

Significance
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a disease that is selective to the right side of the heart and results in heart failure and sudden death. Genetic defects in desmosome components account for approximately 50% of human ARVC cases; in the other 50% of patients, however, the causes remain unknown. We show that inhibitor of apoptosis-stimulating protein of p53 (iASPP) is an important regulator of desmosomes. It interacts with desmoplakin and desmin in cardiomyocytes and regulates desmosome integrity and intermediate filaments. iASPP-deficient mice display pathological features of ARVC and die of sudden death. In human ARVC patients, cardiomyocytes exhibit reduced levels of iASPP at the cell junctions, suggesting that iASPP may be critical in ARVC pathogenesis.
Keywords: ARVC, sudden death, desmosome, iASPP, cell–cell junctions
Abstract
Desmosomes are anchoring junctions that exist in cells that endure physical stress such as cardiac myocytes. The importance of desmosomes in maintaining the homeostasis of the myocardium is underscored by frequent mutations of desmosome components found in human patients and animal models. Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a phenotype caused by mutations in desmosomal components in ∼50% of patients, however, the causes in the remaining 50% of patients still remain unknown. A deficiency of inhibitor of apoptosis-stimulating protein of p53 (iASPP), an evolutionarily conserved inhibitor of p53, caused by spontaneous mutation recently has been associated with a lethal autosomal recessive cardiomyopathy in Poll Hereford calves and Wa3 mice. However, the molecular mechanisms that mediate this putative function of iASPP are completely unknown. Here, we show that iASPP is expressed at intercalated discs in human and mouse postmitotic cardiomyocytes. iASPP interacts with desmoplakin and desmin in cardiomyocytes to maintain the integrity of desmosomes and intermediate filament networks in vitro and in vivo. iASPP deficiency specifically induces right ventricular dilatation in mouse embryos at embryonic day 16.5. iASPP-deficient mice with exon 8 deletion (Ppp1r13lΔ8/Δ8) die of sudden cardiac death, displaying features of ARVC. Intercalated discs in cardiomyocytes from four of six human ARVC cases show reduced or loss of iASPP. ARVC-derived desmoplakin mutants DSP-1-V30M and DSP-1-S299R exhibit weaker binding to iASPP. These data demonstrate that by interacting with desmoplakin and desmin, iASPP is an important regulator of desmosomal function both in vitro and in vivo. This newly identified property of iASPP may provide new molecular insight into the pathogenesis of ARVC.
Desmosomes are anchoring junctions that exist predominantly in cells that endure physical stress such as cardiac myocytes or skin keratinocytes. One of the main functions of desmosomes is to link intermediate filaments to sites of intercellular junctions to form cellular scaffolding that distributes mechanical forces throughout a tissue (1). The importance of desmosomes in maintaining the homeostasis of the myocardium is underscored by frequent mutations of desmosome components found in human patients and animal models suffering from cardiomyopathy. One of the best studied human disorders in which patients harbor mutations in desmosome components is arrhythmogenic right ventricular cardiomyopathy (ARVC) (2). Pathologically, ARVC is characterized by replacement of the myocardium with adipocytes and fibrous deposits; this replacement typically occurs in conjunction with apoptosis (3) and leads to arrhythmia, ventricular failure, and sudden cardiac death (4). Molecular and genetic analyses have shown that mutations in genes encoding several components of desmosomes, such as γ-catenin, plakophilin-2 (PKP2), desmoglein-2, desmocollin-2, and desmoplakin, account for approximately half of human ARVC cases; most of these genes are inherited as an autosomal dominant trait (5). However, the causes of ARVC in the other patients remain unknown (5), suggesting that regulators of desmosomes may contribute to the pathology in this population. Thus, identifying novel regulators of desmosomes is of great interest in improving our understanding of human health and diseases.
Interestingly, a deficiency of inhibitor of apoptosis-stimulating protein of p53 (iASPP), an evolutionarily conserved inhibitor of p53, caused by spontaneous mutation recently has been associated with a lethal autosomal recessive cardiomyopathy in Poll Hereford calves (6, 7) and Wa3 mice (8). However, the molecular mechanisms that mediate this function of iASPP are completely unknown. iASPP belongs to the ASPP family of proteins (ASPP1, ASPP2, and iASPP) that possess a signature sequence of ankyrin repeats, SH3 domain, and proline-rich sequences (9). The C termini of the ASPP family members share high sequence similarity. However, the N terminus of iASPP is different from that of ASPP1 and ASPP2. The ASPP family members originally were identified as binding partners of two transcription factors, p65/NF-κb and p53, in a yeast two-hybrid assay (10, 11). However, little is known about the biological importance of the ASPP-p65/NF-κb interaction. The cocrystal structure of the C terminus of ASPP2 and p53 indicates a strong interaction between the two proteins (12). The best studied function of the ASPP family is regulation of p53’s apoptotic activity (9). In particular, ASPP1 and ASPP2 are common activators of p53 and its family members, p63 and p73 (13, 14). In vivo, ASPP2 is a haploinsufficient tumor suppressor and coactivator of p53 (15). Interestingly, ASPP2 recently was identified as an important regulator of cell polarity and cell proliferation during the development of the central nervous system through its binding to Par3, thus maintaining the integrity of tight/adherens junctions. This function of ASPP2 is mediated by its N terminus independently of p53 (16, 17).
In contrast to ASPP1 and ASPP2, much less is known about the biological importance of iASPP, even though it is the only ASPP family member that exists in Caenorhabditis elegans (Ce-iASPP), and its ability to inhibit p53-mediated apoptosis is conserved from nematodes to humans (9, 18). It remains unclear why iASPP deficiency has a phenotype reminiscent of that seen with a defect in desmosome integrity and in particular why iASPP deficiency causes cardiomyopathy. Here, we identify iASPP as a previously unidentified regulator of desmosomes, and this property of iASPP provides the first molecular explanation (to our knowledge) of how iASPP deficiency can cause cardiomyopathy in Poll Hereford cattle with cardiomyopathy and woolly haircoat (CWH) syndrome and Wa3 mice, suggesting that iASPP could be an ARVC candidate gene.
Results
iASPP Anchors Desmoplakin and Desmin at Intercalated Discs in Postmitotic Cardiomyocytes in Vivo.
