNPA [R(−)propylnorapomorphine] and S(−)propylnorapomorphine differ in their ability to regulate activity of adenylyl cyclases compared with ion channels; RNPA and S(−)propylnorapomorphine were reported as full agonists for activation of Gαi/o, whereas neither compound displayed detectable activity for the activation of G protein–coupled inwardly-rectifying potassium channels (GIRK) through D2Rs (Gay et al., 2004). Notably, blockade of β-arrestin recruitment reportedly is a shared property of antipsychotics that exhibit either antagonist (e.g., haloperidol), or partial agonist (e.g., aripiprazole) activity through Gαi/o-cAMP pathways (Klewe et al., 2008; Masri et al., 2008). This suggests that β-arrestin–biased D2 antagonists might exhibit unique antipsychotic profiles (Masri et al., 2008). In contrast, a study with analogs of the novel antipsychotic aripiprazole suggested that D2 ligands with Gαi/o antagonist and β-arrestin agonist activity may have antipsychotic behavioral activity with reduced extrapyramidal side effects in a mouse model (Allen et al., 2011).
Heightened awareness of the potential benefit of pathway-biased ligands has created the need for methods to efficiently quantify and compare agonist-mediated activity through multiple pathways. Recently described methods have been proposed as tools to assess bias. The new quantitative methods incorporate efficacy and potency to calculate “bias factors” (Rajagopal et al., 2011; Kenakin et al., 2012; Kenakin and Christopoulos, 2013b). The values from these methods reflect the relative activities of a test ligand with that of a reference compound for activating one effector pathway relative to another, such as Gα versus β-arrestin signaling. Activation of more complex signaling pathways downstream of GPCRs may require multiple effectors, and this suggests an additional use of bias analyses. Specifically, comparisons of ligand bias profiles for more immediate effectors versus complex pathways such as ERK phosphorylation, heterologous sensitization, and dynamic mass redistribution (DMR) could provide insight on the relative contribution of the effectors toward the complex signaling pathway.
In the present study, the ability of reference or clinically relevant ligands to activate multiple signaling pathways coupled to D2Rs was examined in a Chinese hamster ovary (CHO) cell line stably expressing the human dopamine D2L receptor (CHO-D2L) cells. Specifically, we analyzed Gαi/o activation, Gβγ activation, β-arrestin recruitment, ERK phosphorylation, heterologous sensitization, and DMR in response to a series of D2R ligands. The results were analyzed using four of the most commonly used methods to measure ligand bias (Rajagopal et al., 2011; Kenakin and Christopoulos, 2013b). The analyses revealed general consistency across several bias models and highlighted the utility of using a single cell line in studies of functional selectivity. Additionally, the dependency of the complex signaling pathways on the immediate effectors of the D2R was also explored by comparing ligand bias profiles.
Materials and Methods
Compounds and Other Chemicals Used.
The following compounds were purchased from Sigma-Aldrich (St. Louis, MO): dopamine hydrochloride, (±)-quinpirole dihydrochloride, pramipexole dihydrochloride, (+)-3-PPP [R(+)-3-(3-hydroxyphenyl)-N-propylpiperidine hydrochloride], (+)-bromocriptine methanesulfonate salt, RNPA, ropinirole hydrochloride, pergolide mesylate, 3-isobutyl-1-methylxanthine (IBMX), and rotigotine hydrochloride. Lisuride maleate and forskolin were purchased from Tocris (Ellisville, MO), and aripiprazole was purchased from Santa Cruz Biotechnology (Dallas, TX). We purchased HEPES and EDTA from Fisher Scientific (Pittsburgh, PA), and MgCl2 and Tris from Sigma-Aldrich.
Cell Culture and Cryopreservation.
CHO cells expressing CHO-D2L in the PathHunter β-Arrestin GPCR assay platform were purchased from DiscoveRx (Freemont, CA). Cells were grown in Ham’s F12 media supplemented with 1 mM l-glutamine (Thermo Scientific, West Palm Beach, FL), 10% fetal bovine serum (Hyclone, Logan, UT), 50 U/ml of penicillin, 50 μg/ml of streptomycin (Life Technologies, Grand Island, NY), 800 μg/ml of G418 (InvivoGen, San Diego, CA), and 300 μg/ml of hygromycin B (Fisher Scientific). Confluent 15-cm dishes of cells were harvested with Cell Dissociation Buffer (Life Technologies) and resuspended in 5 ml of fetal bovine serum containing 10% dimethylsulfoxide (Sigma-Aldrich); 1 ml was added to cryovials and frozen overnight at −80°C in a CoolCell device (BioCision, Larkspur, CA). On the following day, cryovials were stored in liquid N2 until the assay day.
Transient Transfections.
CHO-D2L cells were plated in 15-cm dishes at a confluence of 2.6 × 106 cells/dish with culture medium without selection antibiotics, and were incubated at 37°C in a humidified incubator overnight. On the following day, a 6-ml solution containing 30 μg of rat AC2 plasmid and 60 μl Lipofectamine 2000 (Life Technologies) in Opti-MEM (Life Technologies) was prepared and incubated at room temperature for 45 minutes. The solution was added dropwise to the cells, and transfection was performed for 48 hours. Cells were harvested and cryopreserved as described previously.
Gαi/o Assay.
Cryopreserved CHO-D2L cells were thawed in a 37°C water bath, resuspended in 10 ml of Opti-MEM, and centrifuged at 500g for 5 minutes. The supernatant was aspirated, the cells were resuspended in 1 ml of Opti-MEM and then counted using a Countess automated cell counter (Life Technologies). The cells were diluted to reach a concentration of 3 × 105 cells/ml. We added 10 μl/well of cell suspension to a white, flat-bottomed, tissue culture–treated 384-well plate (PerkinElmer, Shelton, CT), resulting in a final density of 3000 cells/well. The plate was centrifuged for 30 seconds at 100g and incubated in a 37°C humidified incubator for 1 hour.
After incubation, 5 μl/well of D2R ligand was added, followed by the addition of 5 μl/well of forskolin (10 μM final concentration) in 0.5 mM IBMX. The cells were incubated at room temperature for 1 hour, and the cAMP accumulation was measured using Cisbio’s Dynamic 2 kit (Cisbio Bioassays, Bedford, MA) according to the manufacturer’s instructions. The plates were analyzed for fluorescent emissions at 620 nm and 665 nm using 330 nm as the excitation wavelength in a Synergy 4 (BioTek, Winooski, VT), and ratiometric analysis was performed by dividing the 665 nm emission by the 620 nm emission to extrapolate the cAMP concentration from a cAMP standard curve.
β-Arrestin Assay.
Cryopreserved CHO-D2L cells were thawed in a 37°C water bath, resuspended in 10 ml of Opti-MEM, and centrifuged at 500g for 5 minutes. The supernatant was aspirated, and the cells were resuspended in 1 ml of Opti-MEM and counted. Cells were diluted to reach a concentration of 2.5 × 105 cells/ml. We added 10 μl/well of cell suspension to a white, flat-bottomed, low-volume, tissue culture–treated 384-well plate (PerkinElmer), resulting in a final density of 2500 cells/well. The plate was centrifuged for 30 seconds at 100g and incubated in a 37°C humidified incubator overnight. The next day, 2.5 μl/well of D2R ligands or vehicle/buffer control was added to the cells. After the drug addition, the cells were incubated in a 37°C humidified incubator for 1.5 hours.
Recruitment of β-arrestin to the D2R was assessed using the PathHunter assay (DiscoveRx) according to the manufacturer’s instructions. The PathHunter assay uses an enzyme complementation platform in which the GPCR is tagged with ProLink and β-arrestin 2 is tagged with an enzyme acceptor upon interaction between the GPCR and β-arrestin 2. The two fragments complement to generate a functional β-galactosidase that converts substrate to a chemiluminescent signal (Zhao et al., 2008) that was measured in a Synergy 4.
Gβγ Assay.
