

ABSTRACT
Bacterial toxin-antitoxin systems play a critical role in the regulation of gene expression, leading to developmental changes, reversible dormancy, and cell death. Type II toxin-antitoxin pairs, composed of protein toxins and antitoxins, exist in nearly all bacteria and are classified into six groups on the basis of the structure of the toxins. The VapBC group comprises the most common type II system and, like other toxin-antitoxin systems, functions to elicit dormancy by inhibiting protein synthesis. Activation of toxin function requires protease degradation of the VapB antitoxin, which frees the VapC toxin from the VapBC complex, allowing it to hydrolyze the RNAs required for translation. Generally, type II antitoxins bind with high specificity to their cognate toxins via a toxin-binding domain and endow the complex with DNA-binding specificity via a DNA-binding domain. Despite the ubiquity of VapBC systems and their critical role in the regulation of gene expression, few functional studies have addressed the details of VapB-VapC interactions. Here we report on the results of experiments designed to identify molecular determinants of the specificity of the Mycobacterium tuberculosis VapB4 antitoxin for its cognate VapC4 toxin. The results identify the minimal domain of VapB4 required for this interaction as well as the amino acid side chains required for binding to VapC4. These findings have important implications for the evolution of VapBC toxin-antitoxin systems and their potential as targets of small-molecule protein-protein interaction inhibitors.
IMPORTANCE VapBC toxin-antitoxin pairs are the most widespread type II toxin-antitoxin systems in bacteria, where they are thought to play key roles in stress-induced dormancy and the formation of persisters. The VapB antitoxins are critical to these processes because they inhibit the activity of the toxins and provide the DNA-binding specificity that controls the synthesis of both proteins. Despite the importance of VapB antitoxins and the existence of several VapBC crystal structures, little is known about their functional features in vivo. Here we report the findings of the first comprehensive structure-function analysis of a VapB toxin. The results identify the minimal toxin-binding domain, its modular antitoxin function, and the specific amino acid side chains required for its activity.
INTRODUCTION
Toxin-antitoxin (TA) systems were first identified to be addictive genetic elements responsible for the maintenance of plasmid stability during cell segregation (1, 2). Subsequently, chromosomally encoded TA systems were characterized to be involved in several mechanisms of toxin action (3, 4). TA systems typically consist of two elements, a toxin protein that suppresses cell growth by targeting essential cellular metabolic pathways and an antitoxin RNA or protein that binds to and blocks the growth inhibition activity of the toxin. Different types of TA systems have been defined according to the nature and modes of action of antitoxins, and of these, the type II TA systems are the most abundant and well studied (5, 6). The type II toxins-antitoxins are typically encoded in the same operon, where the gene for the antitoxin is located upstream of that for the toxin, except in the case of higBA, which has a reversed gene order (7). The type II antitoxin is a protein that directly interacts with its cognate toxin, resulting in neutralization of toxicity. The antitoxin or the toxin-antitoxin protein complex often acts as a repressor that autoregulates the transcription of TA operons via its ability to bind to the promoter (8). During cellular stress, the antitoxin is hydrolyzed by a protease, such as Lon or Clp, to release the toxin from the protein complex and derepress transcription of the TA operon (7). These properties allow TA systems to play regulatory roles in bacterial gene expression and the establishment and maintenance of dormancy. This nonreplicative state may contribute to latency.
Type II TA systems are classified into several different families on the basis of sequence similarities and the functions of the toxins (7–9). CcdB and ParE inhibit DNA replication by targeting DNA gyrase (10, 11). Doc arrests translation elongation by phosphorylation of elongation factor Tu (12, 13). Kis and HicA cleave mRNA in a ribosome-independent manner (14, 15). MazF targets various RNAs, including mRNA, 16S rRNA, and 23S rRNA (16–19). RelE inhibits translation by cleavage of ribosome-bound mRNAs at the ribosomal A site (20). HipA inhibits translation by phosphorylation of glutamyl-tRNA synthetase (21). VapC toxins from some enteric bacteria cleave the initiator tRNAfMet (22). The functions of a number of VapC toxins from Mycobacterium tuberculosis have been reported. VapC20 (Rv2549c) cleaves 23S rRNA, while VapC1 (Rv0065) and VapC29 (Rv0617) cut single-stranded RNAs in GC-rich sequences (23, 24) and VapC4 (Rv0595c) appears to inhibit translation by binding to mRNAs (25).
In most characterized cases, the type II antitoxins contain two distinct motifs: a DNA-binding motif in the N-terminal region that is responsible for autoregulation of the TA operon and an antitoxin motif in the C-terminal region that binds to and inactivates the toxin activity (26). The DNA-binding motifs in the N-terminal region of the type II antitoxins are classified into at least four classes, including helix-turn-helix (HTH), ribbon-helix-helix (RHH), looped-hinge-helix (AbrB), and Phd/YefM (7). Studies of the antitoxins MazE and Phd indicated that mutations in amino acid residues in the N-terminal region of the antitoxins disrupt their DNA-binding ability, and mutations in amino acid residues in the C-terminal region result in the loss of their antitoxin activity (27, 28).
VapBC is the largest family of the type II TA systems and is defined by the presence of a putative endoribonuclease PIN domain. The PIN domain, a small protein domain consisting of about 100 amino acids, is found in a wide range of prokaryotes and eukaryotes, where it functions as an endoribonuclease involved in pre-rRNA processing, nonsense-mediated mRNA decay, and RNA interference pathways (29–31). The PIN domain contains four conserved negatively charged amino acids that are essential for its endoribonuclease activity. The majority of PIN domain proteins in prokaryotes are thought to be the toxic components in TA operons (32). The analysis of the crystal structure of the VapBC TA complex from Shigella flexneri suggests that 4 aromatic residues in the C-terminal domain of VapB (Trp47, Trp50, Phe51, and Phe60) contact the hydrophobic core of VapC, and 2 residues (Arg64 and Gln66) interact with the conserved negatively charged amino acid residues of the PIN domain (33). Similarly, the crystal structures of VapBC complexes from M. tuberculosis, Neisseria gonorrhoeae, and Rickettsia felis suggest that multiple contacts govern the interactions between the VapB antitoxins and their cognate VapC toxins (34–38). These structures raise the question of how many protein-protein contacts are required for stable VapBC interaction and whether binding is likely to be sensitive to small-molecule protein-protein interaction inhibitors. However, the structural requirements for VapBC toxin-antitoxin interactions have not been systematically tested in vivo. This study addressed the requirements for a VapB antitoxin to specifically interact with and inhibit its cognate toxin, VapC. The experiments identified the minimal domain of M. tuberculosis VapB4 required for this interaction as well as the amino acid side chains required for binding to VapC4. These findings are discussed in regard to the evolution of VapBC toxin-antitoxin systems and their potential as targets of small-molecule protein-protein interaction inhibitors.
