

Abstract
Integration of DNA viruses into the human genome plays an important role in various types of tumors, including hepatitis B virus (HBV)–related hepatocellular carcinoma. However, the molecular details and clinical impact of HBV integration on either human or HBV epigenomes are unknown. Here, we show that methylation of the integrated HBV DNA is related to the methylation status of the flanking human genome. We developed a next-generation sequencing-based method for structural methylation analysis of integrated viral genomes (denoted G-NaVI). This method is a novel approach that enables enrichment of viral fragments for sequencing using unique baits based on the sequence of the HBV genome. We detected integrated HBV sequences in the genome of the PLC/PRF/5 cell line and found variable levels of methylation within the integrated HBV genomes. Allele-specific methylation analysis revealed that the HBV genome often became significantly methylated when integrated into highly methylated host sites. After integration into unmethylated human genome regions such as promoters, however, the HBV DNA remains unmethylated and may eventually play an important role in tumorigenesis. The observed dynamic changes in DNA methylation of the host and viral genomes may functionally affect the biological behavior of HBV. These findings may impact public health given that millions of people worldwide are carriers of HBV. We also believe our assay will be a powerful tool to increase our understanding of the various types of DNA virus-associated tumorigenesis.
Hepatitis B virus (HBV) infects more than two billion people worldwide, and 400 million chronically infected individuals are at high risk of developing active hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) (Gatza et al. 2005; Lupberger and Hildt 2007). HBV carriers with chronic liver disease are at a 100-fold greater risk of developing HCC, which is the third leading cause of cancer-related death worldwide. The HBV genome is integrated into the host genome in 90% of patients with HCC (HBV-HCC) (Gatza et al. 2005; Lupberger and Hildt 2007). HBV-HCCs have been analyzed by comprehensive genome sequencing and high-resolution genome mapping (Kan et al. 2013; Li and Mao 2013; Nakagawa and Shibata 2013). Moreover, the recent deep sequencing of HBV DNA in patients with HCC revealed increased integration events, structural alterations, and sequence variations (Ding et al. 2012; Fujimoto et al. 2012; Jiang et al. 2012; Sung et al. 2012; Toh et al. 2013). A recent study identified a viral-human chimeric fusion transcript, HBx-LINE1, that functions like a long noncoding RNA to promote HCC (Lau et al. 2014). However, the molecular details and clinical impact of HBV integration on the epigenomes of human cells and HBV remain to be defined.
Methylation of exogenous DNA (including viral DNA) that is integrated into the human genome has been studied over the past decade (Doerfler et al. 2001). Within the human genome, cytosine methylation in CpG dinucleotides (CpG sites), which cluster into islands associated with transcriptional promoters, is an important mechanism for regulating gene expression. Additionally, host cells use methylation as a defense mechanism against foreign agents (e.g., viral DNA) (Doerfler 2008; Doerfler et al. 2001). DNA methylation suppresses the expression of viral genes and other deleterious elements incorporated into the host genome over time. Establishment of de novo patterns of DNA methylation is characterized by the gradual spread of methylation (Orend et al. 1991). Another attractive possibility is that DNA methylation camouflages the virus from the immune system (Tao and Robertson 2003; Hilleman 2004), resulting in a DNA methylation–related blockade of viral antigen presentation that allows the virus to escape immune control (Fernandez et al. 2009).
The DNA methylome of HBV in human cells may undergo dynamic changes at different stages of disease (Fernandez et al. 2009). For example, DNA methylation at the HBVgp2 locus, which codes for the S viral proteins, reportedly increases during the progression from asymptomatic lesions to benign lesions, to premalignant disease and malignant tumors. However, because of the significant deletions of the integrated HBV genome detected in this previous study (Fernandez et al. 2009), the DNA methylome of HBV needs to be further characterized. Moreover, the molecular mechanisms involved and the clinical impact of the integration of HBV on the human and HBV epigenomes are unknown. To address these issues, we developed a next-generation sequencing (NGS)–based method for methylation analysis of integrated viral genomes (denoted G-NaVI) and applied this method to the integrative genomic and epigenomic analysis of human hepatoma cell lines and tissues with integrated HBV genomes.