The Gβγ assay uses a regulatory characteristic that is specific for AC2, AC4, and AC7 (Watts and Neve, 1997; Cooper and Crossthwaite, 2006). These isoforms of adenylyl cyclase are insensitive to inhibition by Gαi/o and are conditionally activated by Gβγ subunits from Gαi/o-linked receptors in the presence of direct AC2 activators (Federman et al., 1992). CHO-D2L cells transiently transfected with AC2 as described previously were thawed in a 37°C water bath, resuspended in 10 ml of Opti-MEM, and centrifuged at 500g for 5 minutes. The supernatant was aspirated, and the resuspension and centrifugation steps were repeated. The supernatant was aspirated, and the cells were resuspended in 1 ml of Opti-MEM and counted. Cells were diluted to reach a concentration of 4 × 105 cells/ml, and 5 μl/well of cell suspension was added to a white, low-volume, flat-bottomed, tissue culture–treated 384-well plate, resulting in a final density of 2000 cells/well. The plate was centrifuged for 30 seconds at 100g and incubated in a 37°C humidified incubator for 1 hour. The plates were removed from the incubator, and 2.5 μl/well of D2R ligand was added. Cyclic AMP accumulation was initiated by the addition of 2.5 μl/well of phorbol 12-myristate 13-acetate (PMA) (final concentration of 1 μM) in the presence of 0.5 mM IBMX to specifically stimulate AC2 (Watts and Neve, 1997). The cells were incubated at room temperature for 1 hour, and the cAMP accumulation was measured as described earlier.
Heterologous Sensitization Assay.
Heterologous sensitization assays were performed as previously described elsewhere (Conley et al., 2014). Briefly, cryopreserved CHO-D2L cells were thawed in a 37°C water bath, resuspended in 10 ml of Opti-MEM, and centrifuged at 500g for 5 minutes. The supernatant was aspirated, and the cells were resuspended in 1 ml of Opti-MEM and counted. The cells were diluted to reach a concentration of 3 × 105 cells/ml. We added 10 μl/well of cell suspension to a white, flat-bottomed, tissue culture–treated 384-well plate, resulting in a final density of 3000 cells/well. The plate was centrifuged for 30 seconds at 100g and incubated in a 37°C humidified incubator for 1 hour.
After incubation, 5 μl/well of D2R ligand was added to the cells, and the cells were incubated in a 37°C humidified incubator for 2 hours to accomplish sensitization. After sensitization, 5 μl/well of forskolin in IBMX and spiperone was added to the cells at final concentrations of 10 μM, 0.5 mM, and 1 μM, respectively. The cells were incubated at room temperature for 1 hour, and the cAMP accumulation was measured as described earlier.
ERK Assay.
Cryopreserved CHO-D2L cells were thawed in a 37°C water bath, resuspended in 10 ml of Opti-MEM, and centrifuged at 500g for 5 minutes. The supernatant was aspirated, and the resuspension and centrifugation steps were repeated. The supernatant was aspirated, and the cells were resuspended in 1 ml of Opti-MEM and counted. The cells were diluted to reach a concentration of 2 × 106 cells/ml. We added 8 μl/well of cell suspension in a white, low-volume, flat-bottomed, tissue culture–treated 384-well plate, resulting in a final density of 16,000 cells/well. The plate was centrifuged for 30 seconds at 100g and incubated in a 37°C humidified incubator for 2 hours. After incubation, 4 μl/well of D2R ligand was added to the cells. The cells were incubated for 10 minutes at room temperature, and ERK phosphorylation was measured using the Cellul’erk assay (Cisbio Bioassays) according to the manufacturer’s instructions. Plates were read for fluorescent emissions at 620 nm and 665 nm using 330 nm as the excitation wavelength in a Synergy 4.
Dynamic Mass Redistribution Assay.
DMR assays were performed as described previously elsewhere (Schroder et al., 2011). Briefly, 20 μl of Opti-MEM was added to each well of one quadrant of a fibronectin-coated EnSpire LFC-384 plate (PerkinElmer), which was centrifuged at 500g for 30 seconds. Cryopreserved CHO-D2L cells were thawed, centrifuged, and counted as described previously. The cells were diluted to achieve 3.3 × 105 cells/ml. We added 30 μl/well of this dilution to the plate for a final volume of 50 μl, and the plate was incubated for 16–24 hours in a 37°C humidified incubator. At 1.0–1.5 hours before the assay, the medium was aspirated, and the cells were washed twice with room temperature Hanks’ balanced salt solution (Life Technologies) supplemented with 20 mM HEPES, which served as the assay buffer, using a JANUS MDT Mini (PerkinElmer). The cells were incubated in 40 μl/well assay buffer for 1.0–1.5 hours at ambient temperature.
We performed 10 baseline DMR reads, added 10 μl/well of D2R ligand dissolved in assay buffer, and measured DMR for 200 reads. All DMR measurements were performed using an EnSpire plate reader (PerkinElmer) according to manufacturer’s protocols. After the assay, the wells were visually inspected for confluency, and the wells with significantly reduced cellular density (<60%) were excluded from further analysis. Receptor activation was quantified by calculating the maximum DMR peak intensity achieved during 40 reads, approximately 20 minutes after ligand addition. Receptor activation was also quantified by calculating the area under the curve of the initial DMR peak that occurred during the first 40 reads, with the resultant concentration response curves being indistinguishable from the maximum DMR peak intensity curves (data not shown).
Membrane Preparations.
The cells were grown to confluency in 15-cm dishes. The culture medium was aspirated, replaced with 10 ml of ice-cold lysis buffer (1 mM HEPES, 2 mM EDTA, pH 7.4), and incubated on ice for 10 minutes. The cells were scraped using a sterile cell scraper, suspended in the lysis buffer, triturated by pipetting up and down, and centrifuged at 30,000g for 20 minutes at 4°C. The supernatant was discarded. The pellet was resuspended in receptor-binding buffer (4 mM MgCl2, 50 mM Tris, pH 7.4), homogenized using a Kinematica homogenizer (Kinematica AG, Lucerne, Switzerland) and aliquoted in 1-ml fractions. The aliquots were centrifuged at 12,000g for 10 minutes at 4°C, the supernatant was decanted, and the pellet was frozen and stored in a −80°C freezer until the assay day.
Isotherm-Binding Assay.
The isotherm-binding assays were performed using [3H]methylspiperone (PerkinElmer) as described previously elsewhere (Vidi et al., 2008). Membrane aliquots were thawed on ice and resuspended in receptor-binding buffer at a final concentration of approximately 30 ng/μl of membrane protein. Total binding reactions were performed in receptor-binding buffer containing increasing concentrations of [3H]methyspiperone and membrane suspension in a total volume of 500 μl. Nonspecific binding was assessed in the presence of 5 μM butaclamol. Reactions were incubated for 30 minutes at 37°C and then harvested in a 96-well Packard Filtermate harvester (PerkinElmer) to type B glass-fiber filter plates (Millipore, Billerica, MA). The concentration of radioligand was determined by pipetting [3H]methylspiperone directly onto the wells of the filter plates for “total radioactivity.” The plates were dried overnight, and 40 μl/well of MicroScint 0 scintillation fluid (PerkinElmer) was added. Radioactivity was measured in a Packard TopCount scintillation detector (PerkinElmer).
Competitive Binding Assay.
Competitive binding assays were conducted in the presence of 0.4 nM [3H]methylspiperone. Total and nonspecific binding were determined as described for the isotherm-binding assay, except that the competitive binding reactions were performed in the presence of 75 μM 5′-guanylyl-imidodiphosphoate (GppNHp; Sigma-Aldrich) (Kent et al., 1980). Competitive binding reactions contained increasing concentrations of the test compound, [3H]methylspiperone, GppNHp, membrane suspension, and receptor-binding buffer. The reactions were incubated and harvested, and the radioactivity was quantified as described previously.
Bias and Data Analyses.