MATERIALS AND METHODS
Bacterial strains and growth media.
Escherichia coli LMG194 [F− ΔlacX74 galE thi rpsL ΔphoA (PvuII) Δara714 leu::Tn10] carrying the plasmids indicated below was grown at 37°C in M9 medium supplemented with 0.2% glycerol, 0.2% Casamino Acids, 1 mM thiamine, and appropriate antibiotics. E. coli TOP10 [F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80dlacZΔM15 ΔlacX74 deoR recA1 araD139 Δ(araA-leu)7697 galU galK rpsL endA1 nupG] was grown at 37°C in Luria-Bertani (LB) medium supplemented with antibiotics appropriate for plasmid preparation.
Plasmid construction. (i) pJSB31-sfGFP.
The superfolder green fluorescent protein (sfGFP)-encoding DNA was amplified by PCR using primers OSB1233 and OSB1234 from the SuperFolder GFP expression plasmid (Sandia Biotech). The PCR product was digested with BglII and KpnI and cloned into the same sites of plasmid pJSB31. The pJSB31-sfGFP plasmid was constructed with 6× glycine linkers at the N termini of the superfolder GFP.
(ii) pBAD-VapC4.
VapC4 (Rv0595c)-encoding DNA was PCR amplified using primers OSB863 and OSB864 from M. tuberculosis H37Rv genomic DNA. The PCR product was digested with NcoI and XbaI and ligated into the corresponding sites of pBAD/myc-His B (Invitrogen). The resulting plasmid expresses a C-terminal myc-His-tagged VapC4 in the presence of l-arabinose.
(iii) pJSB31-VapB4-sfGFP.
VapB4 (Rv0596c)-encoding DNA was PCR amplified using primers OSB874 and OSB1235 from M. tuberculosis H37Rv genomic DNA. The resulting PCR product was digested with NcoI and BglII and cloned into the corresponding sites of pJSB31-sfGFP. The resulting plasmid expresses C-terminal sfGFP-tagged VapB4 in the presence of IPTG (isopropyl-β-d-thiogalactopyranoside).
All the plasmids were verified by DNA sequencing analysis.
Serial dilution cell spotting assay.
LMG194 cells were grown at 37°C overnight. The saturated cultures were diluted in sterile water to an optical density at 600 nm (OD600) of 0.02 and then diluted to 2 × 10−3, 2 × 10−4, and 2 × 10−5. An aliquot of 5 μl of each dilution was pipetted onto M9-glycerol (0.2%) medium supplemented with 100 μg/ml ampicillin and 30 μg/ml chloramphenicol in the presence or absence of 0.02% l-arabinose and/or 500 μM IPTG and grown for 16 h at 37°C.
Western blot analysis.
LMG194 cells carrying the indicated plasmids were grown at 37°C to an A600 of 0.3 to 0.5 and induced by the addition of appropriate inducers for 30 min. One milliliter of each cell culture was removed, and the cells were collected by centrifugation at 16,000 × g for 2 min at room temperature. The cell pellet was resuspended in 10 μl of 1× SDS loading buffer (10% SDS, 10 mM 2-mercaptoethanol, 20% glycerol, 0.05% bromophenol blue, 0.2 M Tris-HCl, pH 6.8) and boiled for 5 min. Insoluble material was pelleted by centrifugation at 16,000 × g for 10 min at room temperature. Ten microliters of the soluble proteins in the sample was separated by electrophoresis on a 12.5% SDS-polyacrylamide gel. Proteins were transferred onto a nitrocellulose membrane (Bio-Rad) by electroblotting at 90 mA overnight at 4°C. The proteins were detected using anti-c-myc monoclonal antibody (1:5,000; Clontech) for myc-tagged protein and anti-GFP monoclonal antibody (1:1,000; Roche) for sfGFP-tagged protein. Primary antibodies were detected with anti-mouse IgG (goat) antibody (1:5,000; PerkinElmer) and developed by a Western Lightning Plus ECL system (PerkinElmer). GroEL served as a loading control for E. coli, was detected by anti-GroEL antibody (1:10,000; Sigma-Aldrich) and anti-rabbit IgG (goat) secondary antibody (1:5,000; PerkinElmer), and was developed by a Western Lightning Plus ECL system (PerkinElmer).
Fluorescence assay.
LMG194 cells carrying the indicated plasmids were grown at 37°C to an A600 of 0.1 to 0.2 and induced by the addition of appropriate inducers for 2 h. Two hundred microliters of each culture was added to the wells in a fluorescence-transparent microtiter plate (Costar 3370). The absorbance in each well was measured at 600 nm using a PowerWave XS microplate reader (BioTek). The GFP fluorescence of each culture was analyzed on a Typhoon 9410 imager (GE Biosciences) using a 488-nm blue laser for excitation and a 520BP40 emission filter.
Protein copurification assay.
LMG194 cells carrying the indicated plasmids were grown at 37°C to an A600 of 0.3 to 0.5 and induced by the addition of appropriate inducers for 30 min. Fifteen A600 units of cells was removed, and the cells were collected by centrifugation at 12,000 × g for 10 min at 4°C. The cell pellet was resuspended in 5 ml of lysis buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 10 mM imidazole, 20 mM β-mercaptoethanol, 20 μl bacterial protease inhibitors cocktail [Sigma]). The cells were broken by two passages through a French pressure cell (Thermo Spectronic) at 14,000 lb/in2. The soluble fraction of 1 ml of cell lysate was clarified by centrifugation at 16,000 × g for 10 min at room temperature. The supernatant was transferred into a 1.5-ml tube containing 100 μl of Ni-nitrilotriacetic acid-agarose resin (Qiagen) that had been preequilibrated in wash buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 10 mM imidazole, 20 mM β-mercaptoethanol). The suspension was nutated at room temperature for 1 h. The resin was collected by centrifugation at 16,000 × g for 1 min at room temperature and washed 3 times with wash buffer. The purified proteins were eluted by the addition of 100 μl of elution buffer (50 mM NaH2PO4 [pH 8.0], 300 mM NaCl, 250 mM imidazole, 20 mM β-mercaptoethanol). Five microliters of cell lysate or purified protein from each strain was separated by electrophoresis on a 12.5% SDS-polyacrylamide gel, and the proteins were visualized by Western blotting.