To understand why iASPP deficiency causes cardiomyopathy in mice and cattle (7, 8), we investigated the cellular distribution of iASPP protein in human and mouse hearts. Interestingly, iASPP was detected at the intercalated discs of fully differentiated cardiomyocytes obtained from human postmortem tissues from unaffected donors and from iASPP wild-type mice (Fig. 1A). iASPP protein was not detected in iASPPΔ8/Δ8 myocardial sections, confirming the specificity of the staining (Fig. 1A). The structural integrity of the cardiomyocytes is maintained by the intercalated discs, which contain various junctional complexes including desmosomes, adherens junctions, and gap junctions (19). Desmoplakin is a constituent of the desmosomal plaque; it binds the intermediate filament protein desmin and connects the cytoskeleton to the sarcolemmal membrane in myocytes in vivo (20, 21). Interestingly, in iASPPΔ8/Δ8 cardiomyocytes desmin failed to localize at the intercalated discs even though it was readily detected at the locations of Z-discs (Fig. 1B). Quantification analysis confirmed the dramatic reduction in the number of cells with desmin at the intercalated discs of iASPPΔ8/Δ8 cardiomyocytes (bar graph in Fig. 1B; *P < 0.01). This alteration also was observed in 20-wk-old iASPP-deficient hearts (Fig. S1A). Similarly, desmoplakin largely failed to locate at the intercalated discs of iASPP-deficient cardiomyocytes in 12-wk-old heart tissues even though N-cadherin was readily detected, demonstrating the presence of cell–cell junctions (Fig. 1C). Quantification analysis demonstrated a greater than twofold increase in the number of intercalated discs with no visible junctional desmoplakin I and II in iASPPΔ8/Δ8 sections as compared with wild-type control sections (bar graph in Fig. 1C). These defects are unlikely to be caused by a reduction in protein levels, because equal amounts of desmoplakin and desmin were detected using heart lysates derived from iASPPΔ8/Δ8 and wild-type littermates (Fig. 1D). These results demonstrate that loss of iASPP specifically impairs the location of desmin at the intercalated discs, but not the interactions between desmin and sarcomeric proteins at the Z-discs, in cardiomyocytes in vivo.
Fig. 1.

iASPP anchors desmin and desmoplakin at intercalated discs (IDs) in adult cardiomyocytes in vivo. (A) Ventricular myocardial sections obtained from postmortem human subjects and iASPP+/+ and iASPPΔ8/Δ8 adult mouse hearts were stained using anti-iASPP antibodies, pAb-bp18 or LX128.5 as indicated. Specificity of iASPP staining was confirmed by the lack of signal from iASPPΔ8/Δ8 sections. (B) Myocardial cross-sections obtained from 12-wk-old iASPP+/+ and iASPPΔ8/Δ8 hearts were immunostained for desmin. Desmin signal is concentrated at the area composita in control tissue but is absent in iASPPΔ8/Δ8 hearts (arrowheads). The bar graph shows the reduced percentage of cells expressing desmin at the intercalated discs. iASPP+/+ n = 204; iASPPΔ8/Δ8 n = 198. P < 0.01. (C) Double immunofluorescence staining using a monoclonal antibody against desmoplakin and a polyclonal antibody against the adherens junction marker N-cadherin in 12-wk-old iASPP wild-type and deficient mouse myocardial sections. Junctional N-cadherin staining was used as a marker of intercalated discs. We examined an average of 200 cardiomyocytes in each section and compared the number of intercalated discs that express N-cadherin but are negative for desmoplakin in wild-type (n = 3) and iASPPΔ8/Δ8 (n = 3) myocardial sections. The bar graph shows the fold increase of the intercalated discs lacking desmoplakin signal in iASPPΔ8/Δ8 myocardial sections. (D) Protein levels of iASPP, desmoplakin, and desmin were assessed by Western blot analyses in wild-type and iASPPΔ8/Δ8 hearts. GAPDH was used as a loading control. TO-PRO was used as a nuclear stain. (Scale bars in A–C, 20 µm.)
The effect of iASPP deletion on other junctional components of the intercalated discs was analyzed also. Connexin 43 is the most abundant connexin isotype found in the gap junctions of the heart (22). γ-catenin is a component of both desmosome and adherens junctions (23). N-cadherin binds either β-catenin or γ-catenin and is the major component of adherens junctions (24). In 12-wk-old iASPP wild-type and null heart samples, the protein levels of connexin 43, γ-catenin, N-cadherin, and β-catenin were similar. All were readily detected at the polar ends of cardiomyocytes where the intercalated discs locate (Fig. S1B). Quantification analysis also revealed comparable levels of the various junctional proteins (bar graph in Fig. S1B). Interestingly, although connexin 43, γ-catenin, and N-cadherin were all decreased at the junctions in iASPP-null cardiomyocytes derived from 20-wk-old mice, the adherens junctional protein β-catenin remained localized at the intercalated discs (Fig. S1C). PKP2 also can be found in the desmosome, and it is a common target in ARVC patients (25). To investigate the cellular localization of PKP2, immunofluorescence analysis was carried out using a specific monoclonal anti-PKP2 antibody. iASPP depletion does not affect PKP2 localization (Fig. S1D).
The N Terminus of iASPP Interacts with Desmoplakin and Desmin in Cardiomyocytes.
To understand why iASPP is required for desmoplakin and desmin to locate at the intercalated discs of cardiomyocytes, we first investigated whether iASPP could interact with desmoplakin in an immortalized murine cardiomyocyte cell line (HL-1). Using RNAi of desmoplakin or iASPP, we found that the expression levels of the two proteins were not interdependent (Fig. S2A). We then immunoprecipitated iASPP with two different anti-iASPP antibodies, a rabbit polyclonal antibody (pAbiASPPN1) and a mouse monoclonal (LX049.3), which recognize different parts of the protein (Fig. S2B). Desmoplakin I and II coimmunoprecipitated with iASPP in HL-1 cells (Fig. S2C). In extracts from wild-type mouse hearts, desmoplakin I and II as well as iASPP were coimmunoprecipitated with desmin (Fig. 2A). Under the same conditions, iASPP did not interact with other desmosomal components, such as γ-catenin, or with gap and adherens junction components, such as connexin 43, N-cadherin, α-catenin, β-catenin, and p120 (Fig. S2D). These results show that iASPP can interact with desmoplakin and desmin in cardiomyocytes and are in agreement with earlier observations that iASPP deficiency mainly affects desmoplakin and desmin but has minimal effect on other intercalated disc components.
Fig. 2.
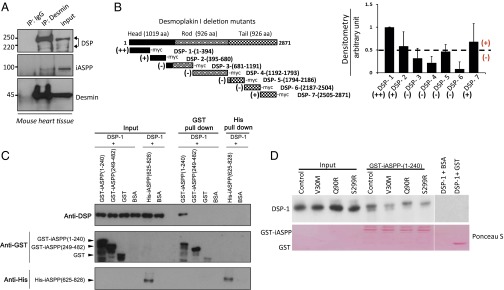
iASPP interacts with desmoplakin and desmin in cardiomyocytes. (A) iASPP interacts with desmoplakin and desmin in adult mouse heart extracts. The coprecipitated desmoplakin (DSP), iASPP, and desmin were detected by blotting with corresponding antibodies as indicated. (B, Left) Schematic representation of different desmoplakin truncated mutants relative to full-length desmoplakin protein and the ability to bind (+), bind very well (++), or not bind (−) iASPP. (Right) The ability of iASPP to bind different desmoplakin truncation constructs was measured using in vitro-translated [35S]methionine-labeled iASPP and different desmoplakin truncation mutants. The signals from three independent experiments were quantified using ImageJ software. The percentage of the different desmoplakin truncated constructs binding to iASPP was normalized against the amount of iASPP immunoprecipitated. Representative immunoblots are shown in Fig. S2E. (C) Binding of recombinant N-terminal GST-iASPP(1–240) and GST-iASPP(249–482) and C-terminal His-iASPP(625–828) to in vitro-translated N-terminal desmoplakin was measured using a pulldown assay as indicated. (D) GST-iASPP(1–240) was pulled down with glutathione beads, and its binding to in vitro-translated ARVC-derived desmoplakin mutants DSP-1(1–394)-V30M, DSP-1(1–394)-Q90R, and DSP-1(1–394)-S299R was assessed by probing with a desmoplakin antibody. Ponceau S staining shows similar protein loading in the samples.