All data were analyzed using GraphPad Prism 6 (GraphPad Software, San Diego, CA). Ligand bias was assessed using four different methods. The equimolar comparison was performed by plotting normalized responses of two signaling pathways for equal concentrations of ligand against each other. Shifts on the plots toward one of the axes in comparison with the reference compound (dopamine) indicated bias for the pathway on that axis.
The equiactive comparison was performed using the ratios of relative activity as previously described elsewhere (Griffin et al., 2007; Ehlert, 2008; Rajagopal et al., 2011) using the following equation:
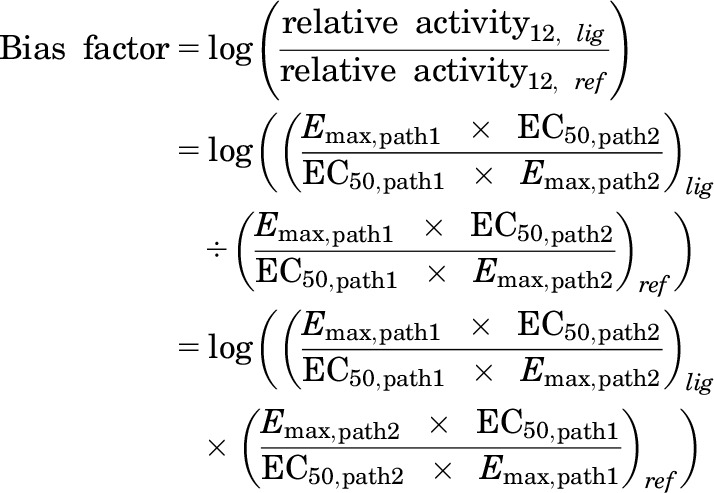 |
where Emax is the maximal effect of the compound, EC50 is the EC50 value of the compound, lig is the compound being analyzed, ref is the reference compound, path1 is one of the pathways being analyzed, and path2 is the other pathway being analyzed.
For the transduction coefficient method, functional data were plotted in the Black and Leff (1983) operational model, and the analysis was performed as previously described elsewhere (Kenakin et al., 2012), except for the standard errors, which were calculated individually for each compound. The following equation was used for this analysis:
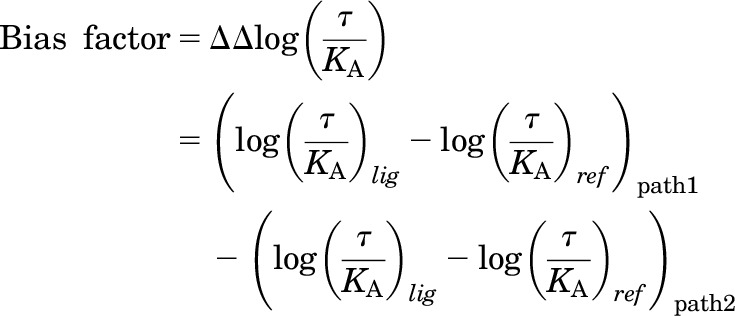 |
where τ is the coupling efficiency and KA is the conditional affinity. Both were obtained by fitting the data to the Black and Leff operational model.
The compounds were also analyzed using the sigma comparison, which was performed by fitting the functional data in the Black and Leff (1983) operational model setting the KA to the ligands’ dissociation constant (Ki) obtained from the competitive binding assays performed with the same CHO-D2L cells used for the functional studies. Data were analyzed as previously described elsewhere (Rajagopal et al., 2011) using the following equations:
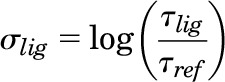 |
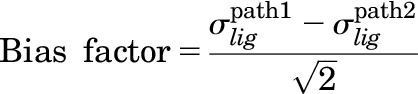 |
where τ is the coupling efficiency and σ is the effective signaling.
For all methods, the natural ligand dopamine was used as the reference compound. Statistical analyses were performed in GraphPad Prism 6 using one-way analysis of variance followed by Dunnett’s post hoc test, with P < 0.05 considered statistically significant.
Results
β-Arrestin Recruitment and Gαi/o Activation.
The first pathway downstream of D2R examined was the recruitment of β-arrestin using the PathHunter assay from DiscoveRx (Fig. 1A; Table 1). Rotigotine and lisuride potently stimulated recruitment of β-arrestin, with EC50 values of 0.2 and 0.4 nM, respectively. The least potent compound was (+)-3-PPP, with an EC50 value of 925 nM. Aripiprazole and lisuride displayed partial agonist activity for the recruitment of β-arrestin, with maximal effects lower than 80% of the effect of dopamine.
Fig. 1.

Activation of three signaling pathways downstream of the D2R in CHO-D2L cells. (A) Recruitment of β-arrestin to the D2R was measured using the PathHunter assay from DiscoveRx. (B) Activation of Gαi/o by the D2R was measured by assessing inhibition of forskolin-mediated cAMP production. (C) Activation of Gβγ by the D2R was assessed by measuring potentiation of PMA-stimulated cAMP accumulation. Data represent the average and S.E.M. of at least three independent experiments.
TABLE 1.
Potency and maximal effects of the compounds tested for downstream effectors of the D2R
Data are an average of at least three individual experiments conducted in duplicate. For EC50 values, the 95% confidence interval is shown in parentheses. Maximal effects are shown as a percentage of dopamine’s maximal response, and S.E.M. is shown in parentheses. Potency ratios of β-arrestin recruitment/Gαi/o activation were also included.
| Compound |
β-Arrestin 2 Recruitment |
Gαi/o Activation |
Gβγ Activation |
Potency (EC50) Ratio |
|||
|---|---|---|---|---|---|---|---|
| EC50 | Max Effect (% Dopamine) | EC50 | Max Effect (% Dopamine) | EC50 | Max Effect (% Dopamine) | β-Arrestin/Gαi/o | |
| nM | nM | nM | |||||
| Dopamine | 124 (100–153) | 100 (± 2) | 1.2 (0.7–2.1) | 100 (± 3) | 171 (72–407) | 100 (± 8) | 103 |
| Quinpirole | 64 (59–70) | 96 (± 1) | 1.2 (0.9–1.6) | 100 (± 2) | 145 (89–236) | 103 (± 4) | 53 |
| Lisuride | 0.4 (0.3–0.6) | 66 (± 2) | 0.07 (0.04–0.14) | 106 (± 5) | 2.2 (0.4–12.0) | 30 (± 6) | 9 |
| Bromocriptine | 2.9 (1.9–4.4) | 101 (± 4) | 1.9 (0.9–4.3) | 104 (± 3) | 0.5 (0.1–2.1) | 34 (± 5) | 2 |
| Aripiprazole | 3.6 (2.5–5.2) | 19 (± 1) | 1.3 (0.5–3.1) | 65 (± 4) | ND | −19 (± 5) | 3 |
| Rotigotine | 0.2 (0.1–0.3) | 111 (± 4) | 0.02 (0.007–0.03) | 95 (± 3) | 1.5 (0.3–6.9) | 54 (± 9) | 10 |
| (+)-3-PPP | 925 (809–1058) | 92 (± 1) | 8.6 (4.9–15.1) | 108 (± 4) | 518 (136–1973) | 57 (± 9) | 108 |
| RNPA | 1.9 (1.5–2.5) | 101 (± 2) | 13 (3–57) | 95 (± 13) | 360 (210–619) | 110 (± 11) | 0.1 |
| Pramipexole | 12 (10–15) | 94 (± 2) | 0.2 (0.1–0.3) | 108 (± 3) | 20 (9–46) | 86 (± 5) | 60 |
| Ropinirole | 49 (37–63) | 92 (± 2) | 0.5 (0.3–0.8) | 111 (± 4) | 100 (34–290) | 56 (± 6) | 98 |
| Pergolide | 7.2 (5.9–8.9) | 90 (± 2) | 0.04 (0.02–0.07) | 101 (± 4) | 2.3 (0.6–9.5) | 38 (± 7) | 180 |
ND, not determined.