Results
DNA methylation levels in PLC/PRF/5 cells and cancerous tissues obtained from HBV-HCC patients
Methylated CpG island (CGI) amplification (MCA) coupled with microarray (MCAM) analysis (Toyota et al. 1999; Oishi et al. 2012) was performed to detect methylated genes in the human PLC/PRF/5 cell line and in six paired specimens of primary HBV-HCC and adjacent tissues. Compared with the DNA methylation of CGIs in the healthy peripheral blood leukocytes of volunteers or the noncancerous tissues, levels of DNA methylation were not remarkable in the PLC/PRF/5 cells and the cancerous tissues obtained from HBV-HCC patients (Supplemental Fig. 1). These results were confirmed by bisulfite pyrosequencing of candidate tumor-related genes.
DNA methylation of CGIs of HBx
We then focused on epigenetic changes in the viral genome. Based on the hidden Markov models for sequence analysis performed on the CpG plugin of bioinformatics software Geneious 5.5.8 (see Methods section), a CpG island was found in only the promoter region of the HBx gene in the HBV genome (Fig. 1A; Supplemental Fig. 2; Durbin et al. 1998; Kearse et al. 2012). Host signal transduction pathways and gene expression are disrupted by the expression of trans-activating factors derived from the HBV genome, such as the HBx protein and PreS2 activators (Gatza et al. 2005; Lupberger and Hildt 2007). Moreover, transgenic mice expressing high levels of HBx in the liver develop HCC (Kim et al.1991; Koike et al. 1994). The DNA methylation levels of the CGIs of HBx were analyzed in 10 HBV-HCC samples and 10 adjacent samples, as well as samples of PLC/PRF/5 cells by bisulfite pyrosequencing (Fig. 1A; Supplemental Fig. 2). We performed advanced methylation quantification in long sequence runs by pyrosequencing on PyroMark Q24 Advanced and PyroMark Q24 instruments. Methylation levels of HBx varied across samples (Fig. 1B,C) and were generally lower in HCC tissues than in the adjacent tissues (Fig. 1B). This finding is consistent with a previous report that most HBV genomes, although globally methylated to a greater extent in malignant samples than in premalignant lesions, retain HBx in an unmethylated state (Fernandez et al. 2009). Because the pyrosequencing results represent the genome-wide average of DNA methylation levels at the particular CpG site, the results could be affected by the HBV integration site. Therefore, genome-wide methylation analysis of the integrated HBV sequence is necessary in relation to the methylation state of the adjacent human genome. We did not detect an association between HBx methylation levels and those of the LINE1 and AluYb8 repeats (Fig. 1B).
Figure 1.
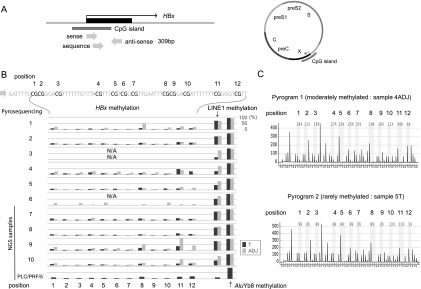
Methylation analysis of the CGI of the HBx gene. (A) Schema of the CGI of the HBx gene. Three arrows show the pyrosequencing primers used for the methylation analysis. (B) DNA methylation levels of the CpGs of the HBx gene, LINE1, and AluYb8 in 10 paired HBV-HCC and adjacent nontumor tissue samples and PLC/PRF/5 DNA were analyzed using bisulfite pyrosequencing. Methylation levels of HBx varied across samples and were generally lower in HCC tissues than in the adjacent nontumor tissues. An association between HBx methylation levels and those of the LINE1 and AluYb8 repeats was not observed. N/A, could not be analyzed. DNAs from four paired HBV-HCC and adjacent nontumor tissue samples (sample nos. 7–10), PLC/PRF5, and HepG2.2.15 were further analyzed using the NGS (G-NaVI method). (C) Representative pyrograms showing DNA methylation levels of the CpGs of the HBx gene. Methylation levels at 12 CpG sites of the HBx gene in adjacent nontumor tissue (sample no. 4ADJ) and tumor tissue (sample no. 5T) are shown.
