

Abstract
Tight junction protein 1 (TJP1), a component of tight junction, has been reported to play a role in protein networks as an adaptor protein, and TJP1 expression is altered during tumor development. Here, we found that TJP1 expression was increased at the RNA and protein levels in TGF-β-stimulated lung cancer cells, A549. SB431542, a type-I TGF-β receptor inhibitor, as well as SB203580, a p38 kinase inhibitor, significantly abrogated the effect of TGF-β on TJP1 expression. Diphenyleneiodonium, an NADPH oxidase inhibitor, also attenuated TJP1 expression in response to TGF-β in lung cancer cells. When TJP1 expression was reduced by shRNA lentiviral particles in A549 cells (A549-sh TJP1), wound healing was much lower than in cells infected with control viral particles. Taken together, these data suggest that TGF-β enhances TJP1 expression, which may play a role beyond structural support in tight junctions during cancer development. [BMB Reports 2015; 48(2): 115-120]
Keywords: TGF-β, TJP1, EMT, Motility, ROS
INTRODUCTION
Transforming growth factor-β (TGF-β) superfamily members, which include well-known growth factors, are involved in a variety of cellular processes, such as cell proliferation and differentiation. TGF-β signaling is initiated by ligand binding to type I, II, and III TGF-β receptors. Then, the type II TGF-β receptor forms a complex and activates the type I TGF-β receptor to initiate intracellular signaling. The type I TGF-β receptor phosphorylates Smad2 and Smad3 directly, which form a complex with Smad4 and accumulate in the nucleus, and regulate gene expression (1-4). In addition to the Smad-dependent canonical pathway, crosstalk and signaling through Smad-independent signaling pathways have been reported (5-7). It is not fully understood how TGF-β signals in these pathways. In advanced cancers, TGF-β displays a tumor-promoting effect by inducing an epithelial-mesenchymal transition (EMT), which enhances invasiveness and metastasis. Generally, EMT is characterized by a loss of cell-cell adhesion and apical-basal polarity and a gain in motility (8).
Epithelial cells allow the separation of different tissues and body compartments by developing cell surface domains called junctions, which are important for the biogenesis, maintenance, and function of epithelia (9-11). Tight junctions, the most apical component of the intercellular junctional complex, form a barrier to the diffusion of molecules from the lumen to the parenchyma and regulate the diffusion of various cellular macromolecules between the apical and basolateral plasma membranes. Thus, a loss of barrier function due to growth factors or cytokines is closely related to pathophysiological processes, leading to tumors and retinopathy, for example (11, 12).
One of the tight junction proteins, “tight junction protein 1” (TJP1, zonula occludens-1, ZO-1), is responsible for the protein networks between actin and integral tight junction proteins, such as occludin and claudins (13, 14), in the tight junction, as well as cell integrity. Thus, a loss of tight junction barrier function due to TJP1 dysfunction has been related to cancer metastasis (9). However, the role of TJP1 expression in cancer has been controversial. Generally, TJP1 is considered a tumor suppressor, supported by various studies (15-17). In contrast, TJP1 expression and localization has also been reported to be altered in pancreatic cancer, colorectal cancer, melanoma, and non-small cell lung cancer (NSCLC) (18-21), suggesting a role for TJP1 in tumor progression. While accumulating data suggests a role for TJP1 in cancer, the detailed mechanism by which TJP1 expression might be regulated in cancer remains unclear.
Here, we investigated the possibility that TGF-β influenced TJP1 expression and sought to identify mediators of TGF-β-induced TJP1 expression in lung cancer cells. We also performed TJP1 knockdown experiments to determine whether TJP1 is involved in cancer cellular processes, such as migration or EMT.
RESULTS
TJP1 is increased in TGF-β-stimulated lung cancer cells
Previously, we showed that TGF-β has an effect on EMT and motility in lung cancer cells (22). To verify the effect of TGF-β on cell adhesion and junctional proteins in A549 cells, we first examined the expression of E-cadherin and N-cadherin in TGF-β-stimulated A549 lung cancer cells (Fig. 1A, B). TGF-β increased N-cadherin expression and decreased E-cadherin expression in A549 lung cancer cells, as expected. In terms of tight junction proteins, TJP1 was increased in response to TGF-β (Fig. 1A), while TJP3 was decreased at the RNA level (data not shown). Because TJP1 decreased in response to TGF-β in various cancer cell lines (15, 23, 24), we conducted additional experiments to confirm the effects of TGF-β on TJP1 expression in A549 lung cancer cells. As shown in Fig. 1C and D, TGF-β induced TJP1 expression, beginning at 10 pM and peaking at 50 pM TGF-β. In time-course experiments, TJP1 expression reached a maximum at 24 h in A549 lung cancer cells. Induction of TJP1 in response to TGF-β was also seen in NCI-H596 and A427 human lung carcinoma cell lines (Fig. 4C), suggesting a role for TGF-β on TJP1 expression in lung cancer.