The second signaling pathway examined was the canonical D2R activation of Gαi/o by measuring the inhibition of forskolin-stimulated cAMP accumulation. These studies were performed in the same line of CHO-D2L cells used for the β-arrestin assay. The compounds displayed a wide range of potencies for activation of Gαi/o (Fig. 1B; Table 1), with the most potent compounds being rotigotine, pergolide, and lisuride with EC50 values of 20, 40, and 70 pM, respectively. The least potent compound was (+)-3-PPP, with an EC50 value of 8.6 nM. Aripiprazole displayed partial agonist activity, with efficacy equal to 65% of the maximal effect of dopamine. The majority of the compounds were more potent at inhibition of cAMP accumulation than recruitment of β-arrestin. Alternatively, RNPA was more potent in stimulating recruitment of β-arrestin (Table 1), suggesting that this compound displays bias for the β-arrestin pathway.
More detailed bias analyses for Gαi/o and β-arrestin were completed employing the previously described methods of measuring ligand bias using dopamine as the reference compound. The equimolar comparison identified apparent biased agonists through qualitative analyses (Fig. 2A). For example, the data points of rotigotine are shifted toward the β-arrestin recruitment axis in comparison with data points from dopamine (Fig. 2A). The qualitative nature of this method precludes statistical analyses. However, in comparison with the current quantitative analyses, the equimolar comparison may be useful for comparing compounds that are antagonists or inverse agonists for one or more of the signaling pathways under investigation.
Fig. 2.

Bias analyses of β-arrestin recruitment in comparison with Gαi/o activation by the D2R. (A) Equimolar comparison. (B) Equiactive comparison. (C) Transduction coefficient. (D) Sigma comparison. Dopamine was used as the reference compound for all the analyses. For the quantitative analyses positive values indicate bias for β-arrestin; negative values indicate bias for Gαi/o. Data represent the average and S.E.M. of at least three independent experiments. *P < 0.05.
The results from the Gαi/o and β-arrestin assays were then analyzed using three recently described quantitative methods that incorporate functional data from concentration response curves. The equiactive comparison model compares the log of the ratios of the relative activity of the test compounds with the reference compound (Griffin et al., 2007; Ehlert, 2008; Rajagopal et al., 2011). When functional data were compared using the equiactive comparison, five compounds (lisuride, bromocriptine, aripiprazole, rotigotine, and RNPA) displayed statistically significant bias for the recruitment of β-arrestin compared with Gαi/o, with RNPA displaying the greatest degree of bias (Fig. 2B). The bias patterns observed here generally reflect the patterns seen in the potency ratios for β-arrestin activity versus Gαi/o (Table 1).
The recently described transduction coefficient was then used to assess bias (Kenakin et al., 2012). The functional data were plotted in the Black and Leff (1983) operational model to calculate values for coupling efficiency (τ) and conditional affinity (KA). The Δlog (τ/KA) ratios for the reference compound were calculated and subtracted from the Δlog (τ/KA) ratios of test compounds at each respective pathway to calculate bias (Kenakin et al., 2012). In agreement with the equiactive comparison, this approach identified the same five compounds as biased for recruitment of β-arrestin (Fig. 2C). In contrast to the equiactive model, the transduction coefficient model identified pergolide as biased for Gαi/o in comparison with β-arrestin recruitment. An inspection of the potency ratios also identifies pergolide as the most Gαi/o-selective compound (Table 1).
A third quantitative model also uses the Black and Leff (1983) operational model with the notable requirement for dissociation constants (Ki) for each ligand. For this analysis, we performed competitive binding assays in membranes prepared from the same CHO-D2L cell line that was used for the functional studies. The affinity values (Ki) of the compounds for the D2R were measured using [3H]methylspiperone (Supplemental Table 1). Competitive binding experiments were performed in the presence of GppNHp to uncouple receptors from G proteins and provide a more homogenous receptor state (Kent et al., 1980). The bias factors from the sigma model were very similar to the bias factors obtained from the other quantitative analyses (Fig. 2D). Four of the five compounds that were identified as biased for β-arrestin recruitment using the other methods were also identified using the sigma model (lisuride, bromocriptine, aripiprazole, and RNPA).
In all three quantitative models, RNPA was identified as the most biased compound for recruitment of β-arrestin. Notably, in each of these quantitative bias analyses aripiprazole was identified as significantly biased for β-arrestin recruitment. These findings appear inconsistent with a simple pharmacologic inspection. For example, aripiprazole was a partial agonist for Gαi/o activation and β-arrestin recruitment, with relative efficacies of 65 and 19%, respectively. Furthermore, the EC50 values of aripiprazole were 1.3 nM for Gαi/o activation versus 3.6 nM for β-arrestin recruitment. Taken together, these values imply aripiprazole is biased for Gαi/o activation in comparison with β-arrestin recruitment due to increased efficacy and potency. This discrepancy can be accounted for by considering that dopamine, the reference compound, is nearly 100 times more potent for Gαi/o activation than β-arrestin recruitment. This highlights the importance of the choice and activity of the reference compound in bias analysis and interpretation. Measures of bias are always considered relative to the activity of the reference compound.
Activation of Gβγ.
The next signaling pathway downstream of D2Rs that was analyzed was the activation of Gβγ subunits. Activation of Gβγ by GPCRs can lead to several different cellular responses, including activation of GIRKs, phospholipase C β2, modulation of N-type calcium channels, and potentiation of AC2 (Lin and Smrcka, 2011). D2R-mediated Gαi/o activation and the subsequent release/rearrangement of Gβγ produce a conditional enhancement of cAMP production by AC2 when the enzyme is activated by protein kinase C via PMA (Watts and Neve, 1997). To examine Gβγ activation by the D2R, CHO-D2L cells were transiently transfected with AC2, and the potentiation of PMA-stimulated cAMP accumulation by agonists was examined.
Consistent with activation of Gβγ, most of the compounds tested elicited an enhanced cAMP response to PMA (Fig. 1C; Table 1). Several compounds [(+)-3-PPP, bromocriptine, ropinirole, pergolide, lisuride, and rotigotine] displayed partial agonist profiles for Gβγ signaling, with maximal activities that were lower than 80% of dopamine’s maximal response. Aripiprazole did not display an agonist response in this assay. The most potent compounds were bromocriptine and rotigotine, with EC50 values of 0.5 and 1.5 nM, respectively, whereas the least potent compound was (+)-3-PPP with an EC50 value of 518 nM. Dopamine and quinpirole were full agonists with EC50 values of 171 and 145 nM, respectively. The majority of the compounds were an order of magnitude (>25-fold) less potent for Gβγ signaling compared with the Gαi/o-mediated response (Table 1). Alternatively, bromocriptine was about 6-fold more potent for Gβγ activity, suggesting that this compound may be biased for Gβγ activation.
Bias analyses were performed to compare Gβγ activation with Gαi/o signaling through D2Rs. The equimolar comparison resulted in apparent bias of aripiprazole for Gαi/o, with a lack of response for Gβγ activation (Fig. 3A). The quantitative methods could not analyze the results from aripiprazole given the lack of efficacy. The equiactive comparison and the transduction coefficient identified bromocriptine as biased for Gβγ activation in comparison with Gαi/o activation (Fig. 3, B and C). Conversely, the sigma comparison suggested that pergolide was biased for Gαi/o activation (Fig. 3D). Bias analyses for activation of Gβγ were also performed in comparison with β-arrestin recruitment (Supplemental Fig. 1). In the equiactive comparison and transduction coefficient, RNPA was the only compound that displayed significant bias for β-arrestin recruitment (Supplemental Fig. 1, B and C). The sigma comparison identified rotigotine as significantly biased for β-arrestin recruitment (Supplemental Fig. 1D).
Fig. 3.