Construction of mutants by overlap PCR.
Double mutants containing mutations in the C-terminal and N-terminal domains of VapB4 were created by overlap PCR amplification in 50 μl of a reaction mixture containing 10 ng DNA template, 0.2 mM deoxynucleoside triphosphates (dNTPs), 0.2 μM each required primer, and 1 unit of iProof polymerase (Bio-Rad) in the reaction buffer supplied with the polymerase. The PCR conditions were 1 cycle of 98°C for 3 min and then 30 cycles of 98°C for 10 s, 60°C for 30 s, and 72°C for 30 s, followed by one cycle of a final extension step at 72°C for 5 min. The PCR products were purified using a PCR purification kit (Qiagen) and served as reverse primers for the second PCRs. The second PCRs were performed using the same conditions used for the first PCRs in a 50-μl reaction mixture containing 10 ng DNA template, 0.2 mM dNTPs, 0.2 μM forward primer, 0.2 μM the first PCR product, and 1 unit of iProof polymerase (Bio-Rad) in the supplied reaction buffer.
Site-directed mutagenesis.
Oligonucleotide-directed site-specific mutagenesis was carried out by a modification of the method of Fisher and Pei (39). The template plasmid was amplified in 50 μl of a reaction mixture containing 10 ng DNA template, 0.2 mM dNTPs, 0.2 μM each primer with the appropriate base changes, and 1 unit of iProof polymerase (Bio-Rad) in the reaction buffer supplied with the polymerase. PCR conditions consisted of one cycle of 98°C for 3 min and then 30 cycles of 98°C for 30 s, 60°C for 30 s, and 72°C for 2 min, followed by one cycle of a final extension step at 72°C for 5 min. The PCR product was digested with 5 units of DpnI at 37°C for 1 h. E. coli TOP10 cells were transformed with 5 μl of the DpnI-treated PCR product. DNA sequencing analysis verified all the constructs.
Genetic screen for antitoxin mutants.
Antitoxin mutations were generated by error-prone PCR. The template plasmid was amplified in 50 μl of a reaction mixture containing 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 0.2 mM dNTPs, 0.2 μM each primer, 3.5 mM MgCl2, 0.5 mM MnCl2, 40 μM dGTP, 0.1 unit of Taq polymerase, and 10 ng DNA template. PCR conditions consisted of one cycle of 94°C for 3 min and 30 cycles of 94°C for 30 s, 55°C for 30 s, and 68°C for 30 s, followed by one cycle of a final extension step at 68°C for 5 min.
The PCR products were digested with NcoI and BglII, and 100 ng was ligated to 1 μg of pJSB31-sfGFP plasmid cut at the same sites. One hundred microliters of LMG194 competent cells carrying pBAD-VapC4 was transformed with 5 μl of the ligation mixture, plated on LB medium supplemented with 100 ng/μl of ampicillin and 50 ng/μl of chloramphenicol, and then grown at 37°C overnight. The transformants were replica plated onto M9 minimal medium supplemented with the same antibiotics, 0.2% glycerol, and 500 μM IPTG or 500 μM IPTG plus 0.02% l-arabinose and grown at 37°C overnight. Colonies that grew only on the plate with IPTG but not on the plate with IPTG and l-arabinose were streaked on the new M9 medium plates with IPTG or IPTG plus l-arabinose to confirm the phenotype. Colony GFP fluorescence was analyzed on a Typhoon 9410 imager (GE Biosciences) using a 488-nm blue laser for excitation and a 520BP40 emission filter. Strains that expressed green fluorescent protein and grew only on the M9 medium with IPTG and not on IPTG plus l-arabinose were selected. The size of the antitoxin-encoding DNA fragment in the selected strains was analyzed by PCR. The DNA sequence of the VapB4 portion of the plasmids was analyzed by DNA sequencing.
RESULTS
Identification of the minimal antitoxin domain of VapB4.
We began our assessment of the specificity determinants of VapB4 (Rv0596c) by determining the amino acid sequences necessary to counteract the toxicity of its cognate toxin, VapC4, in vivo. To facilitate the detection and analysis of VapB4 mutants, we measured antitoxin activity in the context of C-terminal fusions to superfolder GFP (sfGFP) after induction of expression from the tac promoter by IPTG (Fig. 1A). VapC4 was produced by induction of expression of VapC4 from the ara promoter on a compatible plasmid by addition of l-arabinose. Our data showed that in this study 500 μM IPTG and 0.02% l-arabinose were ideal concentrations for expression of the antitoxin and toxin, respectively (data not shown). Growth of the E. coli cells carrying the indicated plasmids was assessed by spotting 10-fold dilutions of cells on the appropriate medium, as described in Materials and Methods. Expression of VapC4 inhibited cell growth, as expected (Fig. 1B). Coinduction of VapB4 or VapB4-sfGFP inhibited the toxicity of VapC4. These results indicate that the antitoxin VapB4 functions to reverse the toxicity of VapC4 equally well with or without the sfGFP fusion (Fig. 1B).
FIG 1.

Expression of the VapB4 (Rv0596c)-myc-6× His or VapB4-sfGFP fusion in trans inhibits the toxicity of VapC4 (Rv0595c). (A) The open reading frame of VapB4 from M. tuberculosis was cloned into the expression plasmid pJSB31 or pJSB31-sfGFP, creating a myc-6× His fusion or a sfGFP fusion, respectively. (B) The growth of E. coli strains carrying the indicated plasmids was compared using the serial dilution cell spotting assay, as described in Materials and Methods.