Fig. 1. TGF-β induces TJP1 expression in A549 lung carcinoma cells. Subconfluent cells were exposed to TGF-β for 24 h, harvested, lysed, and analyzed for TJP1, E-cadherin, N-cadherin, and GAPDH expression by (A) RT-PCR and (B) real-time PCR in A549 cells. Subconfluent cells were exposed to (C) various concentrations of TGF-β for 24 h or to (D) 50 pM TGF-β for various time periods, as indicated, in A549 cells, and analyzed for TJP1, E-cadherin, N-cadherin, and β-actin expression by immunoblotting. All results are representative of at least three independent experiments. Data are expressed as means ± SE. Statistical significance was assessed using paired Student’s t-tests (*P < 0.0001).
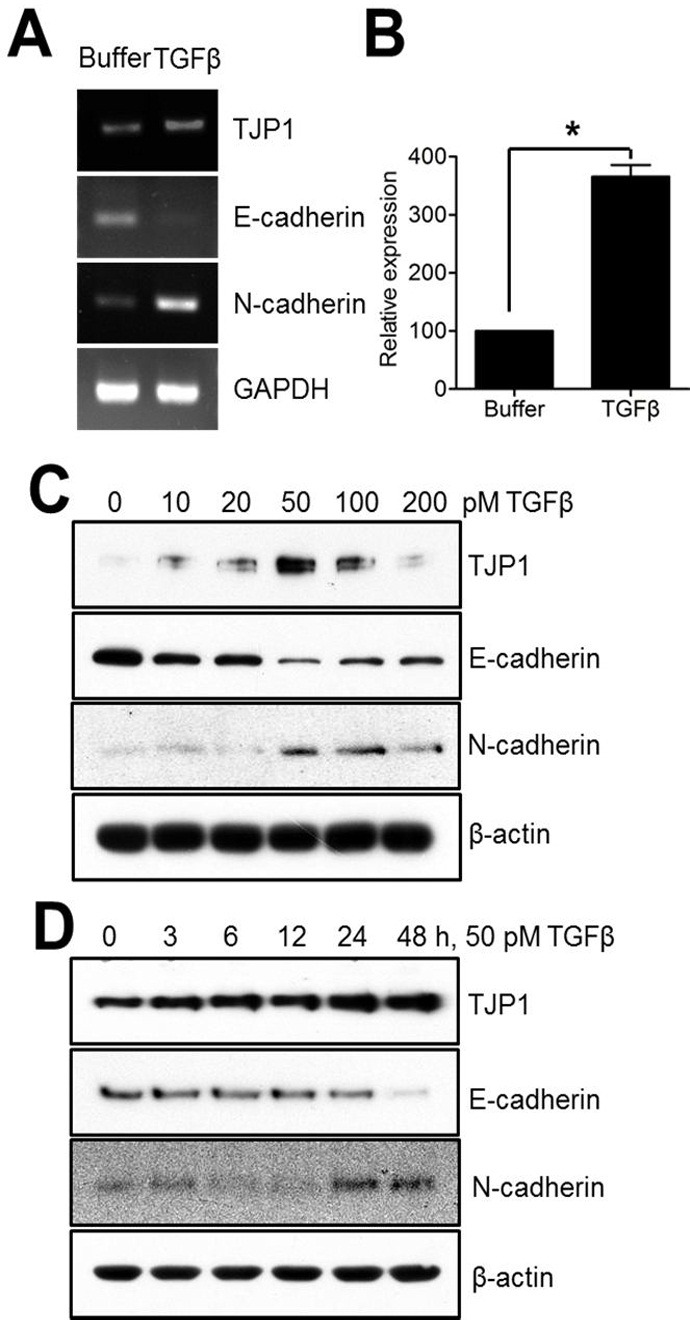
Fig. 4. TJP1 is increased in response to TGF-β in lung cancer cells and contributes to cellular motility. (A) Subconfluent cells were treated with 50 pM TGF-β for 24 h and harvested for immunoblotting. (B) Cells were grown until fully confluent, and a uniform scratch was made down the center of the well. Digital images of the wound were obtained in 12-h intervals until 72 h (top), and the horizontal distance between both sides of the wound was measured and analyzed statistically (bottom). All results shown are representative of at least three independent experiments. Data are expressed as the means ± SE. Statistical significance was assessed using paired Student’s t-tests (*P < 0.0001). (C) A427 and NCI-H596 lung cancer cells were treated with 100 pM TGF-β for 24 h and evaluated for TJP1 expression by immunoblotting. All results shown are representative of at least three independent experiments. (D) Cells were induced to invade through Matrigel- coated Transwell membranes. After 24 h, invaded cells were fixed and stained. Digital images were taken for cell counting. All results shown are representative of at least three independent experiments. Data are expressed as the means ± SE. Statistical significance was assessed using paired Student’s t-tests (P < 0.0001).