Bias analyses of Gβγ in comparison with Gαi/o activation by the D2R. (A) Equimolar comparison. (B) Equiactive comparison. (C) Transduction coefficient. (D) Sigma comparison. Dopamine was used as the reference compound for all the analyses. For the quantitative analyses positive values indicate bias for Gβγ; negative values indicate bias for Gαi/o. Data represent the average and S.E.M. of at least three independent experiments. *P < 0.05.
Heterologous Sensitization of Adenylyl Cyclases.
After measuring the activation of immediate effectors of the D2R, more complex signaling pathways were analyzed. Heterologous sensitization is a cellular adaptive response that is observed after persistent activation of Gαi/o-coupled GPCRs (Watts, 2002). It has been shown that both Gα (which can be blocked by pertussis toxin treatment) and Gβγ subunits (prevented by the expression of βARK-CT) are required for this response (Watts and Neve, 2005; Ejendal et al., 2012). To explore the potential relationship and apparent requirement for both Gα and Gβγ, sensitization studies were completed in CHO-D2L cells used previously.
Heterologous sensitization was induced by pretreating the cells with the D2R ligands for 2 hours followed by measuring forskolin-stimulated cAMP accumulation in the presence of spiperone. This prolonged pretreatment results in an enhancement in cAMP production when compared with vehicle-treated cells (Fig. 4A). The responses observed for heterologous sensitization displayed diversity among the compounds tested (Table 2). Quinpirole and RNPA were more efficacious than dopamine. In contrast, aripiprazole had no detectable response in this assay, and lisuride was an inverse agonist. Most compounds displayed EC50 values that were comparable to those obtained for Gαi/o activation.
Fig. 4.

Activation of complex signaling pathways downstream of the D2R. (A) Heterologous sensitization was assessed by pretreating the cells with the D2R ligand for 2 hours and then stimulating them with forskolin. (B) ERK phosphorylation was measured after treating the cells with the D2R ligand for 10 minutes using Cisbio’s Cellul’erk kit. (C) Dynamic mass redistribution was measured during stimulation with D2 ligands, and the maximal peak height was determined. Data represent the average and S.E.M. of at least three independent experiments.
TABLE 2.
Potency and maximal effects of the compounds tested for the complex signaling pathways of the D2R
Data are an average of at least three individual experiments conducted in duplicate. For EC50 values, the 95% confidence interval is shown in parentheses. Maximal effects are shown as a percentage of dopamine’s maximal response, and S.E.M. is shown in parentheses.
| Compound | Heterologous Sensitization |
ERK Phosphorylation |
DMR (Max Peak Height) |
|||
|---|---|---|---|---|---|---|
| EC50 | Max Effect (% Dopamine) | EC50 | Max Effect (% Dopamine) | EC50 | Max Effect (% Dopamine) | |
| nM | nM | nM | ||||
| Dopamine | 3.3 (0.9–11.6) | 100 (± 8) | 53 (32–86) | 100 (± 5) | 11 (8.6–15) | 100 (± 2) |
| Quinpirole | 3.3 (2.3–4.7) | 147 (± 3) | 44 (24–81) | 95 (± 5) | 8.7 (5.8–12.9) | 97 (± 3) |
| Lisuride | 0.09 (0.04–0.18) | −73 (± 3) | 1.5 (0.7–3.4) | 53 (± 5) | 12 (7.5–19) | 95 (± 4) |
| Aripiprazole | ND | −13 (± 13) | 19 (3–112) | 12 (± 2) | 593 (323–1090) | 57 (± 3) |
| Rotigotine | 0.06 (0.02–0.22) | 98 (± 9) | 1.4 (0.7–2.9) | 80 (± 5) | 6.4 (4.4–9.5) | 120 (± 3) |
| RNPA | 14 (11–18) | 183 (± 8) | 216 (107–433) | 84 (± 6) | 13 (9.0–17) | 111 (± 3) |
| Pergolide | 0.05 (0.02–0.18) | 105 (± 9) | 1.6 (0.7–3.7) | 57 (± 4) | 14 (8.5–25) | 106 (± 5) |
ND, not determined.
Ligand bias analyses were employed to compare heterologous sensitization with activation of the immediate D2R effectors. The initial contrast between the methods was that aripiprazole and lisuride could only be analyzed using the qualitative equimolar comparison. The bias analyses for sensitization versus Gαi/o were model dependent and identified only one biased compound (Fig. 5, A and D; Supplemental Fig. 2A). Specifically, the transduction coefficient method and the sigma comparison revealed no bias, whereas the equiactive comparison identified RNPA as biased for heterologous sensitization. When heterologous sensitization was compared with Gβγ activation, none of the compounds displayed significant bias in any of the methods used (Fig. 5, B and E; Supplemental Fig. 2B). Heterologous sensitization was then compared with β-arrestin recruitment. These bias comparisons revealed that RNPA was biased for β-arrestin recruitment and pergolide was biased for heterologous sensitization in each of the quantitative models (Fig. 5, C and F; Supplemental Fig. 2C). Additionally, the equiactive comparison identified rotigotine as significantly biased for β-arrestin recruitment (Fig. 5C).
Fig. 5.

Bias analyses using the equiactive comparison and transduction coefficients of heterologous sensitization in comparison with effectors of the D2R. (A) Equiactive comparison of heterologous sensitization and Gαi/o activation. (B) Equiactive comparison of heterologous sensitization and Gβγ activation. (C) Equiactive comparison of heterologous sensitization and β-arrestin recruitment. (D) Analyses using the transduction coefficients of heterologous sensitization in comparison with Gαi/o activation. (E) Analyses using the transduction coefficients of heterologous sensitization in comparison with Gβγ activation. (F) Analyses using the transduction coefficients of heterologous sensitization in comparison with β-arrestin recruitment. Dopamine was used as the reference compound for all the analyses. Positive values indicate bias for heterologous sensitization; negative values indicate bias for the D2R effector under analysis. Data represent the average and S.E.M. of at least three independent experiments. *P < 0.05.
ERK Phosphorylation.
The next complex signaling pathway analyzed was D2R-mediated ERK phosphorylation. β-Arrestins are involved in ERK phosphorylation downstream of several GPCRs (Lefkowitz and Shenoy, 2005; Zhu et al., 2013). However, it has been suggested that the D2R-mediated ERK phosphorylation is a G protein–mediated event that is not dependent on β-arrestins (Quan et al., 2008). Moreover, both G protein inactivation with pertussis toxin and sequestration of Gβγ with βARK-CT inhibited D2R-mediated ERK phosphorylation in CHO cells (Oak et al., 2001). For this signaling pathway a subset of the D2R ligands was tested that included dopamine and quinpirole, commonly used reference compounds, and ligands that displayed functional selectivity for the immediate effectors of the D2R, such as aripiprazole, lisuride, rotigotine, RNPA, and pergolide (Fig. 4B; Table 2).
The quantitative bias analyses were then performed comparing ERK phosphorylation with the activation of the immediate effectors of the D2R. Notably, none of the quantitative methods identified significantly biased compounds in the comparisons of ERK phosphorylation with Gαi/o or Gβγ activation (Fig. 6, A, B, D, and E; Supplemental Fig. 3). In contrast, the comparisons with β-arrestin recruitment using the equiactive comparison identified aripiprazole, lisuride, rotigotine, and RNPA as significantly biased for β-arrestin recruitment (Fig. 6C). The transduction coefficient and the sigma comparison were generally consistent, revealing bias trends for aripiprazole, rotigotine, and RNPA for β-arrestin recruitment (Fig. 6F; Supplemental Fig. 3C).
Fig. 6.