We sought to identify the amino acid residues required for the ability of VapB4 to reverse the toxicity of VapC4 in vivo. We screened for VapB4 loss-of-function mutations after error-prone PCR amplification of the gene and expression as described above. Analysis of five mutants identified growth defects caused only by multiple mutations (data not shown). As an alternative strategy, we sought the minimal portion of VapB4 necessary for antitoxin activity by expressing N- and C-terminal deletions of VapB4-sfGFP in the presence of VapC4 (Fig. 2A). Induction of expression of the wild type and the VapB4 deletion mutant revealed that removal of the C-terminal 31 amino acids of VapB4 abolished antitoxin activity (Fig. 2B). In contrast, deletion of the N-terminal 54 amino acids of VapB4 (VapB4 Δ54) had little effect on its antitoxin activity. Measurement of the growth rates of theses mutants in culture confirmed the results of the plate assays. Western blot analysis of the VapB4 and VapC4 proteins in these cells revealed similar levels of expression, except for VapB4 Δ40, which was not stably expressed and had no antitoxin activity (Fig. 2B and C). Similarly, quantification of each of the mutant proteins in vivo by measuring GFP fluorescence confirmed that all but VapB4 Δ40 existed at similar levels in the cells induced for expression of the VapB and VapC proteins (Fig. 2D). We note that deletions to positions 34 and 47 also resulted in polypeptides that did not accumulate in vivo, presumably due to protein instability. We conclude that amino acids 55 to 85 of VapB4 are sufficient for VapB4 antitoxin activity in vivo.
FIG 2.

The C-terminal region of VapB4 (positions 55 to 85) is sufficient for VapB4 antitoxin activity. (A) The open reading frames of VapB4 and its deletion mutants were cloned into the pJSB31-sfGFP plasmid, creating sfGFP fusions. (B) Spotting assay for VapB4 deletion mutants carried out as described in the legend to Fig. 1B. (C) Western blot analysis of cells carrying pJSB31-sfGFP (vector) or pJSB31 carrying the indicated VapB4 alleles and pBAD-VapC4. The cells were grown in M9-glycerol (0.2%) medium at 37°C and induced with 0.02% l-arabinose and 500 μM IPTG for 30 min. Blots were probed with anti-GFP antibody for VapB4 or its mutant proteins and anti-myc antibody for VapC4-myc proteins. Anti-GroEL served as a loading control. The four lanes in each of the panels on the left show a 2-fold dilution series of VapB4-sfGFP, which served as a control for signal linearity. (D) E. coli strain LMG194 carrying the indicated plasmids was grown in M9-glycerol (0.2%) medium supplemented with 50 μg/ml ampicillin and 30 μg/ml chloramphenicol at 37°C to an A600 of 0.1 to 0.2 and induced with 0.02% l-arabinose and 500 μM IPTG for 2 h. GFP fluorescence was measured in a Typhoon 9410 imager and normalized to the OD600 of the culture. Error bars illustrate the standard deviations of triplicate measurements.
VapB5 (Rv0626) is the closest M. tuberculosis homologue to VapB4 (Fig. 3A). The proteins exhibit the most identity and similarity over their first 50 amino acids, likely reflecting their DNA-binding domains, and both belong to the Phd/YefM family of DNA-binding proteins. We switched the DNA-binding domains between the two proteins and tested whether the resultant VapB5-VapB4 and VapB4-VapB5 constructs could function as antitoxins to VapC4 (Fig. 3B and C). We could not test for the effects of these chimeras on VapC5 because it is not toxic when expressed in E. coli. The results showed that VapB5-VapB4 but not VapB4-VapB5 functions as an antitoxin to VapC4 (Fig. 3C). Western blot analysis and fluorescence measurements revealed that cells expressed each of the antitoxin chimeras at similar levels, and neither chimera altered the expression of VapC4 (Fig. 3D and E). Importantly, the wild-type VapB4 and the VapB5-VapB4 construct but not the VapB4-VapB5 construct copurified with VapC4-myc-6× His from lysates of cells expressing the proteins (Fig. 3F). Thus, the VapB4 C-terminal domain functions when fused to either an N-terminal (VapB5) or a C-terminal (sfGFP) heterologous domain. These findings indicate that the C-terminal 31 amino acids of VapB4 are necessary and sufficient for its antitoxin function in vivo.
FIG 3.

VapB5-VapB4 but not VapB4-VapB5 functions as an antitoxin to VapC4. (A) ClustalW alignment of VapB4 (Rv0596c) with the VapB5 (Rv0626) sequence. Colons, nonidentical residues; periods, functionally similar residues; asterisks, identical residues. (B) The open reading frames of VapB4, VapB5, and their domain-swapped mutants were cloned into the expression plasmid pJSB31-sfGFP, creating sfGFP fusions. (C) Spotting assay for E. coli strain LMG194 carrying the indicated plasmids carried out as described in the legend to Fig. 1B. (D) Western blot analysis was performed as described in the legend to Fig. 2C. (E) Fluorescence measurements for VapB4, VapB5, and their domain-swapped mutants were performed as described in the legend to Fig. 2D. (F) Western blot analysis of copurified proteins from E. coli LMG194 cells carrying plasmids expressing the indicated VapB combinations and VapC4-myc-6× His. Cells were induced with 0.02% l-arabinose and 500 μM IPTG for 30 min at 37°C. Western blots were probed with anti-GFP antibody for VapB4 or its mutant proteins and anti-myc antibody for VapC4 proteins.
Identification of mutations that disrupt VapB4 antitoxin activity.
Next, we asked if single amino acid changes could abolish the antitoxin activity of VapB4. PCR mutagenesis of the VapB4 portion of VapB4 Δ54-sfGFP yielded loss-of-function mutations identifiable by screening for cells that grew upon induction of VapB4 Δ54-sfGFP with IPTG but not upon coinduction of VapC4 with arabinose (see Fig. S1 in the supplemental material). Secondary screening for GFP fluorescence eliminated VapB4 Δ54-sfGFP mutants with nonsense mutations, as these should not produce full-length, fluorescent fusion proteins. This method identified 28 single, 15 double, and 4 triple missense alleles that disrupted VapB4 Δ54-sfGFP antitoxin activity (Table 1). Figure 4A and B show the phenotypes and positions of each of the single mutations. All of the VapB4 proteins with single mutations were expressed at levels similar to those at which they were expressed by the wild type and did not decrease the levels of the VapC4 toxin (Fig. 4C; compare lanes 6 and 7). The single mutations occurred at 14 positions in VapB4 (Fig. 4B). The occurrence of multiple alleles at nine of these positions suggests that the method identified the side chains that are the most important for the function of the antitoxin. Eight of these sites (D64, L68, E71, Q78, T79, T80, D81, and D82) were conserved between VapB4 and VapB5, suggesting conservation of antitoxin function. Six positions in VapB4 (I55, V59, L61, G62, T66, and E74) were not conserved between the two antitoxins, yet they were necessary for VapB4 antitoxin function. One of the conserved positions, D64, was changed to N or E. To determine if the effects at this position resulted from the loss of the side chain or the inhibitory effects of the mutant side chains, we changed D64 to alanine in the context of the C-terminal domain. This mutation also led to the loss of the antitoxin function without changing its expression level, thereby supporting our conclusion that position 64 requires aspartate for full antitoxin function (Fig. 4D and E).