p38 kinase, NADPH oxidase, and type I TGF-β receptor inhibitors reverse TGF-β-induced TJP1 expression in A549 lung cancer cells
To assess the regulatory mechanism for TJP1 expression in response to TGF-β in A549 human lung carcinoma cells, cells were treated with TGF-β in the presence of SB431542, a type I TGF-β receptor inhibitor, SB203580, a p38 kinase inhibitor, PB98059, a MEK1 inhibitor, SP600126, a c-Jun amino-terminal kinase (JNK) inhibitor, diphenyleneiodonium (DPI), an NADPH oxidase inhibitor, N-acetyl cysteine (NAC), a reactive oxygen species scavenger, wortmannin, a phosphatidylinositol 3-kinase (PI3-kinase) inhibitor, and dimethyl sulfoxide (DMSO) for 24 h. Inhibitors were added 30 min prior to TGF-β addition. SB431542, SB203580, NAC, and DPI attenuated TJP1 expression in A549 lung carcinoma cells 3 (Fig. 2) while PD98059, SP600125, and wortmannin did not (data not shown). These results suggest that the type I TGF-β receptor, p38 kinase, and ROS may be involved in TJP1 expression in response to TGF-β. To further confirm the role of the type I TGF-β receptor, p38 kinase and ROS in TGF-β signaling-mediated TJP1 expression, we examined downstream signaling activation in TGF-β-stimulated A549 cells (Fig. 3A). As expected, Smad2 was phosphorylated after 10 min and reached a maximum at 60 min in response to TGF-β in A549 lung cancer cells. p38 kinase was also phosphorylated in response to TGF-β, occurring at 30 min and lasting for 3 h. We did not observe any significant change in the phosphorylation status of extracellular signal-regulated kinase (ERK) or JNK in TGF-β-stimulated A549 lung cancer cells (data not shown). To examine any crosstalk between downstream signaling pathways, cells were treated with TGF-β in the presence of SB431542, SB203580, DPI, NAC, or vehicles (DMSO or H2O) for 10 min and then harvested for immunoblotting. SB431542 inhibited Smad2 phosphorylation markedly in response to TGF-β, but it did not affect p38 kinase phosphorylation by TGF-β in A549 lung cancer cells (Fig. 3B). SB203580, a p38 kinase inhibitor, did not affect the phosphorylation of Smad2, indicating that Smad2, as well as p38 kinase, may be involved independently in TGF-β-mediated TJP1 expression in lung cancer cells. Recently, TGF-β was reported to increase cellular ROS with the help of NADPH oxidase 4, contributing to cellular differentiation (25). Here, we observed ROS generation in response to TGF-β in A549 lung cancer cells. Moreover, TGF-β-induced ROS generation was inhibited by DPI, an NADPH oxidase inhibitor and NAC, a ROS scavenger (Fig. 3E, F). Additionally, neither DPI nor NAC altered the phosphorylation status of Smad2 (Fig. 3C), while ROS generation by TGF-β was attenuated by SB431542 and SB203580 (Fig. 3E), indicating that ROS may contribute to TJP1 expression after activation of Smad 2 and p38 kinase in TGF-β-stimulated A549 lung cancer cells.