Fluorescence in situ hybridization (FISH) and Alu PCR analyses of HBV integration
We developed a FISH technique for detecting HBV DNA in the genome of PLC/PRF/5 cells (Supplemental Figs. 3, 4). Twelve specific primer pairs (FISH probes 1–12) were designed based on the HBV sequences integrated into the genome of PLC/PRF/5 cells; amplification from all primer pairs was confirmed (Supplemental Fig. 4A). These results suggest full-length or partial HBV sequences that are covered by the 12 primer pairs were integrated into the genome of the PLC/PRF/5 cells. The FISH probes were labeled with digoxigenin, and FISH was performed using Carnoy-fixed chromosomal and nuclear specimens. Multiple HBV fluorescent signals (green) were detected in the nuclei (Supplemental Fig. 4B) using probes for HBx and its CGI sequences (probes 5 and 6), but not with probes 1–4 or 7–12 (Supplemental Fig. 4C–E). Alu-PCR identified one HBx integration site in PLC/PRF/5 (Supplemental Fig. 5). The integrated HBx sequence was 213 bp and included a promoter region. The HBx gene body was located only 13 bases (ATG GCT GCT AGG T) from the transcription start site and was integrated into a noncoding region of the host genome. There were 200 bases of viral DNA sequence upstream of the HBx transcription start site. According to the human genome reference sequence (GRCh38) published by the Genome Reference Consortium, this integration site was identified as a noncoding region of host Chromosome 5 1,350,106–1,350,478 that is near the telomerase reverse transcriptase (TERT) gene (Supplemental Fig. 5).
NGS analysis of HBV DNA integration site sequences
We developed an NGS analysis technique for sequencing the HBV DNA integration sites (Supplemental Fig. 6A). For efficient genome analysis, we synthesized 12,391 custom baits based on the sequences of the HBV genotypes A to J and on those sequences present in the HBV-transformed PLC/PRF/5 cells that were not related to the human genome sequence (Supplemental Fig. 6B). The average read length was 333.14 bp with a modal length of ∼500 bp (Supplemental Fig. 6C). The average read quality was 31.91, corresponding to >99.9% accuracy. We did not detect a common HBV integration site (Fig. 2). The integration sites in the PLC/PRF/5 genome included intergenic (39%), intronic (39%), promoter (8%), and divergent promoter (15%) regions but not exonic (0%) sequences (Fig. 2). HepG2.2.15 cells, which stably express and replicate HBV in a culture system, are derived from the human hepatoblastoma cell line HepG2 (Sells et al. 1987). In the HepG2.2.15 genome, the integration sites included intergenic (29%), intronic (57%), and other (14%) regions but not promoter (0%), divergent promoter (0%), or exonic (0%) sequences (Fig. 2).
Figure 2.
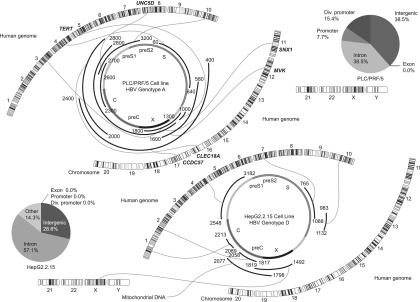
Distribution of the integration sites in the HBV genome and human chromosomes represented by Circos plots of the PLC/PRF/5 genome and the HepG2.2.15 genome. HBV DNA integration was analyzed using the G-NaVI method in the genome of PLC/PRF/5 cells and HepG2.2.15 cells. A common HBV integration site was not detected. Integration sites were not detected in exonic regions of the DNA from cell lines (Venn diagrams). The HBV genes (PreC, Precore; C, Core; PreS, Presurface; S, Surface; X, X) and the 24 human chromosomes are shown.
DNA methylation of the integrated HBV genome as well as the adjacent human genome in cell lines
DNA methylation of the integrated HBV genome, as well as the adjacent human genome, was analyzed by bisulfite pyrosequencing. We detected varying levels of methylation of the HBV sequences integrated into the genome of PLC/PRF/5 cells (Fig. 3; Supplemental Fig. 7). Our data suggest DNA methylation in the integrated HBV genome is related to the methylation status of the integration sites within the human genome. We further characterized the methylation status of the HBV genome and human genome by allele-specific DNA methylation analysis (Fig. 3A), which revealed that the HBV genome often showed significant methylation when integrated into highly methylated sites in the human genome; however, the HBV genome remained largely unmethylated when integrated into unmethylated regions such as promoters (Fig. 3B). Integration of the HBV genome did not affect the methylation status of the human genome, including the promoter regions of the TERT and SNX15 genes. Methylation of HBV DNA integrated into HepG2.2.15 cells transformed with HBV DNA (using a head-to-tail dimer) was further analyzed by bisulfite pyrosequencing, which revealed that the HBV genome generally showed significant methylation when integrated into highly methylated regions of the human genome; however, the HBV genome remains largely unmethylated when integrated into unmethylated regions (Fig. 3A).