The region of desmoplakin that interacts with iASPP was mapped using truncation mutants of desmoplakin synthesized using an in vitro transcription-coupled translation system in the presence of [35S]methionine to label the mutant proteins (Fig. 2B). iASPP and desmoplakin mutants then were coimmunoprecipitated using anti-iASPP antibody as indicated. iASPP interacts mainly with the 394 N-terminal amino acids of desmoplakin, DSP-1(1–394). Occasional interactions with desmoplakin mutants DSP-2(395–680) and DSP-7(2505–2871) were detected (Fig. 2B and Fig. S2 E and F). Binding between three recombinant iASPP mutants, GST-iASPP(1–240), GST-iASPP(249–482), and His-iASPP(625–828) with in vitro-translated DSP-1(1–394) was assessed. Fig. 2C shows that DSP-1(1–394) interacts mainly with GST-iASPP(1–240) but not with iASPP(249–482) or iASPP(625–828) mutants in pulldown assays. Consistent with this finding, DSP-1(1–394) preferentially interacts with iASPP but not with the other ASPP family members, ASPP1 and ASPP2, which share sequence similarity only with the C terminus of iASPP (Fig. S2G). The interaction between the iASPP N terminus and DSP-1(1–394) suggests that iASPP may not compete with desmin to interact with the C-terminal part of desmoplakin (26).
PKP2 is known to interact with the N-terminal amino acids 1–584 of desmoplakin (27); thus, the ability of iASPP to compete with PKP2 to interact with DSP-1(1–394) was tested. As expected, PKP2 interacted with DSP-1(1–394), and this interaction was not affected by the presence of iASPP (Fig. S2H), suggesting that iASPP may regulate desmoplakin function independently of PKP2. A number of ARVC-derived desmoplakin mutations, including V30M, Q90R (28), and S299R (29), are located within the iASPP-interacting region of desmoplakin. ARVC-derived mutations were introduced into DSP-1(1–394) using in situ mutagenesis to generate DSP-1(1–394)-V30M, DSP-1(1–394)-Q90R, and DSP-1(1–394)-S299R mutants. Recombinant GST-iASPP(1–240) and in vitro-translated DSP-1(1–394) or its mutants were pulled down using glutathione beads. ARVC derivate DSP-1(1–394) mutants V30M and S299R, but not Q90R, showed a reduced binding to iASPP(1–240) (Fig. 2D). These results illustrate that iASPP and desmoplakin interact via their N termini and form a protein complex with desmin in cardiomyocytes. The identified iASPP and desmoplakin interaction may contribute to the development of ARVC-like cardiomyopathy.
Loss of iASPP Increases Desmoplakin Phosphorylation and Solubility.
Similar to cardiomyocytes, keratinocytes also require junctional desmoplakin as protection against mechanical stress. Because of difficulties in culturing and manipulating primary cardiomyocytes, we used primary mouse keratinocytes to investigate how iASPP affects desmosome function. Similar to cardiomyocytes, we also observed coimmunoprecipitation of iASPP with desmoplakin in both mouse primary keratinocytes and immortalized mouse oral keratinocytes (IMOKs) (Fig. S3A). Furthermore, colocalization was observed between iASPP and desmoplakin at the cell junctions of keratinocytes using high-resolution light microscopy (∼200-nm resolution; white arrows in Fig. 3A, Inset). Additionally, iASPP accumulates rapidly at the cell border 30 min after Ca2+-dependent cell–cell contacts are induced by switching from low- to high-Ca2+ medium (Fig. 3B). This accumulation is in keeping with other junctional components moving to sites of cell–cell interaction upon the formation of Ca2+-induced junctions, as reported previously (30, 31).
Fig. 3.

Junctional iASPP interacts with desmoplakin and increases its solubility. (A) Junctional localization of iASPP (green) and desmoplakin (red) was detected in wild-type primary mouse keratinocytes using high-resolution confocal microscopy (nuclear stain DAPI is blue). Inset shows yellow colocalization between iASPP and desmoplakin. (B) Translocation of iASPP protein to the cell–cell junctions in response to the Ca2+ switch was investigated in primary keratinocytes. iASPP can be seen at the cell–cell junctions (arrowheads) at 30 min to 24 h after the switch to 1.2 mM Ca2+. (C) The total amount of nonionic detergent-soluble (S) and insoluble (I) junctional components was detected in mouse primary keratinocytes under different culture conditions as indicated. (D) The total amount of nonionic detergent-soluble and insoluble fractions from primary keratinocytes isolated from iASPP CreER mice treated with vehicle or tamoxifen to delete iASPP was loaded on a Phos-tag gel to separate phosphorylated (top band) and unphosphorylated (lower bands) desmoplakin. Phosphorylated and unphosphorylated insoluble desmoplakin are indicated by the black arrows. White arrowheads show that phosphorylated soluble desmoplakin is increased in iASPP-deficient keratinocytes compared with vehicle-treated keratinocytes. β-Actin was used as a loading control. (Scale bars in A and B, 20 μm.)
Keratin 5 is an intermediate filament (IF) protein that binds specifically to desmoplakin in keratinocytes (32). Immunoprecipitation of keratin 5 on IMOK cells in the presence or absence of a desmoplakin siRNA revealed that immunoprecipitated keratin 5 bound much less to iASPP in the presence of a desmoplakin siRNA compared with scrambled siRNA control (Fig. S3B), suggesting that the iASPP–keratin 5 complex is desmoplakin dependent. Because desmin mimics keratin in the cardiomyocytes, we can postulate that the iASPP–desmin complex also would be dependent on desmoplakin. These data also confirm that keratinocytes are a reasonable cell system for exploring the role of iASPP for desmosome function in our studies.
Interestingly, a higher amount of desmoplakin was detected in the Triton-soluble fraction of high-Ca2+–treated iASPP∆8/∆8 keratinocytes (Fig. 3C, compare lanes 9 and 10), even though iASPP status does not affect overall desmoplakin protein levels (Fig. 3C, compare lanes 3 and 4). The Triton-insoluble pool often represents proteins associated with the intermediate filament network (33). Under the same conditions, iASPP deficiency had no impact on the solubility of γ-catenin, desmoglein-1, β-catenin, or E-cadherin (Fig. 3C). These data suggest that in iASPP∆8/∆8 cells desmoplakin is not stably associated with the intermediate filaments.
Phosphorylation of desmoplakin by protein kinase C (PKC) is important for its solubility and desmosome disassembly (34). Keratinocytes lacking all types of keratins have elevated PKC-mediated desmoplakin phosphorylation and accumulation in the cytosol, rendering desmosomes unstable and enhancing susceptibility to mechanical stress (35). Because the iASPP C terminus is known to bind protein phosphatase 1 (PP1), we tested if iASPP is able to influence the phosphorylation status of desmoplakin using a Phos-tag gel that can detect the global phosphorylation status of a protein regardless of its solubility. We observed that although the phosphorylation status of the insoluble desmoplakin from iASPPΔ8/Δ8 primary mouse keratinocytes was not altered [upper blot (shorter exposure) in Fig. 3D], it was greatly enhanced in the soluble desmoplakin pool [white arrowheads on middle blot (longer exposure) in Fig. 3D] compared with iASPP+/+ primary mouse keratinocytes. These data suggest that iASPP may inhibit desmoplakin solubility and cytosolic localization by preventing its phosphorylation.
iASPP Maintains the Integrity of Desmosomes in the Myocardium in Vivo.