Bias analyses using the equiactive comparison and transduction coefficients of ERK phosphorylation in comparison with effectors of the D2R. (A) Equiactive comparison of ERK phosphorylation and Gαi/o activation. (B) Equiactive comparison of ERK phosphorylation and Gβγ activation. (C) Equiactive comparison of ERK phosphorylation and β-arrestin recruitment. (D) Analyses using the transduction coefficients of ERK phosphorylation in comparison with Gαi/o activation. (E) Analyses using the transduction coefficients of ERK phosphorylation in comparison with Gβγ activation. (F) Analyses using the transduction coefficients of ERK phosphorylation in comparison with β-arrestin recruitment. Dopamine was used as the reference compound for all the analyses. Positive values indicate bias for ERK phosphorylation; negative values indicate bias for the immediate D2R effector under analysis. Data represent the average and S.E.M. of at least three independent experiments. *P < 0.05.
Dynamic Mass Redistribution.
The final measure of complex signaling after stimulation with D2R ligands employed in this study was DMR. DMR is a label-free phenotypic measure of ligand-receptor signaling that temporally monitors changes in intracellular mass via the optical characteristics of the cells, namely, the refractive index (Fang et al., 2006). DMR has been used previously to study and characterize a number of GPCRs (Schroder et al., 2011). Ligands characterized using this assay were dopamine and quinpirole as reference compounds and the immediate effector-biased compounds lisuride, pergolide, rotigotine, RNPA, and aripiprazole. Of the compounds tested, only aripiprazole’s responses were markedly different than that of dopamine (Fig. 4C; Table 2). Rotigotine was the most potent compound tested with an EC50 value of 6.4 nM. Aripiprazole was the only compound tested that exhibited significantly lower efficacy as it only elicited 57% of dopamine’s response. Aripiprazole was also the least potent compound tested with an EC50 value of 593 nM (Table 2).
DMR was then compared with the immediate effectors of the D2R using the quantitative bias analyses. Notably, in contrast to the other complex signaling pathways, the comparisons against all immediate D2R effectors resulted in significantly biased compounds (Fig. 7; Supplemental Fig. 4). The equiactive comparison identified aripiprazole, lisuride, rotigotine, and pergolide as significantly biased for Gαi/o, and RNPA as significantly biased for DMR in comparison with Gαi/o (Fig. 7A). The transduction coefficient resulted in a similar pattern, except that RNPA was not significantly biased (Fig. 7D). The sigma comparison resulted in only one significantly biased compound in the analyses against Gαi/o. Pergolide was biased for Gαi/o (Supplemental Fig. 4A). The analyses of DMR versus Gβγ activation also resulted in significantly biased compounds. The equiactive comparison identified lisuride, rotigotine, and pergolide as biased for Gβγ (Fig. 7B). The transduction coefficient identified only lisuride as biased for Gβγ (Fig. 7E). And the sigma comparison did not result in any significantly biased compounds (Supplemental Fig. 4B).
Fig. 7.

Bias analyses using the equiactive comparison and transduction coefficients of DMR in comparison with effectors of the D2R. (A) Equiactive comparison of DMR and Gαi/o activation. (B) Equiactive comparison of DMR and Gβγ activation. (C) Equiactive comparison of DMR and β-arrestin recruitment. (D) Analyses using the transduction coefficients of DMR in comparison with Gαi/o activation. (E) Analyses using the transduction coefficients of DMR in comparison with Gβγ activation. (F) Analyses using the transduction coefficients of DMR in comparison with β-arrestin recruitment. Dopamine was used as the reference compound for all the analyses. Positive values indicate bias for DMR; negative values indicate bias for the immediate D2R effector under analysis. Data represent the average and S.E.M. of at least three independent experiments. *P < 0.05.
All quantitative methods of analyzing ligand bias resulted in significantly biased ligands in the comparisons between DMR and β-arrestin recruitment. The equiactive comparison identified aripiprazole, lisuride, rotigotine, RNPA, and pergolide as significantly biased for β-arrestin recruitment (Fig. 7C). A similar pattern was found in the analyses using the transduction coefficient, except that pergolide was not significantly biased (Fig. 7F). The analyses using the sigma comparison identified rotigotine, aripiprazole, and RNPA as significantly biased for β-arrestin recruitment (Supplemental Fig. 4C).
Discussion
Ligand bias is increasingly appreciated as a potential strategy for development of drugs with improved efficacy and/or reduced side effects. However, debate currently exists with regard to the most appropriate methods for assessing bias (Kenakin and Christopoulos, 2013a; Rajagopal, 2013). In the present study, the ability of several agonists to activate multiple signaling pathways coupled to D2Rs was examined. The most commonly used methods to measure ligand bias were employed and evaluated.
Bias analyses may be influenced by variability in cell-to-cell expression levels of receptors and other signaling proteins that may lead to inconsistent results. For instance, the potency and efficacy of aripiprazole, a D2R partial agonist, for activation of Gαi/o are very cell line–dependent (Lawler et al., 1999; Burris et al., 2002; Shapiro et al., 2003), making the choice of an appropriate cellular model an important first step. In the present CHO-D2L model, factors such as receptor-expression levels and the expression level of immediate signaling transduction proteins, such as Gα, Gβγ, and β-arrestin, were constant in an effort to ensure that the observed bias only reflects interactions between ligand and receptor signaling complex.
Each of the methods used to assess ligand bias was effective in identifying pathway-biased ligands. Although the equimolar comparison is logical and easy to perform, its qualitative nature limits its use when comparing multiple compounds in structure activity relationships or screening studies (Rajagopal et al., 2011; Kenakin and Christopoulos, 2013b). The relative merits and limitations of quantitative approaches to measure ligand bias have been reviewed elsewhere (Rajagopal et al., 2011; Kenakin and Christopoulos, 2013b). The equiactive method uses EC50 values and the maximal effects of the compounds from standard four-parameter sigmoid curve-fitting approaches to calculate relative activities (Griffin et al., 2007; Ehlert, 2008; Rajagopal et al., 2011). Both the sigma comparison and transduction coefficient methods fit agonist data according to the Black-Leff operational model, but they differ in defining the agonist affinity parameter KA. The sigma comparison uses affinity values derived from radioligand-binding experiments. This analysis, therefore, requires additional experiments or literature mining to obtain the affinity constants of the drug for the receptor. Furthermore, debate exists as to the design and appropriateness of using binding assay–derived affinity values, as agonists often have different affinity values for multiple conformational states of the receptor (Kenakin and Christopoulos, 2013b; Nygaard et al., 2013; Kenakin, 2014). The transduction coefficient model uses functionally derived KA values, which incorporate the interaction between ligand, receptor, and transducer (Kenakin et al., 2012), although the meaningfulness of these KA values has also been questioned (Rajagopal, 2013).
Despite the differences in the analyses, the present results revealed similar trends between the quantitative models, especially the equiactive and transduction coefficient comparisons. The relationship between the equiactive and transduction coefficient results was expected because the methods become nearly identical when the slopes of the concentration response curves are close to unity (Kenakin and Christopoulos, 2013b). Similarly, if the experimentally determined affinity constants obtained from competitive binding assays (Ki) are the same as the conditional affinities obtained via the transduction coefficient method (KA), the bias factors from the sigma comparison and the transduction coefficient would also show good agreement (Kenakin and Christopoulos, 2013b).
In contrast, poor agreement between the measured Ki value and calculated KA value leads to inconsistent bias results. For example, bromocriptine was not biased for Gβγ signaling in comparison with Gαi/o activation in the sigma comparison, but it was identified as biased in both the equiactive comparison and the transduction coefficient (Fig. 3). This inconsistency can be explained by the constraint added by the measured Ki value to the fit of the data in the Black and Leff operational model (Kenakin and Christopoulos, 2013b). Specifically, the Ki value of the compound was nearly 10 times larger than the KA value obtained from the transduction coefficient, yielding a suboptimal fit (R2 = 0.56), resulting in the lack of bias in the sigma comparison.
It also appears that suboptimal fitting of the data can also increase the noise in the calculated bias factors. This was illustrated by examining RNPA in the analyses of Gβγ signaling using the sigma comparison (Supplemental Fig. 2B). The added constraint to the sigma comparison by the use of the measured Ki value (which for this example was nearly 100 times lower that the KA value and 28 times lower than the EC50 value) resulted in a poor fit (R2 = 0.49) of the data, increasing the noise in the bias analysis.