Fig. 2. SB431542, a type I TGF-β receptor inhibitor, SB203580, a p38 kinase inhibitor, DPI, an NADPH oxidase inhibitor, and NAC, a ROS scavenger, attenuated TGF-β-mediated TJP1 expression in A549 cells. Subconfluent cells were treated with 50 pM TGF-β for 24 h in the presence of the indicated amounts of buffer, SB431542, SB203580, DPI, or NAC and evaluated for TJP1 expression by immunoblotting. All results shown are representative of at least three independent experiments.

Fig. 3. p38 kinase, ROS, and type I TGF-β receptor pathways did not show significant crosstalk in A549 lung carcinoma cells. (A) Subconfluent cells were treated with TGF-β (50 pM) for the time periods indicated and evaluated by immunoblotting to analyze TGF-β-induced signaling in A549 cells. (B, C) Subconfluent cells were treated with TGF-β (50 pM) for 10 min in the presence of various inhibitors (10 μM SB431542, 10 μM SB203580, 1 mM NAC, 5 μM DPI) or vehicles (DMSO, H2O), and evaluated by immunoblotting. (D) Subconfluent cells were treated with TGF-β for 0-30 min. DCF fluorescence was measured using a FACSCalibur. (E, F) Subconfluent cells were starved and treated with TGF-β (50 pM) for 30 min in the presence of SB431542, SB203580, DPI, NAC, or vehicles (DMSO, H2O). Inhibitors were added 30 min prior to TGF-β treatment. All results shown are representative of at least three independent experiments. Data are expressed as means ± SE. Statistical significance was assessed using paired Student’s t-tests (*P < 0.0001, **P < 0.01, ***P < 0.05).

TGF-β induces TJP1 expression in lung cancer cells, contributing to cell motility
To investigate the role of TJP1 in TGF-β-mediated signaling in A549 lung cancer cells, we generated A549-sh control and A549-sh TJP1 cell lines, expressing control and TJP1 targeting shRNA, respectively. TGF-β treatment altered the expression of E-cadherin as well as N-cadherin in both A549-sh control and A549-sh TJP1 cells, suggesting that TJP1 knockdown caused no significant effect on E-cadherin or N-cadherin expression in TGF-β-treated A549 lung cancer cells (Fig. 4A). TJP1 knockdown showed delayed wound healing in wound healing assays (Fig. 4B), suggesting a role for TJP1 in cell motility. To further confirm a role of TJP1 in cell migration and invasion, A549-sh control and A549-sh TJP1 cells were treated with TGF-β or buffer for 24 h, after which each group of cells was induced to invade through Matrigel-coated Transwell membranes (Fig. 4D). Consistent with the wound healing assay, A549-sh Control treated with TGF-β for TJP1 induction showed more invaded cells than A549-sh Control cells treated with buffer. A specific role for TJP1 in cell invasion in A549 cells was supported in that there were fewer invaded cells in A549-sh TJP1 (with TGF-β, no induction of TJP1) than A549-sh Control (with TGF-β, TJP1 induction). Also, TGF-β induced TJP1 expression in other lung cancer cell lines, such as A427 and NCIH596 (Fig. 4C). Together, these results support a role for TJP1 in TGF-β-induced cancer progression in lung cancer.
DISCUSSION
The main finding of this study was that TJP1 increased in response to TGF-β through Smad-dependent, p38 kinase, and ROS pathways, and that it contributed to cell motility in lung cancer cells. TJP1 functions as a scaffold protein at tight junctions, which are important for cellular polarity, especially in epithelia (9-11). Thus, expression of TJP1 is believed to be one of the characteristics of epithelial cells and is reported to decrease or disappear during EMT and cancer cell development. However, some studies suggest a role for TJP1 during cancer progression. For example, Balda and Matter reported that ‘ZO-1-associated nucleic acid-binding protein’ (ZONAB) was an interacting protein for TJP1 (26). In this study, ZONAB, a Y box transcription factor, was shown to mediate ErbB-2 expression with the help of TJP1 in vivo, suggesting that TJP1 may act as more than just a structural protein in a variety of cellular processes through its binding proteins. Additionally, TJP1 is overexpressed in pancreatic ductal adenocarcinoma, suggesting a role for TJP1 in cancer progression, especially in metastasis (19). In human melanoma, TJP1 was shown to be upregulated and to contribute to invasion by association with N-cadherin (21). Gilles et al. reported that TJP1 also played a role in the regulation of interleukin-8, a CXC chemokine family member, in breast cancer cells (27). These studies suggest a role for TJP1 in cancer invasion and metastasis; however, it remains to be determined how TJP1 might be involved in cancer cell malignancy. Recently, a role for TJP1 in mouse embryonic stem cells was explored by inactivating the TJP1 locus through homologous recombination, suggesting a role for TJP1 in mouse embryonic stem cell self-renewal and differentiation under certain conditions (28). These studies caused us to hypothesize that TJP1 might be increased in certain cancers, thus contributing to disease progression. Although a few studies have shown a role for TGF-β on TJP1 expression, they did not show the crosstalk between Smad-dependent and independent pathways and TJP1 expression in TGF-β-stimulated lung cancer cells. They also did not clarify the regulatory mechanism by which TGF-β increases TJP1 expression (15, 24). Here, we provide a regulatory mechanism by which TGF-β affects TJP1 expression in three human NSCLC cell lines: A549, HCI-H596. and A427 cells.