TABLE 1.
VapB4 Δ54 antitoxin loss-of-function mutations
| Type | Mutations |
|---|---|
| Single | D82G, L61F, T66A, T66P, L68Q, D81G, T80S, T80K, L61T, L68P, Q78R, I55V, T80K, D82N, T66S, D81A, Q78R, D64N, T80M, G62S, D64E, E71A, E74V, D64E, V59E, D64N, T79A, I55V |
| Double | T80P, R84Q; R60H, D64V; R73C, T75S; N67D, T79A; E74G, T75S; T66I, T75S; T65N, L76Q; N67D, V83A; T75I, L68P; D64G, L76Q; P63L, E71G; Q78H, T84G; Q78R, D82G; R60H, D81G; R60C, N67Y |
| Triple | E70D, Q78R, T80S; I55F, L76Q, T77M; P63H, L72P, D82E; G56C, T79I, V83A |
FIG 4.

Single mutations disrupt the antitoxin activity of VapB4 Δ54. (A) Spotting assay for E. coli strain LMG194 carrying the indicated plasmids carried out as described in the legend to Fig. 1B. (B) Single amino acid changes in the C-terminal 31-amino-acid domain of VapC4 and comparison with the same portion of VapB5. Each amino acid residue listed above VapB4 protein sequence represents a single mutation isolated from the screen. (C) Western blot analysis was carried out as described in the legend to Fig. 2C. The asterisk marks the position of an anti-GFP-positive band. (D) Spotting assay for E. coli strain LMG194 carrying the indicated plasmids carried out as described in the legend to Fig. 1B. (E) Western blot analysis was carried out as described in the legend to Fig. 2C.
Analysis of effect of VapB4 mutations in the context of full-length antitoxin.
Next, we asked if the C-terminal domain mutations affect antitoxin activity in the context of full-length VapB4. Full-length alleles were constructed by overlap PCR, sequenced, and tested for antitoxin activity. However, none of the mutations affected the ability of the full-length antitoxin to counter the toxicity of VapC4 expression (see Fig. S2 in the supplemental material). These findings suggest that some portion of VapB4 outside the terminal 31 amino acids might make contact with portions of VapC4 and that this stabilizes the interaction between the proteins and compensates for the weakened interactions caused by mutations in the C-terminal domain. This suppression also indicates that the mutations in the C-terminal domain do not cause a loss of function by irreversible unfolding of the polypeptide.
To identify the region in VapB4 that suppresses the defects caused by the C-terminal mutations, we carried out PCR mutagenesis on the VapB4 D64A allele and screened for mutants with defective antitoxin function. In addition to the original D64A mutation, the screen yielded single mutations W48R (twice) and V54E, as well as three double mutations and one triple mutation (Fig. 5A). All of the mutants with multiple mutations had changes in a 7-amino-acid region that included W48, L49, E53, and V54, and they included two additional W48R mutations. The D64A, V54E, and W48R mutations by themselves did not cause defects in antitoxin activity in full-length VapB4, and their expression levels were similar to those of wild-type VapB4 (Fig. 5B and C). Mutation of W48 to G alone had no effect on antitoxin activity, but it rendered full-length VapC4 D64A inactive, suggesting that the defect caused by W48R results from the loss of the tryptophan rather than its replacement with a charged side chain (Fig. 5B). In contrast, the V54G mutation had no effect on antitoxin activity alone or in the context of D64A (Fig. 5B). However, reducing the level of VapB4 V54G D64A expression by lowering the amount of the inducer IPTG to 100 μM caused the mutant antitoxin to function poorly compared to the wild type (Fig. 5D). Consistent with these findings, VapB4 V54G D64A copurified with VapC4-myc-6× His less efficiently than wild-type VapB4 did (Fig. 5E). Neither VapB4 L49S D64A nor VapB4 E53G D64A disrupted antitoxin activity, suggesting that the defect seen with the original L49S and E53G isolates reflects the contribution of the other mutations (Fig. 5A and E). These findings support our conclusion that W48 and V54 of VapB4 play specific roles in stabilizing the interaction between the antitoxin and VapC4.
FIG 5.
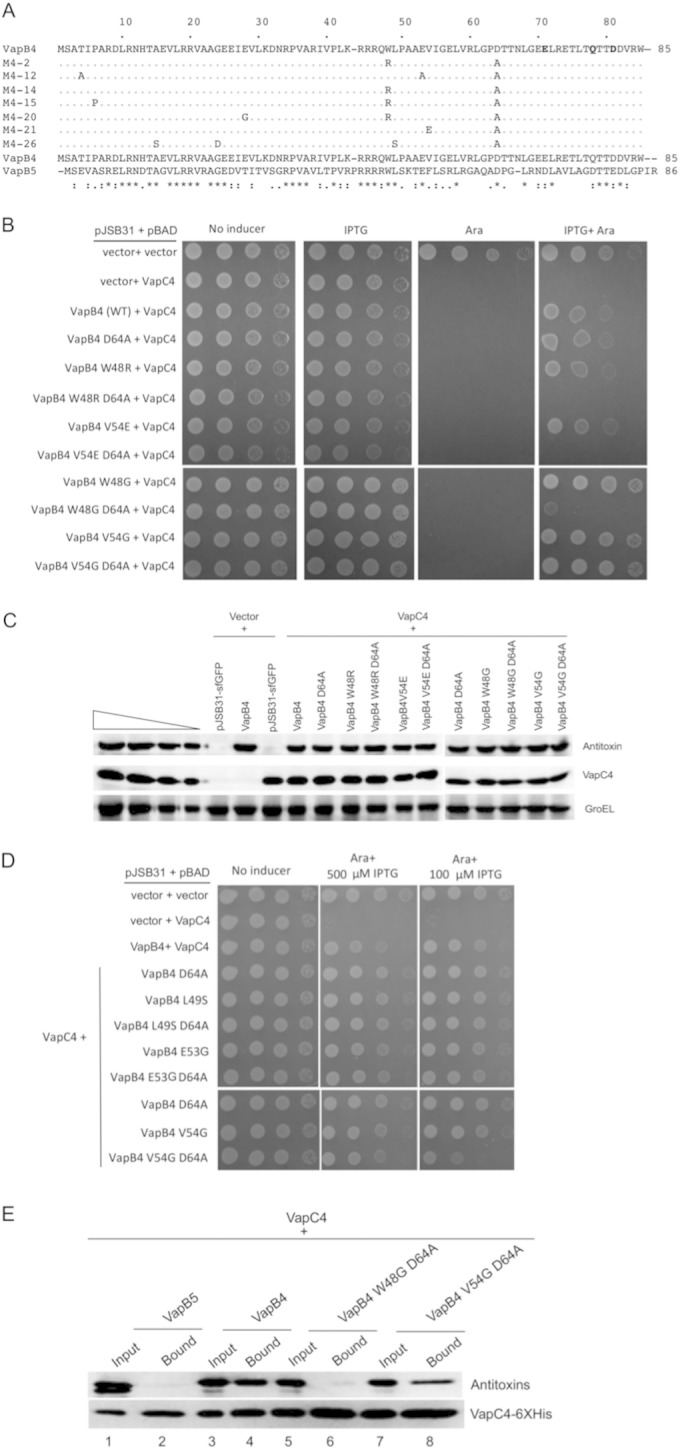
Mutation of W48 results in the loss of antitoxin activity of full-length VapC4 D64A. (A) Comparison of the sequences of VapB4 with those of the defective mutants isolated from screening. (B) Spotting assay for E. coli strain LMG194 carrying the indicated plasmids carried out as described in the legend to Fig. 1B. (C) Western blot analysis was carried out as described in the legend to Fig. 2C. (D) E. coli strain LMG194 carrying the indicated plasmids was grown and spotted with 10-fold dilutions on M9 medium supplemented with 100 μg/ml ampicillin, 30 μg/ml chloramphenicol, and 0.2% glycerol in the presence or absence of 0.02% l-arabinose and 500 μM (or 100 μM) IPTG and grown for 16 h at 37°C. (E) Western blot analysis of copurified proteins from E. coli LMG194 cells carrying plasmids expressing VapB5 or the indicated VapB4 alleles and VapC4-myc-6× His. Cells were induced with 0.02% l-arabinose and 500 μM IPTG for 30 min at 37°C. Western blots were probed with anti-GFP antibody for VapB4 or its mutant proteins and anti-myc antibody for VapC4 proteins.