To provide evidence that iASPP controls desmosome function by maintaining desmosome integrity, electron microscopic evaluation of the myocardium obtained from 12-wk-old iASPP+/+ and iASPPΔ8/Δ8 mice was performed. In iASPPΔ8/Δ8 cardiomyocytes, irregularly shaped intercalated discs in conjunction with widened gaps between the two-electron dense desmosomal plates were observed (Fig. 4, yellow arrowhead). Moreover, the interaction between the desmin-containing intermediate filaments was disrupted in most of the desmosomes analyzed (Fig. 4, red arrows).
Fig. 4.

iASPP deficiency weakens desmosome function in vivo. Transmission electron microscopy of iASPP+/+ and iASPPΔ8/Δ8 hearts shows cross-sections of myocardial intercalated discs. The yellow arrowhead indicates widening of the desmosomal plate. Red arrows indicate the disarrangement and disruption of the intermediate filament network. (Scale bar, 200 nm.)
To determine precisely the location of iASPP within the intercalated disc area, we carried out immunogold particle labeling of frozen sections of myocardium from 12-wk-old mice. The specific anti-iASPP mouse monoclonal antibody LX049.3 and a secondary antibody labeled with 5-nm gold particles were used to determine the localization of iASPP. Fig. S4A shows that the gold particles are concentrated at the site of intercalated disc (yellow arrowheads). These data suggest that, by interacting with desmoplakin and desmin, iASPP is able to maintain the integrity of desmosomes. We also observed less insoluble desmoplakin in the lysate from the right ventricle of iASPPΔ8/Δ8 than in lysate from the right ventricle of iASPP+/+ mice; however, no differences were observed in desmin (Fig. S4B). This finding is consistent with the localization of only a very small fraction of desmin at the cell junctions, where it would interact with iASPP and desmoplakin. Thus, loss of iASPP would have minimal effects on desmin solubility.
iASPP Deficiency Specifically Causes Right Ventricular Dilatation in E16.5 Embryo Hearts.
To understand how iASPP deficiency causes ventricular dilatation, we examined the morphological and structural integrity of iASPP mutant hearts during mouse development. Hearts were dissected from mouse embryos at 14.5 (E14.5 iASPP+/+, n = 1; iASPPΔ8/Δ8, n = 6), 16.5 (E16.5 iASPP+/+, n = 3; iASPPΔ8/Δ8, n = 3), and 18.5 (E18.5 iASPP+/+, n = 7; iASPPΔ8/Δ8, n = 4) days post coitum and were analyzed using high-resolution episcopic microscopy (HREM) and 3D reconstruction software (36). Of the six E14.5 hearts analyzed, five appeared to be entirely normal, and only one showed mild right ventricular dilatation and possible thinning of the right ventricular wall (Fig. S5A, arrowheads). However, at both E16.5 and E18.5 there was clear evidence of abnormal organ size and shape in all the iASPP mutant hearts analyzed (Fig. 5 and Fig. S5B). The most profound effect was the erosion and thinning of the right ventricular wall (Fig. 5, yellow arrowheads) associated with severe disruption of the myocardial structure, with a variegated pattern of muscle damage (black arrowheads). Fig. 5 also shows a blood clot that replaced part of the right ventricular wall (red arrowheads). By E18.5, loss of myocardial integrity encompassed the interventricular septum, and right ventricular dilation resulted in severe distortion of the heart’s morphology. The orthogonal plane sections of the iASPP-depleted E18.5 heart show severe dilatation of the right ventricular chamber associated with a loss of cardiac muscle in the right ventricular wall (Fig. S5B, arrowheads). In all E16.5 and E18.5 hearts, loss and damage of the myocardium was associated only with thinner right ventricular walls and dilatation of the right ventricular chambers; thus, iASPPΔ8/Δ8 embryos suffer from cardiac pathology that mainly involves the right ventricle.
Fig. 5.

iASPP-deficient embryos exhibit cardiac right ventricular dilatation. Three-dimensional reconstruction of E16.5 iASPP+/+ and iASPPΔ8/Δ8 embryonic hearts from HREM shows progressive heart erosion in iASPPΔ8/Δ8 hearts. Note the thinning of the right ventricular wall (yellow arrowheads), the variegated muscle damage (black arrowheads), and the blood clot accumulation which replaces part of the right ventricular wall (red arrowheads).
iASPP Deficiency Causes Sudden Death with Features of ARVC.
To determine if the right ventricular cardiomyopathy observed in utero is sustained in adult iASPPΔ8/Δ8 mice, we used cardiac-gated MRI on sex- and age-matched 12-wk-old mutant (n = 3) and wild-type (n = 3) mice. Interestingly, the structural abnormalities described in iASPPΔ8/Δ8 embryonic hearts were tightly linked with impaired cardiac performance in both the left and right ventricles. In particular, both the left and right ventricles of iASPP-depleted hearts were dilated, and contraction was minimal, as shown by minimal change in the cross-sectional area between end-diastolic and end-systolic frames at midpapillary level (Fig. 6A). This dilatation resulted in a significant increase in the end-systolic volumes and a consistent decrease in stroke volume, ventricular ejection fraction, and cardiac output (Fig. S6A). These results demonstrate that young adult iASPP-null mice have profoundly impaired cardiac function in both the left and right ventricles.
Fig. 6.

iASPP-deficient adult mice exhibit cardiac right ventricular dilatation and phenotypic features of ARVC. (A) Representative long-axis MRI images of wild-type and transgenic iASPP hearts show the abnormal size, shape, and position within the chest cavity of the mutant heart. Two midpapillary slices from two different iASPP+/+ and iASPPΔ8/Δ8 12-wk-old hearts are shown at diastole. The area covered by the lumen of the left ventricle of the same slice at systole is shown in black. (B) Cross-sections from iASPP+/+ and iASPPΔ8/Δ8 hearts, stained using Oil Red O, show that fibrotic deposits are infiltrated with fat droplets and adipocytes in iASPPΔ8/Δ8 sections only (arrowheads). (Scale bar, 10 µm.) (C, Upper) ECG recordings from iASPP+/+ and iASPPΔ8/Δ8 animals demonstrate that rhythm and conduction abnormalities are present in adult iASPPΔ8/Δ8 mice. Normal sinus rhythm was recorded in all iASPP+/+ mice (n = 5). In contrast, recordings from iASPPΔ8/Δ8 mice (n = 5), revealed frequent ventricular ectopy and episodes of spontaneous NSVT. In addition, adult iASPPΔ8/Δ8 mice displayed other rhythm disturbances, including aberrant junctional rhythm and ventricular conduction. (Lower) Table shows ECG recorded parameters ± SEM.