Figure 3.

Allele-specific methylation analysis of the PLC/PRF/5 genome and the HepG2.2.15 genome. (A) A schema of allele-specific methylation analysis. (B) The methylation levels of the HBV and human genomes for the integrated and unintegrated alleles. Detailed results of the HBV integrants (PreC, Precore; C, Core; PreS, Presurface; S, Surface; X, X) and flanking host genomes (position, chromosome, location of the genome, and gene names) are shown. DNA methylation of the integrated HBV genome as well as the flanking human genome was examined by allele-specific DNA methylation analysis using bisulfite pyrosequencing. The HBV genome often showed significant methylation when integrated into highly methylated sites in the human genome; however, the HBV genome remained largely unmethylated when integrated into unmethylated regions. Methylation levels of orthologous loci in HepG2.2.15 cells and in PBLs of a healthy volunteer were examined and compared to the methylation levels at the same (empty) target sites of PLC/PRF/5 cells. Methylation levels of orthologous loci in HepG2.2.15 cells and PBLs were generally similar to those of PLC/PRF/5 cells. Similarly, methylation levels of orthologous loci in PLC/PRF/5 cells and in PBLs of a healthy volunteer were examined and compared to the methylation levels at the same (empty) target sites of HepG2.2.15 cells. Methylation levels of orthologous loci in PLC/PRF/5 cells and PBLs were generally similar to those of HepG2.2.15 cells. (×) The desired quantitative methylation levels were not obtained because of technical difficulties with the sequences that were being analyzed.
DNA methylation levels in orthologous loci
We examined methylation levels of orthologous loci in HepG2.2.15 cells and in peripheral blood lymphocytes (PBLs) of a healthy volunteer and compared them to the methylation levels at the same (empty) target sites of PLC/PRF/5 cells. Methylation levels of orthologous loci in HepG2.2.15 cells and PBLs were generally similar to those of PLC/PRF/5 cells (Fig. 3B). Similarly, we examined methylation levels of orthologous loci in PLC/PRF/5 cells and in PBLs of a healthy volunteer and compared them to the methylation levels at the same (empty) target sites of HepG2.2.15 cells. Methylation levels of orthologous loci in PLC/PRF/5 cells and PBLs were also generally similar to those of HepG2.2.15 cells (Fig. 3B).
DNA methylation of the integrated HBV genome and the adjacent human genome in HCC tissues
To determine whether our results are relevant to human tumors, we used bisulfite pyrosequencing to investigate the methylation status of the HBV and human genomes in surgical specimen pairs of HCC and adjacent nontumor tissues. We detected no common HBV integration site (Fig. 4; Supplemental Fig. 8). Recurrent HBV integration into the SLC6A13 gene was observed in cancerous tissues. Integration sites were rarely detected in exonic regions of the DNA from HBV-HCC samples (Fig. 4; Supplemental Fig. 8). Similar to the results obtained from the PLC/PRF/5 and HepG2.2.15 cells, our analysis revealed that the HBV genome became significantly methylated when integrated into highly methylated human genome regions but not when integrated into unmethylated human genome regions (Fig. 4).
Figure 4.

Allele-specific methylation analysis of the tumor (T) and adjacent nontumor (ADJ) sample genomes. The methylation levels of the HBV and human genomes for the integrated and unintegrated alleles in four paired tumor and adjacent nontumor samples (sample nos. 7–10) are shown. Detailed results of the HBV integrants (PreC, Precore; C, Core; PreS, Presurface; S, Surface; X, X) and flanking host genomes (position, chromosome, location of the genome, and gene names) are shown. The HBV genome became significantly methylated when integrated into highly methylated human genome regions, but not when integrated into unmethylated human genome regions. (×) The desired quantitative methylation levels were not obtained because of technical difficulties with the sequences that were being analyzed.