The requirement for W48 to stabilize the interaction of the VapB4 D64A mutant suggests that it plays an important role in binding to the toxin. Consistent with this idea, W48 is conserved in the closely related VapB5, which, like VapB4, is a Phd family antitoxin. VapB antitoxins from R. felis and S. flexneri also have tryptophans in similar positions that interact closely with their VapC partners (33, 37). The VapBC5 crystal structure contains 34 amino acids from the C-terminal region of VapB5 which wrap around VapC5 and terminate near the active site (Fig. 6A and B). W48 nestles into a depression on the surface of VapC5 (Fig. 6B). This interaction and the homology between VapC4 and VapC5 suggest that W48 of VapB4 likely plays a similar role in its binding to VapC4. As noted above, we attempted to express a polypeptide extending from W48 to the C terminus, but it did not accumulate in the cell. Instead, we asked if the interaction of VapC4 with other VapB4 C-terminal mutants requires W48. Interestingly, W48G affects the antitoxin activity only of the T66A, L72V, Q78R, and T80S mutants and does so most effectively only when induction of the antitoxins is lowered by the use of 100 μM IPTG, in contrast to the strong effect of W48G on D64A at 500 μM IPTG (Fig. 6C). We asked whether the inability of the W48G mutation to act synergistically with the other mutations might result from the higher levels of expression of these double mutant proteins. Measurement of their GFP fluorescence in the presence of 100 μM IPTG revealed that all of the strains express the mutant antitoxins at concentrations similar to the concentration at which the wild-type antitoxin is expressed (Fig. 6D).
FIG 6.

Mutation of W48 results in the loss of antitoxin activity of full-length VapC4 D64A. (A) Amino acid sequence alignment of VapB4 and VapB5, with their corresponding predicted secondary structure elements assigned. Secondary structure assignments for VapB4 were from the Phyre2 server (43), and those for VapB5 were from reference 37. (B) Structure of the VapB5 (orange)-VapC5 (white) heterodimer (37). The structure with PDB accession number 3DBO was generated by use of the PISA program (44). (C) Spotting assay for E. coli strain LMG194 carrying the indicated plasmids carried out as described in the legend to Fig. 5D. (D) Fluorescence measurements for VapB4 mutants that were assayed for growth and for which the results are presented in panel C. Measurements were taken directly from the third dilution spot on the plate with 100 μM IPTG after determination that all spots in the dilution were in the linear range of detection of the Typhoon 9410 imager. The columns report the average fluorescence intensity from three biological replicates, and error bars illustrate standard deviations. WT, wild type.
DISCUSSION
We began the experiments reported here to determine if single amino acid substitutions could abrogate the ability of a VapB antitoxin to bind and inhibit its cognate toxin. The answer sheds light on two important issues regarding VapB antitoxin function. First, small-molecule inhibitors of the toxin-antitoxin interaction could prove useful for studies of the function of VapBC and other TA systems, as well as antimicrobial drugs (40). However, the strength and number of contacts between the two proteins may limit the potential efficacy of such inhibitors. Second, since single missense mutations that disrupt VapBC pairs would inhibit cell growth, natural selection might favor the evolution of TA pairs for which only multiple changes would destabilize binding. These properties would diminish the likelihood of identifying effective small-molecule inhibitors because they would need to disrupt multiple contacts between the proteins. Initially, we screened for missense alleles of full-length VapB4 that disrupted its ability to inhibit the toxicity of VapC4 but never found mutants with single mutations with this phenotype. However, deletion analysis and a more targeted screen for deleterious mutations identified the minimal C-terminal region necessary for VapB activity and the amino acids required for the ability of the truncated protein to inhibit VapC4. Interestingly, these mutations disrupted antitoxin function only in the absence of the N-terminal 54 amino acids, despite the fact that activity did not require this portion of the antitoxin. We found that the loss of VapB4 activity apparently requires at least two mutations and that the critical contacts do not lie adjacent to one another. This redundancy may protect M. tuberculosis from the possibility that single mutations in VapB4 would lead to growth arrest and, potentially, cell death. It may also limit the spectrum of molecules capable of interfering with the binding of VapB4 to VapC4.