Because iASPP deficiency specifically affects the right ventricle in embryonic hearts, we hypothesized that a loss of iASPP might induce ARVC, which primarily affects the right ventricle. However, left ventricular involvement is reported in 76% of ARVC cases and usually is age dependent (3, 5, 37). Histological analysis of 12-wk-old iASPPΔ8/Δ8 hearts confirmed the dilatation of both ventricles (Fig. S6B), and extensive myocardial regions were associated with positive Masson’s trichrome blue staining, myocardial disarray, and increased intercellular spaces between myocytes (Fig. S6B, Lower). Loss of myocardium and replacement by fibrotic tissue is not specific to ARVC; it also occurs in dilated cardiomyopathy (20). However, adipocytic deposition is observed only in ARVC patients (3). Thus, to confirm further that iASPP loss causes ARVC, the accumulation of fat droplets was investigated in iASPPΔ8/Δ8 hearts using Oil Red O staining. Importantly, in all iASPP-null hearts examined (n = 3) accumulation of fat droplets was only observed at the sites of fibrosis (Fig. 6B, white arrowheads). Although the fatty infiltration was not extensive in iASPPΔ8/Δ8 hearts, it was undetectable in wild-type controls. Recently it has been suggested that PPARγ activation is important in adipogenesis and ARVC pathogenesis (38). We observed a clear increase in the protein levels of PPARγ and its target gene adiponectin in hearts from iASPP-deficient mice compared with wild-type control hearts (Fig. S6C), which suggests that PPARγ activation may be the primary pathway for adipogenesis in iASPP-deficient mice. This phenotype resembles that of desmoplakin-deficient mice (39), and these results show that iASPPΔ8/Δ8 hearts exhibit many of the classic histopathological features of human ARVC.
We next looked for the presence of rhythm and conduction abnormalities in adult mice using surface ECG recordings. All the control iASPP+/+ mice examined demonstrated normal sinus rhythm. In contrast, young adult iASPPΔ8/Δ8 mice demonstrated frequent multifocal ventricular ectopy, as well as spontaneous runs of nonsustained ventricular tachycardia (NSVT) (Fig. 6C). Finally, to confirm that iASPP deficiency causes sudden death, life expectancy was monitored in a cohort of age- and sex-matched iASPP-deficient and wild-type mice. Beginning at 10 wk of age, iASPPΔ8/Δ8 mice displayed a more rapid mortality, with a median survival of 40 wk, whereas iASPP heterozygous mice showed an intermediate survival rate of 57 wk (Fig. S6D). In contrast, all iASPP+/+ mice survived beyond 90 wk. All the iASPPΔ8/Δ8 and iASPPΔ8/+ mice died suddenly, with no obvious sign of prior illness. The presence of multiple spontaneous rhythm abnormalities, particularly episodes of NSVT, in association with sudden death is strongly suggestive of an arrhythmic mode of death in iASPPΔ8/Δ8 mice. Taken together, these results demonstrate that iASPPΔ8/Δ8 mice die of ARVC.
Human ARVC Cardiomyocytes Exhibit Reduction or Loss of iASPP at Intercalated Discs and Decreased Interaction with Known ARVC Mutations in Desmoplakin.
To determine if the iASPP protein expression profile was altered in ARVC patients, we examined iASPP levels and localization in the cardiomyocytes of six ARVC patients and three normal controls. iASPP was specifically detected at the intercalated discs in the cardiomyocytes of normal controls (Fig. 7). Reduced iASPP levels were detected at the intercalated discs in cardiomyocytes derived from four of the six ARVC samples examined (Fig. 7 and Fig. S7A). Intercalated discs were not lost completely, because N-cadherin was detected at intercalated discs in most ARVC cardiomyocytes examined (Fig. 7 and Fig. S7A). In two of the six cases, levels of iASPP at intercalated discs were reduced or lost in ARVC cardiomyocytes in which γ-catenin was present (Fig. 7 and Fig. S7A). In one case, the reduction of iASPP levels was more pronounced in cardiomyocytes from the right ventricle than in those from the left ventricle of the same individual (Fig. S7B). These results suggest that iASPP contributes to the integrity of intercalated discs and that iASPP deficiency may contribute to the development of ARVC-like cardiomyopathy. The presence or absence of iASPP at intercalated discs of cardiomyocytes therefore may serve as a previously unidentified diagnostic marker for ARVC. Taken together, these results suggest that either decreased iASPP levels or disrupted iASPP and desmoplakin interactions may contribute to the development of ARVC.
Fig. 7.

Representative images show iASPP staining in unaffected and ARVC-affected myocardial sections. Note that the ARVC samples showed immunoreactivity to N-cadherin and γ-catenin. (Scale bar, 20 μm.)
Discussion
Here, for the first time to our knowledge, we identify that, by forming a ternary complex with desmoplakin and desmin, iASPP is a key regulator of desmosome and a gatekeeper of ARVC. This newly identified property of iASPP places it into a crucial position in the pathogenesis of ARVC. The heart undergoes a remodeling process during embryonic and early postnatal development, and this process requires proliferative and apoptotic signals (40). Neonatal cardiomyocytes do not form proper intercalated disc structures until day 15 of mouse postnatal development (41). Additionally, the establishment of survival signals during hemodynamic changes or cardiac injuries is required to protect nonproliferative, fully differentiated cardiomyocytes (42). Therefore, in embryonic cardiomyocytes in which intercalated discs are not formed or in adult hearts subjected to stress conditions, iASPP may locate at the cytoplasm or even in the nucleus, instead of at cell–cell junctions where it can interact with p53 and p63 to prevent apoptosis (43–45). The molecular mechanism for this ventricular-specific effect of iASPP is currently unknown. However, that iASPP deletion specifically causes right vertical dilatation in E16.5 embryos and that adult iASPP-deficient mice exhibited features of human ARVC argue for the importance of iASPP as a gatekeeper of ARVC.
Many of the pathological features of ARVC, including right ventricular dilatation and adipogenesis, were present in our iASPP-deficient mice (Fig. 6B and Fig. S6B). PPARγ signaling previously has been implicated in adipogenesis in ARVC (38, 46), and its activation also was present in iASPP-deficient hearts (Fig. S6C). This finding is in keeping with cardiac-specific PPARγ transgenic animals presenting with cardiac dysfunction and heart failure (47). Because iASPP is a known inhibitor of p53, it is possible that enhanced p53 activity in iASPP-deficient mice may result in PPARγ activation through p53’s ability to increase the levels of LPIN 1 (48), a protein that forms a complex with PPARγ to regulate the expression of genes involved in fatty acid oxidation (49).
The precise mechanism that regulates iASPP’s translocation is unknown. We recently have shown that cyclin B/cyclin-dependent kinase 1 phosphorylation of serines 84 and 113 on the N terminus of iASPP prevents an N- and C-terminal self-interaction (50). iASPP's N terminus phosphorylation exposes the C terminus, which contains a RanGDP binding code in its ankyrin repeats, and enables its nuclear entry via the newly identified RaDAR-mediated nuclear import pathway (51). In addition, the unmasking of the iASPP C terminus also reveals p53 and PP1 binding sites (52, 53). When the N terminus of iASPP interacts with desmoplakin (Fig. 2C), the PP1 binding site at the C terminus of iASPP may be exposed to enable iASPP to recruit PP1 to the iASPP/desmoplakin complex to dephosphorylate desmoplakin. Consistent with this notion, microcystin LR (an inhibitor of PP1) is known to increase desmoplakin phosphorylation and desmosome disassembly (54). Further studies are needed to test this hypothesis.