There are still many questions to be addressed, in terms of cancer selectivity and correlation to cancer stage, among others. Together, our data show that TGF-β upregulates the expression of TJP1, an adaptor protein that contributes to various cellular functions, including cell migration in lung cancer cells.
MATERIALS AND METHODS
Materials and plasmids
DMEM and RPMI 1640 were purchased from Hyclone (Logan, UT, USA). McCoy’s 5A and defined fetal bovine serum (FBS) were from GIBCO (Life Technologies Corp., Grand Island, NY, USA). SB431542, NAC, SB203580, wortmannin, and diphenyleneiodonium (DPI) were purchased from Calbiochem (La Jolla, CA, USA). TGF-β was from R&D Systems, Inc. (Minneapolis, MN, USA). The mouse monoclonal antibody for β-actin was from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Rabbit polyclonal antibodies against TJP1, E-cadherin, N-cadherin, phospho-p38 kinase, p38 kinase, and HRP-conjugated anti-mouse and anti-rabbit antibodies were from Cell Signaling Technology Inc. (Beverly, MA, USA). Rabbit monoclonal antibodies specific for Smad2, and phospho-Smad2 were from Cell Signaling Technology Inc. Short hairpin (sh) RNA-lentiviral particles against human TJP1 and control lentiviral particles were from Santa Cruz Biotechnology Inc.
Cell culture
Human lung carcinoma A549 cells (CCL-185), A427 (HTB-53), and human lung adenosquamous carcinoma NCI-H596 (HTB-178) cells were obtained from the American Type Culture Collection. A549 and NCI-H596 cells were maintained in RPMI 1640 media supplemented with 10% FBS. A427 cells were maintained in DMEM supplemented with 10% FBS. All cells were grown at 37℃ in a humidified 5% CO2 atmosphere.
Isolation of RNA, RT-PCR, and real-time PCR
Cells were treated with TGF-β for the indicated time periods and harvested. Total cellular RNA was extracted with RNeasy kit (Qiagen, Valencia, CA, USA). The RNA was quantified by UV scanning, and samples (5 μg) were reverse-transcribed at 42℃ for 60 min in 50 μl buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl, 5 mM MgCl2, and 1 mM each of dATP, dCTP, dGTP, and dTTP) in the presence of oligo(dT) primer. The TJP1 sense primer 5’-GGAGAGGTGTTCCGTGTTGT-3’ and antisense primer 5’-GAGCGGACAAATCCTCTCTG-3; (GenBank Accession No.: NM_175610.2) were used to generate a 253-bp product. The E-cadherin sense primer 5’-TGGAGAGACACTGCCAACTG-3’ and antisense primer 5’-GGCTTTGGATTCCTCTC-ACA-3′ (GenBank Accession No.: NM_004360) were used to generate a 251-bp product. To amplify the 248-bp glyceraldehyde 3-phosphate dehydrogenase (GAPDH) product, specific primers were used: sense primer 5’-GAGTCAACGGATTTGGTCGT-3’ and antisense primer 5’-TTGATTTTGGAGGGATC-TCG-3’ (GenBank Accession No.: NM_002046). The PCR products were subjected to electrophoresis, visualized with ethidium bromide, and photographed using the GelDoc program (Bio-Rad, Chicago, IL, USA).