Correlation between the methylation pattern of the integrated HBV DNA and the human genome
DNA fragments, including 200 bp of the HBV DNA and 200 bp of the human genome around the boundary, were analyzed for average methylation, GC content, and repetitive sequences. A statistically significant correlation was observed between the average methylation of the HBV DNA and that of the human genome in cell lines and clinical samples (Fig. 5A–C; Supplemental Table 2). In contrast, average methylation did not correlate with GC content or repetitive sequences in the human and viral genome (Fig. 5D,E; Supplemental Table 2).
Figure 5.
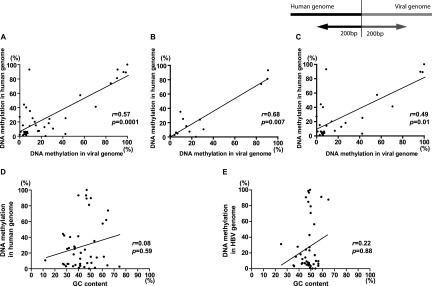
Correlation analysis between the methylation pattern of the integrated HBV DNA and that of the human genome. DNA fragments, including 200 bp of the HBV DNA and 200 bp of the human genome around the boundary, were analyzed for average methylation and GC content. (A) A correlation between the average methylation of the HBV DNA and that of the human genome in combined two cell lines and eight clinical samples (n = 40, r = 0.57, P = 0.0001, 95%CI = 0.3091–0.7545). (B) A correlation between the average methylation of the HBV DNA and that of the human genome in two cell lines (n = 14, r = 068, P = 0.007, 95%CI = 0.2233–0.8946). (C) A correlation between the average methylation of the HBV DNA and that of the human genome in eight clinical samples (n = 26, r = 0.49, P = 0.01, 95%CI = 0.1222–0.7463). (D) No correlation between the average methylation and GC contents in the human genome in the combined two cell lines and eight clinical samples (n = 45, r = 0.08, P = 0.59, 95%CI = −0.2253–0.3745). (E) No correlation between the average methylation and GC contents in the viral genome in the combined two cell lines and eight clinical samples (n = 47, r = 0.22, P = 0.88, 95%CI = −0.3151–0.2751).
Using Bander software, we analyzed the chromatin structure at the integrated HBV site in PLC/PRF/5 and HepG2.2.15. Open chromatin and heterochromatin were observed more frequently at the integrated HBV in PLC/PRF/5 and HepG2.2.15, respectively (Supplemental Table 3). The difference may reflect the fact that PLC/PRF/5 is a naturally derived HBV-positive cell line and HepG2.2.15 is an HBV DNA-transfected cell line.
Discussion
We developed an NGS-based method for structural methylation analysis of integrated viral genomes. This method is a novel approach that enables the enrichment of viral fragments for sequencing using unique baits based only on the sequence of the HBV genome. We detected all regions of the human genome that harbored integrated HBV genomes without conducting unnecessary sequencing of regions where the HBV genome was not integrated. Because this technique only requires sequencing a small region of DNA around the integrated HBV sequences, a sufficient number of sequence reads can be acquired.
Methylation of viral DNA in infected cells may alter the expression patterns of viral genes related to infection and transformation (Burgers et al. 2007; Fernandez et al. 2009) and may clarify why certain infections are either cleared or persist with or without progression to precancer (Mirabello et al. 2012). To the best of our knowledge, we have, for the first time, established that the de novo patterns of DNA methylation in the integrated HBV genome are related to the methylation status of the integration sites within the human genome. A statistically significant correlation between the average methylation of the HBV DNA and that of the human genome in cell lines and clinical samples has greatly substantiated our findings. It is possible that the HBV genome becomes inactivated by methylation, when it is integrated into highly methylated host sites; therefore, HBV methylation may not contribute to tumor development. However, after integration into unmethylated human genome regions such as promoters, the HBV DNA remains unmethylated and may eventually play an important role in tumorigenesis (Fig. 6). Because multiple HBV integration sites were present in each of the analyzed samples, there remains the possibility of an association between methylation and viral transcript levels. The biological impact of methylation on viral transcript levels or viral function, induced by viral insertions, also needs to be further addressed.
Figure 6.