ARVC is the main cardiac component in recessive cardiocutaneous disorders and also is inherited in isolation as an autosomal dominant condition. Mutations in five known genes, which are intimately involved in maintaining the integrity of desmosomal function, account for ∼50% of ARVC patients (www.ncbi.nlm.nih.gov/omim/), illustrating the importance of desmosomal integrity in heart homeostasis. It is possible that mutations in the regulators of desmosomes might contribute to the development of ARVC in the remaining 50% of cases. The identification of iASPP as a binding partner of desmoplakin and as a regulator of desmosomal function suggests that mutations in the iASPP gene, PPP1R13L, may contribute to ARVC in at least some patients who do not carry mutations in the five known desmosomal genes. Because iASPP heterozygous mice have a shorter life expectancy than wild-type mice and die suddenly (Fig. S6D), it is possible that reduced iASPP protein levels may predispose individuals to ARVC. A defect in the iASPP interactions with desmoplakin and desmin also may predict susceptibility to ARVC. Desmosomes are intercellular junctions that connect intermediate filaments to the cell surface and mediate strong cell–cell adhesion. They are particularly prominent in stratified squamous epithelia and myocardium (55, 56). Accordingly, the junctional localization of iASPP is crucial for desmin intermediate filaments to locate at the intercalated discs in cardiomyocytes. Further studies are needed to elucidate the precise mechanisms by which iASPP can anchor desmoplakin and desmin to the intercalated discs in cardiomyocytes. Nonetheless, this newly identified property of iASPP provides a molecular explanation for why iASPP deficiency causes the cardiomyopathy that occurs spontaneously in Wa3 mice and Poll Hereford calves. It also suggests that iASPP may be a potential ARVC candidate gene and that, as with γ-catenin, a lack of iASPP at the intercalated discs in cardiomyocytes may help in the diagnosis of ARVC (57).
Materials and Methods
Human samples were handled according to the guidelines of the National Research Ethics Committee. All animals were handled according to Institutional Animal Care and Use Committee guidelines. Procedures were carried out following the Home Office Animal (Scientific Procedures) Act 1986 guidelines (license PPL 30/2862).
Mouse Colonies.
iASPPΔ8/Δ8 mice were generated in a mixed C57BL/6Jx129vJ background as previously described (45). Genotyping of iASPPΔ8/Δ8 mice was performed by PCR analysis using the primers FLP2 (5′-CCGAATTGGAGAAGTGAAGC-3′), I8-2 (5′-CCGAATTGGAGAAGTGAAGC-3′), and E8-2 (5′-AGAGCAGCCTCAGAGCATGG-3′) (45).
Cell Culture.
The immortalized mouse cardiomyocyte HL-1 cells were cultured according to the manufacturer’s recommendations (Claycomb Lab). Isolation and culture of primary keratinocytes from iASPPΔ8/Δ8 and iASPPflox/flox; Cre+ERT mice were performed as described. Briefly, skin was isolated from pups at postnatal day 2 and floated over 0.25% trypsin-EDTA (Gibco) overnight at 4 °C. The following day, the epidermis was peeled off and triturated in calcium-free Eagle’s Minimum Essential Medium with 8% (vol/vol) calcium stripped FBS, 0.05 mM CaCl2, and 50 μg/mL gentamicin (low-calcium medium). The cells were isolated from the resulting suspension and plated on plastic dishes or coverslips coated with rat collagen type I (BD Bioscience). iASPP deletion was induced by adding 1 μM 4-hydroxytamoxifen to the cells for 4 d. The IMOKs were maintained in the same medium as primary keratinocytes.
Histology and Tissue Section Staining.
Mouse hearts at various developmental stages were dissected and processed for histological examination as previously described (16). For immunofluorescence staining, tissues sections were incubated overnight at 4 °C with mouse monoclonal antibodies against iASPP (LX128.5) and desmoplakin I and II (Progen Scientific Ltd) or with rabbit polyclonal antibodies against desmin, γ-catenin, N-cadherin, and connexin 43 (Abcam), followed by either biotinylated or Alexa Fluor (1:400; Molecular Probes)-conjugated secondary antibody for 30 min at room temperature. The sections were mounted, viewed, and photographed with a confocal microscope under the same conditions. A bright-light microscope was used to visualize stains performed using peroxide substrate solution diaminobenzidine (DAB) (Vector Laboratories). TO-PRO or DAPI (Invitrogen) was used to stain nucleic acids. Masson’s trichrome staining was performed on deparaffinized heart sections from iASPP+/+ and iASPPΔ8/Δ8 mice according to the manufacturer’s recommendations (Sigma-Aldrich). Oil Red O staining was performed on frozen myocardial sections fixed in 10% neutral formalin buffer (Sigma-Aldrich), washed in distilled water, and placed in 100% propylene glycol for 2 min. Slides then were immersed in Oil Red solution for 1 h at 60 °C, rinsed in distilled water, and counterstained with Mayer’s hematoxylin (Sigma-Aldrich).
HREM.
High-resolution episcopic images of mouse hearts at E14.5, E16.5, and E 18.5 were obtained as previously described (58). We used the episcopic procedures for sectioning and image capture using a previously described arrangement of microtome and magnification optic (59).
Transmission Electron Microscopy.
Endocardial biopsy samples were fixed in 4% glutaraldehyde in cacodylate buffer at 4 °C overnight and were post-fixed in buffered 1% osmium tetroxide for 1 h. Samples then were dehydrated and embedded in epoxy resin. Areas of interest were identified by light microscopy in 1-μm sections. Thin (70-nm) sections of suitable areas were cut and stained with uranyl acetate and lead citrate before examination in the electron microscope.
Immunogold Labeling and Imaging.
Myocardial cryosections were prepared by the Tokuyasu method (60) and were immunolabeled with mouse monoclonal antibody against iASPP (LX128.5). Mouse monoclonal antibodies were detected with rabbit anti-mouse (Cappel/ICN) and protein A-gold particles (61). Immunolabeled sections were examined in a FEI Tecnai electron microscope with CCD camera image acquisition.
Immunoprecipitation and Western Blotting.