One limitation of all these methods is that the nature of the data transformations precludes the traditional quantitative analyses from incorporating data from antagonists or inverse agonists. Furthermore, weak partial agonists may have poor fits when using the Black and Leff equation, limiting their analysis by these methods. For example, due to the lack of a significant agonist response for aripiprazole in the Gβγ activation assay, a bias factor could not be determined using the quantitative models.
The studies presented identified several biased compounds via both quantitative and qualitative measures. For example, aripiprazole had no agonist response for Gβγ activation while retaining partial agonist activity for all the other immediate receptor effectors analyzed (Fig. 1; Table 1). These results can be explained by differences in stimulus-response coupling efficiencies (i.e., most ligands were more potent for Gαi/o activation and β-arrestin recruitment than they were for Gβγ activation). Nevertheless, these data are in agreement with a previous study demonstrating that aripiprazole was inactive for stimulation of GIRK channels in MES-23.5 cells stably expressing the D2R (Shapiro et al., 2003). Both the activation of GIRK channels and the potentiation of AC2 result from the activation of Gβγ (Watts and Neve, 1997; Cooper and Crossthwaite, 2006; Lin and Smrcka, 2011). Furthermore, we have recently demonstrated in human embryonic kidney cells that aripiprazole fully antagonizes dopamine’s Gβγ response through the D2R (Brust et al., 2015). In contrast, RNPA was an agonist for Gβγ activation in our assays, although it has been shown that the compound does not activate GIRK-mediated K+-currents in CHO cells stably expressing the D2R (Gay et al., 2004). Aripiprazole also failed to display any detectable responses for heterologous sensitization. This finding is consistent with the requirement of Gβγ activation for heterologous sensitization (Ejendal et al., 2012). Furthermore, lisuride was an apparent inverse agonist for heterologous sensitization and an agonist for all the other signaling pathways. This apparent functional selectivity profile may be due to pseudo-irreversible binding as a result of the high affinity of lisuride for the receptor (see Supplemental Table 1) (Watts and Neve, 1996).
Rotigotine, a dopamine agonist approved for treatment of Parkinson’s disease, was among the most potent compounds for all signaling pathways, while (+)-3-PPP was the least potent compound. The remaining compounds showed similar rank orders across the assays (Table 1). The maximal effects of the compounds for the different signaling pathways also varied, but this was not specifically associated with bias. For instance, although lisuride was a full agonist for Gαi/o and a partial agonist for β-arrestin recruitment, the quantitative analyses indicated bias for β-arrestin recruitment. This can be explained, in part, by the reference compound used for the bias analyses. The EC50 value of dopamine for activation of Gαi/o was nearly 100-fold lower than its EC50 value for β-arrestin recruitment, a profile that is similar to that of the prototypical D2R agonist quinpirole (Table 1). However, for lisuride the magnitude of the change in the EC50 values from Gαi/o to β-arrestin was about 10-fold (Table 1). A similar bias profile was attributed to aripiprazole, which had a relative efficacy of 65% for Gαi/o activation and 19% for β-arrestin recruitment. These results highlight the strong influence that potency has in these analyses compared with fairly large differences in efficacy (approximately 40%).
Bias analyses may also be useful in providing mechanistic insight that underlies the activation of the more complex signaling pathways downstream of a GPCR. Specifically, the comparisons of heterologous sensitization and ERK phosphorylation with the immediate effectors of the D2R suggest that the recruitment of β-arrestin is not an essential event for the activation of those two signaling pathways. This is consistent with the results from assays using pertussis toxin, in which pertussis treatment fully inhibited quinpirole-mediated heterologous sensitization and ERK phosphorylation (Supplemental Fig. 5).
For heterologous sensitization, none of the analyses found significant bias in the comparisons with Gβγ activation. The comparisons with Gαi/o resulted in only one biased compound, RNPA, in the equiactive comparison. In contrast, the analyses comparing heterologous sensitization with β-arrestin recruitment resulted in several significantly biased compounds in all of the quantitative bias analyses. This is consistent with previous studies suggesting that G proteins but not β-arrestins are associated with heterologous sensitization (Bohn et al., 2000; Watts and Neve, 2005). For ERK phosphorylation, none of the compounds analyzed were significantly biased in the comparisons with Gαi/o or Gβγ activation. However, all the quantitative bias analyses resulted in significantly biased compounds in the comparisons with β-arrestin recruitment. These results suggest that in our model ERK phosphorylation is mediated by G proteins and not by β-arrestins. These results are in agreement with previous studies that suggested that D2R-mediated ERK phosphorylation was not dependent on β-arrestins, and that inhibition of G protein signaling with pertussis toxin or βARK-CT also inhibited ERK phosphorylation (Oak et al., 2001; Quan et al., 2008). The comparisons of DMR with the immediate effectors revealed multiple biased compounds for all comparisons, except for the comparisons between DMR and Gβγ activation in the sigma comparison. These results could be interpreted to suggest that the immediate effectors have only limited contributions to the DMR response for those biased compounds. Alternatively, it is probably more reasonable to propose that multiple effectors have significant contributions to DMR. This is consistent with the idea that DMR is an integrated cellular response. However, there is evidence that the most intense DMR peak is caused by activation of Gα (Schroder et al., 2011; Ferrie et al., 2014), and treatment with pertussis toxin fully inhibited our quinpirole-mediated DMR response (Supplemental Fig. 5D).
Biased ligands have the potential of becoming very important tools to improve the safety and specificity of current drug therapies. Many studies have already suggested scenarios where biased ligands are desired (DeWire and Violin, 2011; Whalen et al., 2011). Quantification of ligand bias is a key parameter to guide medicinal chemists in the design of new pathway biased/functionally selective compounds. Here, a single cellular model was used to measure activation of each signaling pathway, and the different equations used to quantify ligand bias were used and compared (Rajagopal et al., 2011; Kenakin and Christopoulos, 2013b). We observed that there was good overall consistency between the equiactive and transduction coefficient bias analyses when they were used in a single cellular model. Additionally, the present studies suggest that quantitative bias analyses can be used to offer mechanistic insight into complex GPCR signaling pathways.
Supplementary Material
Acknowledgments
The authors thank Dr. Meng Wu, the center director of the University of Iowa High-Throughput Screening (UIHTS), Heidi Morgan for technical assistance in establishing the DMR assay, and the reviewers for insightful suggestions.
Abbreviations
- AC2
adenylyl cyclase 2
- CHO
Chinese hamster ovary
- CHO-D2L
CHO cells expressing the human dopamine D2L receptor
- D2R
dopamine D2 receptor
- DMR
dynamic mass redistribution
- ERK
extracellular signal-regulated kinase
- GIRK
G protein–coupled inwardly-rectifying potassium channel
- GPCR
G protein–coupled receptor
- GppNHp
5′-guanylyl-imidodiphosphoate
- IBMX
3-isobutyl-1-methylxanthine
- PMA
phorbol 12-myristate 13-acetate
- (+)-3-PPP
R(+)-3-(3-hydroxyphenyl)-N-propylpiperidine hydrochloride
- RNPA
R(−)propylnorapomorphine
Authorship Contributions
Participated in research design: Brust, Hayes, Roman, Burris, Watts.
Conducted experiments: Brust, Hayes.
Performed data analysis: Brust, Hayes, Watts.
Wrote or contributed to the writing of the manuscript: Brust, Hayes, Roman, Burris, Watts.
Footnotes
This research was supported by the National Institutes of Health National Institute of Mental Health [Grant R01-MH060397]; the National Institutes of Health National Institute of General Medicine [Grant T32-GM067795]; the National Institutes of Health National Center for Research Resources [Grant S10-RR029274]; the American Foundation for Pharmaceutical Education; and the Purdue Research Foundation.