For real-time PCR quantification, reactions were conducted using the LightCycler 480 SYBR Green I Master (Roche Diagnostics Corp., Indianapolis, IN, USA) following the manufacturer’s instruction with various amounts of template cDNA in a 20-μl final volume for 40 cycles. Samples were normalized to GAPDH and the ΔΔCt methods was used to calculate fold expression changes of mRNA (29).
Immunoblotting
Protein samples were subjected to SDS-PAGE, followed by transfer to polyvinylidene difluoride membranes. Membranes were then blocked, incubated for 2 h with primary antibody, followed by 1 h with HRP-conjugated anti-mouse or rabbit antibody. The blots were developed with an enhanced chemiluminescence kit (West-ZOL plus, Intron Biotechnology Inc., South Korea).
Measurement of intracellular ROS by flow cytometry
Subconfluent cells were treated with 50 pM TGF-β for the time periods indicated. Then, cells were washed with warm PBS, trypsinized, and quickly analyzed for green fluorescence by flow cytometry, as described previously (30). For ROS detection, 2’,7’dichlorodihydrofluorescein diacetate (H2DCFDA, 10 μM) was added 10 min before harvest, and the analysis was performed with a FACSCalibur (Becton-Dickinson, Mountain View, CA, USA) with the NCC FACS operator. In some experiments, cells were pretreated with inhibitors such as SB431542, SB203580, DPI, or DMSO 30 min prior to TGF-β treatment. The arithmetic/geometric mean fluorescence channel (Geo MFC) was derived with CellQuest.
shRNA-mediated silencing of human TJP1 in A549 cells
For stable expression of shRNA against TJP1 through lentiviral infection, subconfluent A549 cells were incubated with Polybrene (5 μg/ml) for 1 h before the addition of the lentiviral vector. After 24 h, the medium was replaced and cells were grown for 1 day. A549-sh control and A549-sh TJP1 stable cell lines were selected in 5 μg/ml puromycin dihydrochloride. To avoid clonal variation, we pooled individual infectants for each stable cell line produced by infection.
Wound-healing assay
A549-sh control and A549-sh TJP1 cells were grown in 6-well plates for 48 h until the cells were fully confluent. A uniform scratch was made down the center of the well using a sterile micropipette tip, followed by washing with PBS to remove non-adherent cells. Digital images of the wound were obtained every 12 h at ×10 magnification using the NCC cell observer system (Axiovert 200M; Carl Zeiss Inc., Germany). The horizontal distance between both wound sides was measured.
Invasion assay
First, the upper compartment of an 8-μm Transwell (6.5 mm diameter; Costar Corp., Cambridge, MA, USA) was coated with Matrigel (1 mg/ml) before starting the assay. Cells (104 cells) were suspended in RPMI 1640 and placed in the upper compartment of the Transwell and the lower compartment was filled with RPMI 1640 supplemented with 10% FBS. After 24 h, the filter was washed with PBS and fixed with methanol. Migrated cells on the filter membrane were stained using a Diff-Quik Stain Kit. Each assay was conducted at least three times, and three random fields from a ×20 magnification were analyzed for each filter membrane.
Statistical analyses
All data are expressed as percentages of the control and shown as means ± standard error (SE). Statistical comparisons between groups were made using paired t-tests with the Prism 5.0 statistical software (GraphPad. Software Inc., San Diego, CA, USA). Values of P < 0.05 were considered to indicate statistical significance.
Acknowledgments
This work was supported in part by grants from the National Cancer Center to HJ You (NCC1410120) and in part by the Basic Science Research Program through the National Research Foundation of Korea (No. 2010-0022961, to HJ You).