The existing crystal structures of VapBC pairs show that the C-terminal portions of the VapB antitoxins wrap around and bind in grooves on the surface of their VapC partners (33, 35–38). Secondary structure prediction indicates the presence of two α helices (α2 and α3) in the C terminus of VapB4 at positions which match the positions of the two helices that grip VapC5 in the VapBC5 structure (37) (Fig. 6A). Considering the high degree of similarity between VapC4 and VapC5 and between their antitoxins, VapB4 and VapB5, respectively, it is likely that the VapBC4 complex shares some critical features with VapBC5. Comparison of the homology of VapB4 and VapB5 in view of the VapBC5 structure sheds light on the effects of the VapB4 mutations reported here. The fact that the loss of W48 inhibited the activity of many of the C-terminal mutants strongly supports its importance in binding of VapB4 to VapC4. Analysis of 49 annotated M. tuberculosis VapB antitoxins in the UniProt database indicated that W48 exists only in VapB4 and VapB5. However, tryptophan 48 fits into a hydrophobic pocket in VapC5 in the VapBC5 crystal structure (Fig. 6B), and tryptophans at position 47 in VapB antitoxins from R. felis and S. flexneri do so as well with their VapC partners (33, 37). These considerations support our conclusion that W48 plays a critical role in stabilizing the interaction between VapB4 and VapC4.
Our findings also support the conclusion that amino acid side chains in the C-terminal domain of VapB4 represent at least three classes of interactions. First, D64 likely contributes the most of any of the single side chains to the binding to VapC4 since its mutation is the most sensitive to the loss of W48 (Fig. 6C). Second, I55, L58, L61, G62, L68, E71, E74, T79, D81, and D82 each contribute relatively weak interactions, on the basis of the fact that the loss of W48 does not affect VapB4 with mutations at these positions. Third, the fact that the loss of W48 antagonizes antitoxin function upon mutation of T66, L72, Q78, and T80, but only at levels of expression lower than those seen for D64, suggests that these side chains each contribute to antitoxin binding at a level intermediate to the levels for the first two classes (Fig. 6C).
None of the mutations identified in our screen pointed toward a clear requirement for the DNA-binding module of VapB4 in antitoxin function. However, the fact that inclusion of the N-terminal domain suppresses the defect caused by some of the mutations in the C-terminal domain suggests that features in the DNA-binding module might function to stabilize the interaction between the toxin and antitoxin. Indeed, crystal structures of VapBC pairs indicate that dimerization of VapB toxin-antitoxin pairs involves interactions between the DNA-binding modules of the antitoxins (35–38). These contacts certainly contribute to the formation of heteromeric complexes seen in the crystal structures and may help stabilize the interaction of some of the VapB4 mutants with VapC4.
Previous studies on the function of bacterial type II antitoxins revealed the bipartite nature of the proteins, composed of an N-terminal DNA-binding domain and a C-terminal toxin-binding domain (27, 28, 41). Analysis of the properties of the Phd antitoxin from the P1 plasmid addiction module provided the first evidence for the modular organization of protein antitoxins (41). Mutational analysis demonstrated separable structural requirements for the DNA-binding and toxin-binding activities of Phd and revealed that single amino acid substitutions in the antitoxin could disrupt its ability to counter the toxic effects of its cognate toxin, Doc (27). Binding of the E. coli antitoxin MazE to its cognate toxin, MazF, requires a 37-amino-acid portion near the C terminus of MazE, and a double mutation in this region abrogates binding (28). These findings confirmed the predicted modularity of type II antitoxins and support the hypothesis that these proteins evolve by recombination events that trade DNA-binding and toxin-binding modules (27).
The VapBC family of toxin-antitoxin pairs clearly shows evidence of a modular organization of antitoxin functional domains, and it appears that VapB antitoxins employ most of the known DNA-binding motifs found among type II TA systems (42). VapB proteins use these diverse DNA-binding modules for autogenous control of VapBC gene pair transcription. In VapB antitoxins, the DNA-binding domain is coupled to a conserved C-terminal α-helix–spacer–α-helix motif that binds to the conserved structures of the VapC toxins. The findings reported here constitute the first confirmation of this model in vivo. However, the results indicate that, unlike the case for Phd, the VapB4 toxin-binding module employs redundant features required for its stable binding to and inactivation of its cognate toxin, VapC4. Whether this reflects a general property of VapB antitoxins requires further experimentation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants AI082027 and GM099731 (to J.S.B.) and AI073772 (to M.S.P.) from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02508-14.
REFERENCES
- 1.Gerdes K, Molin S. 1986. Partitioning of plasmid R1. Structural and functional analysis of the parA locus. J Mol Biol 190:269–279. [DOI] [PubMed] [Google Scholar]
- 2.Ogura T, Hiraga S. 1983. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A 80:4784–4788. doi: 10.1073/pnas.80.15.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerdes K. 2000. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J Bacteriol 182:561–572. doi: 10.1128/JB.182.3.561-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayes F. 2003. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301:1496–1499. doi: 10.1126/science.1088157. [DOI] [PubMed] [Google Scholar]
- 5.Aakre CD, Phung TN, Huang D, Laub MT. 2013. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol Cell 52:617–628. doi: 10.1016/j.molcel.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unterholzner SJ, Poppenberger B, Rozhon W. 2013. Toxin-antitoxin systems: biology, identification, and application. Mob Genet Elements 3:e26219. doi: 10.4161/mge.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerdes K, Christensen SK, Lobner-Olesen A. 2005. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 8.Bukowski M, Rojowska A, Wladyka B. 2011. Prokaryotic toxin-antitoxin systems—the role in bacterial physiology and application in molecular biology. Acta Biochim Pol 58:1–9. [PubMed] [Google Scholar]
- 9.Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, Van Melderen L. 2011. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res 39:5513–5525. doi: 10.1093/nar/gkr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang Y, Pogliano J, Helinski DR, Konieczny I. 2002. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44:971–979. doi: 10.1046/j.1365-2958.2002.02921.x. [DOI] [PubMed] [Google Scholar]
- 11.Miki T, Park JA, Nagao K, Murayama N, Horiuchi T. 1992. Control of segregation of chromosomal DNA by sex factor F in Escherichia coli. Mutants of DNA gyrase subunit A suppress letD (ccdB) product growth inhibition. J Mol Biol 225:39–52. [DOI] [PubMed] [Google Scholar]