Portions of this work were presented previously as a poster presentation at the following conference: Brust TF, Hayes MP, Roman DL, and Watts VJ (2014) G protein contribution to complex signaling pathways downstream of the dopamine D2L receptor [FASEB J 28(Suppl):662.10]. Experimental Biology 2014; 2014 Apr 26–30; San Diego, CA.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, et al. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci USA 108:18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. (1983) Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220:141–162. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. (2004) Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol 66:106–112. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. (2000) Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408:720–723. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286:2495–2498. [DOI] [PubMed] [Google Scholar]
- Brust TF, Hayes MP, Roman DL, Watts VJ. (2015) New functional activity of aripiprazole revealed: robust antagonism of D2 dopamine receptor-stimulated Gβγ signaling. Biochem Pharmacol 93:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris KD, Molski TF, Xu C, Ryan E, Tottori K, Kikuchi T, Yocca FD, Molinoff PB. (2002) Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 302:381–389. [DOI] [PubMed] [Google Scholar]
- Conley JM, Brust TF, Xu R, Burris KD, Watts VJ. (2014) Drug-induced sensitization of adenylyl cyclase: assay streamlining and miniaturization for small molecule and siRNA screening applications. J Vis Exp 83:e51218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DM, Crossthwaite AJ. (2006) Higher-order organization and regulation of adenylyl cyclases. Trends Pharmacol Sci 27:426–431. [DOI] [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, Walker JK. (2008) Beta-arrestins specifically constrain beta2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J 22:2134–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Violin JD. (2011) Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res 109:205–216. [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. (2008) On the analysis of ligand-directed signaling at G protein-coupled receptors. Naunyn Schmiedebergs Arch Pharmacol 377:549–577. [DOI] [PubMed] [Google Scholar]
- Ejendal KF, Dessauer CW, Hébert TE, Watts VJ. (2012) Dopamine D(2) receptor-mediated heterologous sensitization of AC5 requires signalosome assembly. J Signal Transduct 2012:210324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Ferrie AM, Fontaine NH, Mauro J, Balakrishnan J. (2006) Resonant waveguide grating biosensor for living cell sensing. Biophys J 91:1925–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federman AD, Conklin BR, Schrader KA, Reed RR, Bourne HR. (1992) Hormonal stimulation of adenylyl cyclase through Gi-protein beta gamma subunits. Nature 356:159–161. [DOI] [PubMed] [Google Scholar]
- Ferrie AM, Sun H, Zaytseva N, Fang Y. (2014) Divergent label-free cell phenotypic pharmacology of ligands at the overexpressed β2-adrenergic receptors. Sci Rep 4:3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay EA, Urban JD, Nichols DE, Oxford GS, Mailman RB. (2004) Functional selectivity of D2 receptor ligands in a Chinese hamster ovary hD2L cell line: evidence for induction of ligand-specific receptor states. Mol Pharmacol 66:97–105. [DOI] [PubMed] [Google Scholar]
- Griffin MT, Figueroa KW, Liller S, Ehlert FJ. (2007) Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J Pharmacol Exp Ther 321:1193–1207. [DOI] [PubMed] [Google Scholar]
- Kenakin T. (2014) What is pharmacological ‘affinity’? Relevance to biased agonism and antagonism. Trends Pharmacol Sci 35:434–441. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. (2013a) Measurements of ligand bias and functional affinity. Nat Rev Drug Discov 12:483. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. (2013b) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12:205–216. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci 3:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent RS, De Lean A, Lefkowitz RJ. (1980) A quantitative analysis of beta-adrenergic receptor interactions: resolution of high and low affinity states of the receptor by computer modeling of ligand binding data. Mol Pharmacol 17:14–23. [PubMed] [Google Scholar]
- Klewe IV, Nielsen SM, Tarpø L, Urizar E, Dipace C, Javitch JA, Gether U, Egebjerg J, Christensen KV. (2008) Recruitment of beta-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology 54:1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler CP, Prioleau C, Lewis MM, Mak C, Jiang D, Schetz JA, Gonzalez AM, Sibley DR, Mailman RB. (1999) Interactions of the novel antipsychotic aripiprazole (OPC-14597) with dopamine and serotonin receptor subtypes. Neuropsychopharmacology 20:612–627. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. (2005) Transduction of receptor signals by beta-arrestins. Science 308:512–517. [DOI] [PubMed] [Google Scholar]
- Lin Y, Smrcka AV. (2011) Understanding molecular recognition by G protein βγ subunits on the path to pharmacological targeting. Mol Pharmacol 80:551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. (2008) Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA 105:13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. (2013) The dynamic process of β(2)-adrenergic receptor activation. Cell 152:532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oak JN, Lavine N, Van Tol HHM. (2001) Dopamine D-4 and D-2L receptor stimulation of the mitogen-activated protein kinase pathway is dependent on transactivation of the platelet-derived growth factor receptor. Mol Pharmacol 60:92–103. [DOI] [PubMed] [Google Scholar]
- Quan W, Kim JH, Albert PR, Choi H, Kim KM. (2008) Roles of G protein and beta-arrestin in dopamine D2 receptor-mediated ERK activation. Biochem Biophys Res Commun 377:705–709. [DOI] [PubMed] [Google Scholar]
- Rajagopal S. (2013) Quantifying biased agonism: understanding the links between affinity and efficacy. Nat Rev Drug Discov 12:483. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Ahn S, Rominger DH, Gowen-MacDonald W, Lam CM, Dewire SM, Violin JD, Lefkowitz RJ. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol 80:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder R, Schmidt J, Blättermann S, Peters L, Janssen N, Grundmann M, Seemann W, Kaufel D, Merten N, Drewke C, et al. (2011) Applying label-free dynamic mass redistribution technology to frame signaling of G protein-coupled receptors noninvasively in living cells. Nat Protoc 6:1748–1760. [DOI] [PubMed] [Google Scholar]
- Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R. (2003) Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 28:1400–1411. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13. [DOI] [PubMed] [Google Scholar]
- Vidi PA, Chemel BR, Hu CD, Watts VJ. (2008) Ligand-dependent oligomerization of dopamine D(2) and adenosine A(2A) receptors in living neuronal cells. Mol Pharmacol 74:544–551. [DOI] [PubMed] [Google Scholar]
- Wang WC, Mihlbachler KA, Brunnett AC, Liggett SB. (2009) Targeted transgenesis reveals discrete attenuator functions of GRK and PKA in airway beta2-adrenergic receptor physiologic signaling. Proc Natl Acad Sci USA 106:15007–15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts VJ. (2002) Molecular mechanisms for heterologous sensitization of adenylate cyclase. J Pharmacol Exp Ther 302:1–7. [DOI] [PubMed] [Google Scholar]
- Watts VJ, Neve KA. (2005) Sensitization of adenylate cyclase by Gα i/o-coupled receptors. Pharmacol Ther 106:405–421. [DOI] [PubMed] [Google Scholar]
- Watts VJ, Neve KA. (1996) Sensitization of endogenous and recombinant adenylate cyclase by activation of D2 dopamine receptors. Mol Pharmacol 50:966–976. [PubMed] [Google Scholar]
- Watts VJ, Neve KA. (1997) Activation of type II adenylate cyclase by D2 and D4 but not D3 dopamine receptors. Mol Pharmacol 52:181–186. [DOI] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med 17:126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Jones A, Olson KR, Peng K, Wehrman T, Park A, Mallari R, Nebalasca D, Young SW, Xiao SH. (2008) A homogeneous enzyme fragment complementation-based beta-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J Biomol Screen 13:737–747. [DOI] [PubMed] [Google Scholar]
- Zhu W, Tilley DG, Myers VD, Coleman RC, Feldman AM. (2013) Arginine vasopressin enhances cell survival via a G protein-coupled receptor kinase 2/β-arrestin1/extracellular-regulated kinase 1/2-dependent pathway in H9c2 cells. Mol Pharmacol 84:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.