References
- 1.Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. (2006);6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- 2.Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta. (2008);1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Macias-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFbeta receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell. (1996);87:1215–1224. doi: 10.1016/S0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- 4.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. (2000);103:295–309. doi: 10.1016/S0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 5.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. (2005);118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 6.Song K, Wang H, Krebs TL, Danielpour D. Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J. (2006);25:58–69. doi: 10.1038/sj.emboj.7600917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. (2001);276:14466–14473. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- 8.Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. (2012);586:1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 9.Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. (2009);1788:872–891. doi: 10.1016/j.bbamem.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Matter K, Balda MS. Epithelial tight junctions, gene expression and nucleo-junctional interplay. J Cell Sci. (2007);120:1505–1511. doi: 10.1242/jcs.005975. [DOI] [PubMed] [Google Scholar]
- 11.Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. (2012);1258:9–18. doi: 10.1111/j.1749-6632.2012.06613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harhaj NS, Antonetti DA. Regulation of tight junctions and loss of barrier function in pathophysiology. Int J Biochem Cell Biol. (2004);36:1206–1237. doi: 10.1016/j.biocel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol. (1999);147:1351–1363. doi: 10.1083/jcb.147.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuomi S, Mai A, Nevo J, et al. PKCepsilon regulation of an alpha5 integrin-ZO-1 complex controls lamellae formation in migrating cancer cells. Sci Signal. (2009);2:ra32. doi: 10.1126/scisignal.2000135. [DOI] [PubMed] [Google Scholar]
- 15.Chen HH, Zhou XL, Shi YL, Yang J. Roles of p38 MAPK and JNK in TGF-beta1-induced human alveolar epithelial to mesenchymal transition. Arch Med Res. (2013);44:93–98. doi: 10.1016/j.arcmed.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Martin TA, Watkins G, Mansel RE, Jiang WG. Loss of tight junction plaque molecules in breast cancer tissues is associated with a poor prognosis in patients with breast cancer. Eur J Cancer. (2004);40:2717–2725. doi: 10.1016/j.ejca.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Hoover KB, Liao SY, Bryant PJ. Loss of the tight junction MAGUK ZO-1 in breast cancer: relationship to glandular differentiation and loss of heterozygosity. Am J Pathol. (1998);153:1767–1773. doi: 10.1016/S0002-9440(10)65691-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaihara T, Kawamata H, Imura J, et al. Redifferentiation and ZO-1 reexpression in liver-metastasized colorectal cancer: possible association with epidermal growth factor receptor-induced tyrosine phosphorylation of ZO-1. Cancer Sci. (2003);94:166–172. doi: 10.1111/j.1349-7006.2003.tb01414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleeff J, Shi X, Bode HP, et al. Altered expression and localization of the tight junction protein ZO-1 in primary and metastatic pancreatic cancer. Pancreas. (2001);23:259–265. doi: 10.1097/00006676-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Ni S, Xu L, Huang J, et al. Increased ZO-1 expression predicts valuable prognosis in non-small cell lung cancer. Int J Clin Exp Pathol. (2013);6:2887–2895. [PMC free article] [PubMed] [Google Scholar]
- 21.Smalley KS, Brafford P, Haass NK, Brandner JM, Brown E, Herlyn M. Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am J Pathol. (2005);166:1541–1554. doi: 10.1016/S0002-9440(10)62370-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park MK, You HJ, Lee HJ, et al. Transglutaminase-2 induces N-cadherin expression in TGF-beta1-induced epithelial mesenchymal transition via c-Jun-N-terminal kinase activation by protein phosphatase 2A down-regulation. Eur J Cancer. (2013);49:1692–1705. doi: 10.1016/j.ejca.2012.11.036. [DOI] [PubMed] [Google Scholar]
- 23.Brown KA, Aakre ME, Gorska AE, et al. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. (2004);6:R215–R231. doi: 10.1186/bcr778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halder SK, Beauchamp RD, Datta PK. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia. (2005);7:509–521. doi: 10.1593/neo.04640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiraga R, Kato M, Miyagawa S, Kamata T. Nox4-derived ROS signaling contributes to TGF-beta-induced epithelial-mesenchymal transition in pancreatic cancer cells. Anticancer Res. (2013);33:4431–4438. [PubMed] [Google Scholar]
- 26.Balda MS, Matter K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. (2000);19:2024–2033. doi: 10.1093/emboj/19.9.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brysse A, Mestdagt M, Polette M, et al. Regulation of CXCL8/IL-8 expression by zonula occludens-1 in human breast cancer cells. Mol Cancer Res. (2012);10:121–132. doi: 10.1158/1541-7786.MCR-11-0180. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, Lim SB, Ng MY, et al. ZO-1 regulates Erk, Smad1/5/8, Smad2, and RhoA activities to modulate self-renewal and differentiation of mouse embryonic stem cells. Stem Cells. (2012);30:1885–1900. doi: 10.1002/stem.1172. [DOI] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. (2001);25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.You HJ, Lee JW, Yoo YJ, Kim JH. A pathway involving protein kinase Cdelta up-regulates cytosolic phospholipase A(2)alpha in airway epithelium. Biochem Biophys Res Commun. (2004);321:657–664. doi: 10.1016/j.bbrc.2004.07.022. [DOI] [PubMed] [Google Scholar]