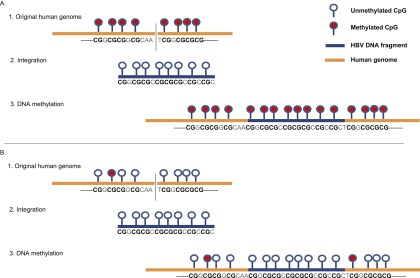
Schema of DNA methylation at HBV integrants and flanking human genomic sequences. (A) DNA hypermethylation was seen in both the integrated HBV fragment and human genome (original human genome shows dense methylation). The HBV genome often showed significant methylation when integrated into highly methylated host sites. (B) DNA hypermethylation was rarely seen in the integrated HBV fragment and human genome (original human genome shows low methylation). The HBV genome remained largely unmethylated when integrated into unmethylated host sites.
Methylation levels of orthologous loci in other samples at the same (empty) target sites of PLC/PRF/5 were generally similar to those of PLC/PRF/5. Similar results were observed in HepG2.2.15. These data suggest that a “before and after” relationship exists between methylation levels at preexisting target sites and those within viral insertions. At the same time, we cannot rule out the possibility that the integration of the virus subsequently affects the methylation established at the flanking target site, perhaps by acting in trans on the empty target site-containing allele. Therefore, this issue needs to be further addressed.
Differences in the integrated viral sequences could have a direct impact on the amount of cytosine methylation observed. In cases where the integration site is a highly active promoter, comparisons of methylation statuses may not be informative. Additional studies, using a large number of samples, are needed to address this issue.
Our results are notable because other studies have detected a statistically significant enrichment of HBV integration into regulatory regions, particularly promoters, in tumors (Sung et al. 2012; Toh et al. 2013); this observation may be explained by the relatively open chromatin structure of promoter regions. Average methylation did not correlate with GC content or repetitive sequences in the human and viral genomes. The relationship between methylation of HBV sequences and chromatin structure remains to be clarified because of the limitation of the Bander software used in this study. Although the mechanism needs clarification, the significant enrichment of HBV integration into regulatory regions would favor integrated HBV nonmethylation and lead to tumorigenesis. Alternatively, while the integration of HBV into the host genome may be random, HBV integration into regulatory regions is positively selected during tumorigenesis (Toh et al. 2013).
The dynamic changes in DNA methylation described here have a major functional impact on the biological behavior of HBV and underlie the molecular mechanisms that control infection or enable tumorigenesis. These findings may significantly impact public health given that millions of people worldwide are carriers of HBV. Distinct DNA methylation profiles may exist, for example, between primary HCCs in Japanese patients and those of other nationalities. Additional studies are needed to address this issue, and research into the influence of other environmental factors is required.
Increased viral DNA methylation is present in cancers associated with DNA viruses, including human papilloma virus types 16 and 18 (HPV 16 and 18) (Fernandez et al. 2009; Mirabello et al. 2012), Epstein-Barr virus (Uozaki and Fukayama 2008; Fernandez et al. 2009), and human T-lymphotropic virus 1 (Taniguchi et al. 2005). An analysis of the haplotype-resolved genome and epigenome of the aneuploid HeLa cervical cancer cell line revealed that an amplified, highly rearranged region of chromosome 8q24.21 harboring an integrated HPV18 genome likely represents the tumor-initiating event (Adey et al. 2013). Whether the dynamic changes in DNA methylation observed in cells with integrated HBV genomes also occur in human cells infected by other viruses is an interesting question for further study. We anticipate that our assay will be a powerful tool for this purpose and have successfully detected integrated HPV sequences in the genomes of cervical cancer cell lines (Y Watanabe, H Yamamoto, F Itoh, and N Suzuki, unpubl.).
This study provides novel mechanistic insights into HBV-mediated hepatocarcinogenesis, which may have preventive and therapeutic applications for carriers of HBV and patients with HBV-HCC, as it suggests that epigenetic alterations provide candidate biochemical markers and therapeutic targets. This study, together with a recent global survey of HBV integration events (Ding et al. 2012; Fujimoto et al. 2012; Jiang et al. 2012; Sung et al. 2012; Toh et al. 2013), provides a foundation for the further experimentation and mechanistic understanding of HBV-HCC.