Adult mouse hearts were homogenized and lysed in radioimmunoprecipitation assay buffer. Equal amounts (1 mg) of protein extracts were used for each immunoprecipitation. Immunoprecipitates were resolved on SDS/PAGE gels and then transferred to nitrocellulose membranes (Protran). Different mutants of desmoplakin I and a full-length iASPP were cloned into a pCDNA3.1-expressing vector and then were in vitro translated and labeled with [35S]methionine using the TNT T7 Quick coupled transcription-translation system (Promega). The lysates containing the indicated proteins were incubated at 30 °C for 1 h. The antibody to V5 immobilized on protein G beads was added to the binding reaction mixtures and incubated on a rotating wheel at 4 °C overnight. Beads were washed in immunoprecipitation buffer, and the bound proteins were released in SDS gel sample buffer and analyzed by SDS/PAGE. Results were visualized by autoradiography. To calculate the percentage of mutant desmoplakin in complex with iASPP, densitometry values were obtained using ImageJ software normalized against the amount of iASPP immunoprecipitated. Western blots were visualized using the Enhanced Chemi-Luminescence system (Amersham Pharmacia Biotech). The input lane contains 20 μg of protein extract. Monoclonal anti-iASPP (LX049.3) or polyclonal anti-iASPP (pAbAbiASPPN1) (62) was used to immunoprecipitate iASPP. Mouse or rabbit IgG was used as a negative control. Keratinocytes were rinsed in ice-cold PBS, and the soluble protein fraction was obtained by lysing cells in NETN buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 1mM EDTA, 1% Nonidet P-40 + 0.1 mM Na3VO4, protease and phosphatase inhibitors] with shaking at 4 °C for 30 min. Cell lysate was pelleted for 15 min at 14,000 × g, and the insoluble pool of proteins was obtained by solubilizing the pellet in urea buffer (8 M urea, 1 M thiourea, 0.5% CHAPS, 50 mM DTT, 24 mM spermine, and protease inhibitor). SDS sample buffer was added to both the soluble and the insoluble pool, and samples were boiled for 10 min before being subjected to SDS/PAGE on a 6% gel or when indicated on Phos-tag gel.
Statistical Analysis.
Kaplan–Meier survival analysis was performed with Prism 4 software (GraphPad Software). Differences were considered significant at a value of P < 0.05. The t-test was used to calculate the statistical significance between two measurements.
Supplementary Material
Acknowledgments
We thank Dr. Claire Beveridge, Evelyn Harvey, and Dr. Michael White for critical reading of the manuscript; Mark Shipman for technical assistance with confocal image microscopy; Dr. Houman Ashrafian for scientific discussions; Dr. Katja Gehmlich for technical advice; and the members of the X.L. group for stimulating discussions. The work was funded predominantly by the Ludwig Institute for Cancer Research Ltd. T.J.M. and L.D. are funded by Medical Research Council Grant U117562103. A.O., C.A.C., and K.C. are funded by the British Heart Foundation. A.Y. was supported by a Wellcome Trust Research Training Fellowship, and H.W received support from the British Heart Foundation Centre of Research Excellence and the Wellcome Trust.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1408111112/-/DCSupplemental.
References
- 1.Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778(3):572–587. doi: 10.1016/j.bbamem.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 2.Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004;13(4):185–194. doi: 10.1016/j.carpath.2004.03.609. [DOI] [PubMed] [Google Scholar]
- 3.Corrado D, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J Am Coll Cardiol. 1997;30(6):1512–1520. doi: 10.1016/s0735-1097(97)00332-x. [DOI] [PubMed] [Google Scholar]
- 4.McRae AT, 3rd, Chung MK, Asher CR. Arrhythmogenic right ventricular cardiomyopathy: A cause of sudden death in young people. Cleve Clin J Med. 2001;68(5):459–467. doi: 10.3949/ccjm.68.5.459. [DOI] [PubMed] [Google Scholar]
- 5.Lombardi R, Marian AJ. Molecular genetics and pathogenesis of arrhythmogenic right ventricular cardiomyopathy: A disease of cardiac stem cells. Pediatr Cardiol. 2011;32(3):360–365. doi: 10.1007/s00246-011-9890-2. [DOI] [PubMed] [Google Scholar]
- 6.Whittington RJ, Cook RW. Cardiomyopathy and woolly haircoat syndrome of Poll Hereford cattle: Electrocardiographic findings in affected and unaffected calves. Aust Vet J. 1988;65(11):341–344. doi: 10.1111/j.1751-0813.1988.tb14260.x. [DOI] [PubMed] [Google Scholar]
- 7.Simpson MA, et al. A mutation in NFkappaB interacting protein 1 causes cardiomyopathy and woolly haircoat syndrome of Poll Hereford cattle. Anim Genet. 2009;40(1):42–46. doi: 10.1111/j.1365-2052.2008.01796.x. [DOI] [PubMed] [Google Scholar]
- 8.Herron BJ, et al. A mutation in NFkB interacting protein 1 results in cardiomyopathy and abnormal skin development in wa3 mice. Hum Mol Genet. 2005;14(5):667–677. doi: 10.1093/hmg/ddi063. [DOI] [PubMed] [Google Scholar]
- 9.Trigiante G, Lu X. ASPP [corrected] and cancer. Nat Rev Cancer. 2006;6(3):217–226. doi: 10.1038/nrc1818. [DOI] [PubMed] [Google Scholar]
- 10.Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci USA. 1994;91(13):6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang JP, Hori M, Sanda T, Okamoto T. Identification of a novel inhibitor of nuclear factor-kappaB, RelA-associated inhibitor. J Biol Chem. 1999;274(22):15662–15670. doi: 10.1074/jbc.274.22.15662. [DOI] [PubMed] [Google Scholar]
- 12.Gorina S, Pavletich NP. Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science. 1996;274(5289):1001–1005. doi: 10.1126/science.274.5289.1001. [DOI] [PubMed] [Google Scholar]
- 13.Bergamaschi D, et al. ASPP1 and ASPP2: Common activators of p53 family members. Mol Cell Biol. 2004;24(3):1341–1350. doi: 10.1128/MCB.24.3.1341-1350.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuels-Lev Y, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8(4):781–794. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 15.Vives V, et al. ASPP2 is a haploinsufficient tumor suppressor that cooperates with p53 to suppress tumor growth. Genes Dev. 2006;20(10):1262–1267. doi: 10.1101/gad.374006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sottocornola R, et al. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev Cell. 2010;19(1):126–137. doi: 10.1016/j.devcel.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Cong W, et al. ASPP2 regulates epithelial cell polarity through the PAR complex. Curr Biol. 2010;20(15):1408–1414. doi: 10.1016/j.cub.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 18.Bergamaschi D, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet. 2003;33(2):162–167. doi: 10.1038/ng1070. [DOI] [PubMed] [Google Scholar]
- 19.Franke WW, Borrmann CM, Grund C, Pieperhoff S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol. 2006;85(2):69–82. doi: 10.1016/j.ejcb.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 21.Leung CL, Green KJ, Liem RK. Plakins: A family of versatile cytolinker proteins. Trends Cell Biol. 2002;12(1):37–45. doi: 10.1016/s0962-8924(01)02180-8. [DOI] [PubMed] [Google Scholar]
- 22.Saffitz JE. Desmosome mutations in arrhythmogenic right ventricular cardiomyopathy: Important insight but only part of the picture. Circ Cardiovasc Genet. 2009;2(5):415–417. doi: 10.1161/CIRCGENETICS.109.909366. [DOI] [PubMed] [Google Scholar]