- 12.Castro-Roa D, Garcia-Pino A, De Gieter S, van Nuland NA, Loris R, Zenkin N. 2013. The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat Chem Biol 9:811–817. doi: 10.1038/nchembio.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz JW, Rothenbacher FP, Maehigashi T, Lane WS, Dunham CM, Woychik NA. 2014. Doc toxin is a kinase that inactivates elongation factor Tu. J Biol Chem 289:7788–7798. doi: 10.1074/jbc.M113.544429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jorgensen MG, Pandey DP, Jaskolska M, Gerdes K. 2009. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J Bacteriol 191:1191–1199. doi: 10.1128/JB.01013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz-Gomez AJ, Lemonnier M, Santos-Sierra S, Berzal-Herranz A, Diaz-Orejas R. 2005. RNase/anti-RNase activities of the bacterial parD toxin-antitoxin system. J Bacteriol 187:3151–3157. doi: 10.1128/JB.187.9.3151-3157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schifano JM, Edifor R, Sharp JD, Ouyang M, Konkimalla A, Husson RN, Woychik NA. 2013. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proc Natl Acad Sci U S A 110:8501–8506. doi: 10.1073/pnas.1222031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vesper O, Amitai S, Belitsky M, Byrgazov K, Kaberdina AC, Engelberg-Kulka H, Moll I. 2011. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 147:147–157. doi: 10.1016/j.cell.2011.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Zhang J, Hara H, Kato I, Inouye M. 2005. Insights into the mRNA cleavage mechanism by MazF, an mRNA interferase. J Biol Chem 280:3143–3150. doi: 10.1074/jbc.M411811200. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M. 2003. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell 12:913–923. doi: 10.1016/S1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 20.Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. 2003. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112:131–140. doi: 10.1016/S0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 21.Germain E, Castro-Roa D, Zenkin N, Gerdes K. 2013. Molecular mechanism of bacterial persistence by HipA. Mol Cell 52:248–254. doi: 10.1016/j.molcel.2013.08.045. [DOI] [PubMed] [Google Scholar]
- 22.Winther KS, Gerdes K. 2011. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc Natl Acad Sci U S A 108:7403–7407. doi: 10.1073/pnas.1019587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahidjo BA, Kuhnert D, McKenzie JL, Machowski EE, Gordhan BG, Arcus V, Abrahams GL, Mizrahi V. 2011. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One 6:e21738. doi: 10.1371/journal.pone.0021738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenzie JL, Duyvestyn JM, Smith T, Bendak K, Mackay J, Cursons R, Cook GM, Arcus VL. 2012. Determination of ribonuclease sequence-specificity using Pentaprobes and mass spectrometry. RNA 18:1267–1278. doi: 10.1261/rna.031229.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharp JD, Cruz JW, Raman S, Inouye M, Husson RN, Woychik NA. 2012. Growth and translation inhibition through sequence-specific RNA binding by a Mycobacterium tuberculosis VAPC toxin. J Biol Chem 287:12835–12847. doi: 10.1074/jbc.M112.340109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goeders N, Van Melderen L. 2014. Toxin-antitoxin systems as multilevel interaction systems. Toxins (Basel) 6:304–324. doi: 10.3390/toxins6010304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKinley JE, Magnuson RD. 2005. Characterization of the Phd repressor-antitoxin boundary. J Bacteriol 187:765–770. doi: 10.1128/JB.187.2.765-770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, Zhang Y, Inouye M. 2003. Characterization of the interactions within the mazEF addiction module of Escherichia coli. J Biol Chem 278:32300–32306. doi: 10.1074/jbc.M304767200. [DOI] [PubMed] [Google Scholar]
- 29.Clissold PM, Ponting CP. 2000. PIN domains in nonsense-mediated mRNA decay and RNAi. Curr Biol 10:R888–R890. doi: 10.1016/S0960-9822(00)00858-7. [DOI] [PubMed] [Google Scholar]
- 30.Fatica A, Tollervey D, Dlakic M. 2004. PIN domain of Nob1p is required for D-site cleavage in 20S pre-rRNA. RNA 10:1698–1701. doi: 10.1261/rna.7123504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gatfield D, Unterholzner L, Ciccarelli FD, Bork P, Izaurralde E. 2003. Nonsense-mediated mRNA decay in Drosophila: at the intersection of the yeast and mammalian pathways. EMBO J 22:3960–3970. doi: 10.1093/emboj/cdg371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arcus VL, McKenzie JL, Robson J, Cook GM. 2011. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng Des Sel 24:33–40. doi: 10.1093/protein/gzq081. [DOI] [PubMed] [Google Scholar]
- 33.Dienemann C, Boggild A, Winther KS, Gerdes K, Brodersen DE. 2011. Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. J Mol Biol 414:713–722. doi: 10.1016/j.jmb.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das U, Pogenberg V, Subhramanyam UK, Wilmanns M, Gourinath S, Srinivasan A. 2014. Crystal structure of the VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin-antitoxin assembly. J Struct Biol 188:249–258. doi: 10.1016/j.jsb.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Mate MJ, Vincentelli R, Foos N, Raoult D, Cambillau C, Ortiz-Lombardia M. 2012. Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res 40:3245–3258. doi: 10.1093/nar/gkr1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattison K, Wilbur JS, So M, Brennan RG. 2006. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing PIN domains and ribbon-helix-helix motifs. J Biol Chem 281:37942–37951. doi: 10.1074/jbc.M605198200. [DOI] [PubMed] [Google Scholar]
- 37.Miallau L, Faller M, Chiang J, Arbing M, Guo F, Cascio D, Eisenberg D. 2009. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J Biol Chem 284:276–283. doi: 10.1074/jbc.M805061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min AB, Miallau L, Sawaya MR, Habel J, Cascio D, Eisenberg D. 2012. The crystal structure of the Rv0301-Rv0300 VapBC-3 toxin-antitoxin complex from M. tuberculosis reveals a Mg(2)(+) ion in the active site and a putative RNA-binding site. Protein Sci 21:1754–1767. doi: 10.1002/pro.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fisher CL, Pei GK. 1997. Modification of a PCR-based site-directed mutagenesis method. Biotechniques 23:570–571, 574. [DOI] [PubMed] [Google Scholar]
- 40.Williams JJ, Hergenrother PJ. 2012. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol 20:291–298. doi: 10.1016/j.tim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith JA, Magnuson RD. 2004. Modular organization of the Phd repressor/antitoxin protein. J Bacteriol 186:2692–2698. doi: 10.1128/JB.186.9.2692-2698.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makarova KS, Wolf YI, Koonin EV. 2009. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct 4:19. doi: 10.1186/1745-6150-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 44.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.