Methods
Cell lines and primary tissues
The PLC/PRF/5 (Alexander) human hepatoma cell line was obtained from the Japanese Collection of Research Bioresources (JCRB). HepG2.2.15 cells, kindly gifted by Professor Stephan Urban at University Hospital Heidelberg, was derived from HepG2 cells transfected with a plasmid carrying four 5′-3′ tandem copies of the HBV genome (Koike et al. 1994). Cell lines were maintained in appropriate media containing 10% fetal bovine serum in plastic culture plates. Primary tissues from tumor and adjacent tissues were obtained at the time of the clinical procedures. Informed consent was obtained from all the patients before specimen collection. This study was approved by the institutional review board. DNA was extracted using the standard phenol–chloroform method. The concentration and quantity of extracted DNA were measured using a NanoDrop spectrophotometer (NanoDrop Technologies).
MCAM analysis
MCAM analysis was conducted as previously described (Oishi et al. 2012). A detailed protocol of MCA was previously described (Toyota et al. 1999). We used a custom human promoter array (G4426A-02212; Agilent Technologies) comprising 36,579 probes corresponding to 9021 unique genes. The probes on the array were selected to recognize SmaI/XmaI fragments mainly derived from sequences near gene transcription start sites. Five micrograms of genomic DNA was digested with 100 U of methylation-sensitive restriction endonuclease SmaI (New England Biolabs) for 24 h at 25°C, which cleaves unmethylated DNA leaving blunt ends (CCC/GGG). Subsequently, the DNA was digested with 20 U of methylation-insensitive restriction endonuclease XmaI for 6 h at 37°C, creating sticky ends (C/CCGGG). Five hundred milligrams of digested DNA was ligated using 50 μL of RMCA12 (5′-CCGGGCAGAAAG-3′)/RMCA24 (5′-CCACCGCCATCCGAGCCTTTCTGC-3′) primers and T4 DNA ligase (TaKaRa Bio) for 16 h at 16°C. After filling in the overhanging ends of the ligated DNA fragments at 72°C, the DNA was amplified for 5 min at 95°C followed by 25 cycles of 1-min incubation at 95°C and 3-min incubation at 77°C using 100 pmol of RMCA24 primer. MCA products were labeled with Cy5 (red) for DNA from hepatoma samples (both tumor and adjacent normal) and Cy3 (green) for DNA from human blood mixture of three healthy volunteers using a randomly primed Klenow polymerase reaction (Invitrogen) for 3 h at 37°C. Human CpG island arrays (4 × 44 K) were purchased from Agilent Technologies. Microarray protocols, including labeling, hybridization, and post-hybridization washing procedures, are provided at http://www.agilent.com/. Labeled samples were then hybridized to arrays in the presence of human Cot-1 DNA for 24 h at 65°C. After washing, arrays were scanned using an Agilent DNA microarray scanner and analyzed using Agilent Feature Extraction software (FE version 9.5.1.1, Agilent Technologies) at St. Marianna University School of Medicine. We used GeneSpring software (Agilent) for choosing candidate genes after normalization of the raw data.
DNA methylation analysis
Hidden Markov models have been successfully used to partition genomes into segments of comparable stochastic structure (Durbin et al. 1998). Using these models for sequence analysis performed on the CpG plugin of bioinformatics software Geneious 5.5.8 (Biomatters), CpG islands were searched in the HBV genome (Kearse et al. 2012). Bisulfite PCR was performed using an EpiTect Bisulfite Kit (Qiagen) according to the manufacturer’s protocol. One microliter of bisulfite-treated DNA was used as a template. The primers used for amplifying CpG sequences in the HBx gene are described in Supplemental Table 1. After PCR, the biotinylated strand was captured on streptavidin-coated beads (Amersham Bioscience) and incubated with sequencing primers (Supplemental Table 1). The pyrosequencing reactions were performed using the PyroMark Q24 and/or PyroMark Q24 advanced (Qiagen). Pyrosequencing quantitatively measures the methylation status of several CpG sites in a given sequence. These adjacent sites usually show highly concordant methylation. Therefore, the mean percentage of methylation at detected sites was used as a representative value for each sequence.