- 23.Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol. 2000;1(3):208–216. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- 24.Baum B, Georgiou M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J Cell Biol. 2011;192(6):907–917. doi: 10.1083/jcb.201009141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerull B, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36(11):1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 26.Kowalczyk AP, Bornslaeger EA, Norvell SM, Palka HL, Green KJ. Desmosomes: Intercellular adhesive junctions specialized for attachment of intermediate filaments. Int Rev Cytol. 1999;185:237–302. doi: 10.1016/s0074-7696(08)60153-9. [DOI] [PubMed] [Google Scholar]
- 27.Kowalczyk AP, et al. The head domain of plakophilin-1 binds to desmoplakin and enhances its recruitment to desmosomes. Implications for cutaneous disease. J Biol Chem. 1999;274(26):18145–18148. doi: 10.1074/jbc.274.26.18145. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99(6):646–655. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- 29.Rampazzo A, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71(5):1200–1206. doi: 10.1086/344208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Godsel LM, et al. Desmoplakin assembly dynamics in four dimensions: Multiple phases differentially regulated by intermediate filaments and actin. J Cell Biol. 2005;171(6):1045–1059. doi: 10.1083/jcb.200510038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marrs JA, Nelson WJ. Cadherin cell adhesion molecules in differentiation and embryogenesis. Int Rev Cytol. 1996;165:159–205. doi: 10.1016/s0074-7696(08)62222-6. [DOI] [PubMed] [Google Scholar]
- 32.Kouklis PD, Hutton E, Fuchs E. Making a connection: Direct binding between keratin intermediate filaments and desmosomal proteins. J Cell Biol. 1994;127(4):1049–1060. doi: 10.1083/jcb.127.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasdar M, Nelson WJ. Kinetics of desmosome assembly in Madin-Darby canine kidney epithelial cells: Temporal and spatial regulation of desmoplakin organization and stabilization upon cell-cell contact. I. Biochemical analysis. J Cell Biol. 1988;106(3):677–685. doi: 10.1083/jcb.106.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amar LS, Shabana AH, Oboeuf M, Martin N, Forest N. Involvement of desmoplakin phosphorylation in the regulation of desmosomes by protein kinase C, in HeLa cells. Cell Adhes Commun. 1999;7(2):125–138. doi: 10.3109/15419069909034396. [DOI] [PubMed] [Google Scholar]
- 35.Kröger C, et al. Keratins control intercellular adhesion involving PKC-α-mediated desmoplakin phosphorylation. J Cell Biol. 2013;201(5):681–692. doi: 10.1083/jcb.201208162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weninger WJ, Mohun T. Phenotyping transgenic embryos: A rapid 3-D screening method based on episcopic fluorescence image capturing. Nat Genet. 2002;30(1):59–65. doi: 10.1038/ng785. [DOI] [PubMed] [Google Scholar]
- 37.Azaouagh A, Churzidse S, Konorza T, Erbel R. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: A review and update. Clin Res Cardiol. 2011;100(5):383–394. doi: 10.1007/s00392-011-0295-2. [DOI] [PubMed] [Google Scholar]
- 38.Kim C, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494(7435):105–110. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Gras E, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116(7):2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savolainen SM, Foley JF, Elmore SA. Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol Pathol. 2009;37(4):395–414. doi: 10.1177/0192623309335060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angst BD, et al. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res. 1997;80(1):88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- 42.van Empel VP, De Windt LJ. Myocyte hypertrophy and apoptosis: A balancing act. Cardiovasc Res. 2004;63(3):487–499. doi: 10.1016/j.cardiores.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 43.Xiong S, Van Pelt CS, Elizondo-Fraire AC, Fernandez-Garcia B, Lozano G. Loss of Mdm4 results in p53-dependent dilated cardiomyopathy. Circulation. 2007;115(23):2925–2930. doi: 10.1161/CIRCULATIONAHA.107.689901. [DOI] [PubMed] [Google Scholar]
- 44.Rouleau M, et al. TAp63 is important for cardiac differentiation of embryonic stem cells and heart development. Stem Cells. 2011;29(11):1672–1683. doi: 10.1002/stem.723. [DOI] [PubMed] [Google Scholar]
- 45.Notari M, et al. Inhibitor of apoptosis-stimulating protein of p53 (iASPP) prevents senescence and is required for epithelial stratification. Proc Natl Acad Sci USA. 2011;108(40):16645–16650. doi: 10.1073/pnas.1102292108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Djouadi F, et al. A potential link between peroxisome proliferator-activated receptor signalling and the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Res. 2009;84(1):83–90. doi: 10.1093/cvr/cvp183. [DOI] [PubMed] [Google Scholar]
- 47.Son NH, et al. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117(10):2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Assaily W, et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. 2011;44(3):491–501. doi: 10.1016/j.molcel.2011.08.038. [DOI] [PubMed] [Google Scholar]
- 49.Finck BN, et al. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4(3):199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 50.Lu M, et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell. 2013;23(5):618–633. doi: 10.1016/j.ccr.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 51.Lu M, et al. A code for RanGDP binding in ankyrin repeats defines a nuclear import pathway. Cell. 2014;157(5):1130–1145. doi: 10.1016/j.cell.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Llanos S, et al. Inhibitory member of the apoptosis-stimulating proteins of the p53 family (iASPP) interacts with protein phosphatase 1 via a noncanonical binding motif. J Biol Chem. 2011;286(50):43039–43044. doi: 10.1074/jbc.M111.270751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skene-Arnold TD, et al. Molecular mechanisms underlying the interaction of protein phosphatase-1c with ASPP proteins. Biochem J. 2013;449(3):649–659. doi: 10.1042/BJ20120506. [DOI] [PubMed] [Google Scholar]
- 54.Toivola DM, Goldman RD, Garrod DR, Eriksson JE. Protein phosphatases maintain the organization and structural interactions of hepatic keratin intermediate filaments. J Cell Sci. 1997;110(Pt 1):23–33. doi: 10.1242/jcs.110.1.23. [DOI] [PubMed] [Google Scholar]
- 55.Bolling MC, Jonkman MF. Skin and heart: Une liaison dangereuse. Exp Dermatol. 2009;18(8):658–668. doi: 10.1111/j.1600-0625.2009.00908.x. [DOI] [PubMed] [Google Scholar]
- 56.Bazzi H, Christiano AM. Broken hearts, woolly hair, and tattered skin: When desmosomal adhesion goes awry. Curr Opin Cell Biol. 2007;19(5):515–520. doi: 10.1016/j.ceb.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asimaki A, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 58.Weninger WJ, Meng S, Streicher J, Müller GB. A new episcopic method for rapid 3-D reconstruction: Applications in anatomy and embryology. Anat Embryol (Berl) 1998;197(5):341–348. doi: 10.1007/s004290050144. [DOI] [PubMed] [Google Scholar]
- 59.Mohun TJ, Leong LM, Weninger WJ, Sparrow DB. The morphology of heart development in Xenopus laevis. Dev Biol. 2000;218(1):74–88. doi: 10.1006/dbio.1999.9559. [DOI] [PubMed] [Google Scholar]
- 60.Tokuyasu KT. Immunochemistry on ultrathin frozen sections. Histochem J. 1980;12(4):381–403. doi: 10.1007/BF01011956. [DOI] [PubMed] [Google Scholar]
- 61.Slot JW, Geuze HJ. A new method of preparing gold probes for multiple-labeling cytochemistry. Eur J Cell Biol. 1985;38(1):87–93. [PubMed] [Google Scholar]
- 62.Bergamaschi D, et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat Genet. 2006;38(10):1133–1141. doi: 10.1038/ng1879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.