LINE1 and AluYb8 methylation analysis
The LINE1 and AluYb8 methylation levels, as measured by pyrosequencing, are good indicators of the cellular levels of 5-methylcytosine (i.e., the global DNA methylation level). To quantify relatively high LINE1 and AluYb8 methylation levels, we used pyrosequencing technology (Igarashi et al. 2010). PCR and subsequent pyrosequencing for LINE1 and AluYb8 were performed using the PyroMark kit (Qiagen). This assay amplifies a region of the LINE1 or AluYb8 elements that includes three CpG sites. The PCR was conducted as follows: 45 cycles for 20 sec at 95°C, for 20 sec at 50°C, and for 20 sec at 72°C, followed by 5 min at 72°C. The biotinylated PCR product was purified and converted to single strands to serve as a template for the pyrosequencing reaction using the pyrosequencing vacuum prep tool (Qiagen). The pyrosequencing reactions were performed using the PyroMark Q24 and/or PyroMark Q24 advanced (Qiagen). The percentage of Cs relative to the total sum of the Cs and Ts at each CpG site was calculated. The average of the percentages of Cs at the three CpG sites was used to represent the overall LINE1 and AluYb8 methylation levels in each sample.
FISH analysis of HBV integration
We developed a FISH analysis method to detect HBV DNA and demonstrate its presence in PLC/PRF/5 cells (Supplemental Fig. 3). The slides were pretreated with hydrogen peroxide and rinsed in 1× phosphate-buffered saline (PBS) to minimize background and quench endogenous peroxidase activity. To remove the excess cytoplasm, the slides were treated with pepsin and then fixed with 1% formaldehyde in PBS/MgCl2. The slides were then dehydrated in an ethanol series (70%, 90%, and 100%) for 3 min at each step. The probes were designed based on the reference HBV sequence in PLC/PRF/5 DNA that is available from the Methylyzer 1.0 website (http://gbrowse.bioinfo.cnio.es/cgi-bin/VIRUS/HBV/). The FISH probes were prepared by combining the PCR-labeled probes (Supplemental Table 1), human Cot-1 DNA, and salmon sperm DNA. The probes were precipitated and mixed with hybridization buffer, and the probe DNA cocktail was denatured for 5 min at 95°C. The DNA on the slides was denatured by soaking in 70% formamide/2× SSC for 3 min at 74°C. The slides were immediately immersed in freshly prepared ice-cold 70% ethanol for 3 min, followed by 3-min immersions in 90% and then 100% ethanol. The denatured probe DNA was applied to the dry denatured slides and covered with a coverslip. The hybridization was performed for 16 h at 37°C.
Tyramide signal amplification (TSA)–FISH
TSA (tyramide signal amplification) detection kits were obtained from PerkinElmer. TSA-FISH detection was performed following the manufacturer’s protocols with minor modifications. High stringency washes (0.1× SSC) were used to reduce the background, and TNT buffer (0.1 M Tris-HCl at pH 7.5, 0.15 M NaCl, 0.05% Tween 20) was adjusted to pH 7.0–7.5. The biotin- or DIG-labeled probes were detected using streptavidin-HRP or anti-DIG-HRP in TNB (0.1 M Tris-HCl at pH 7.5, 0.15 M NaCl, 0.05% blocking reagent [supplied in the kit]) for 30 min at room temperature and washed twice for 5 min each in TNT buffer. For the tyramide amplification procedure, the slide was covered with tyramide solution (Tyr-Bio, 1:50) for 10 min at room temperature. The tyramide solution was removed, and the slides were washed twice for 5 min each with TNT at room temperature. Fluorochrome-conjugated streptavidin (stAv-Alexa 488) diluted in TNB was used to detect the Tyr-Bio. The slides were incubated for 30 min at room temperature, washed with TNT buffer twice for 5 min each at room temperature, and covered with an anti-fade reagent containing DAPI (Speel et al. 1997; Schriml et al. 1999).
Identification of the chromosomal locations of viral–host junctions
The viral–host junctions were amplified using primers specific for human AluYb8 repetitive sequences and HBV X regions (Supplemental Table 1; Minami et al. 1995; Murakami et al. 2004). One microliter of genomic DNA solution served as a template in the subsequent PCR. We used touchdown PCR for most of the assays. All PCR assays included a denaturation step for 30 sec at 95°C, followed by an annealing step at various temperatures for 30 sec and an extension step for 30 sec at 72°C. PCR products were analyzed using electrophoresis through 1% agarose gels. PCR products were ligated to pCR-XL-TOPO vector DNA (TOPO XL PCR Cloning kit; Invitrogen) and transformed into competent cells. Positive colonies were selected and isolated using a QIA prep Spin Miniprep Kit (Qiagen). Direct sequence analysis of TOPO-TA cloning products was performed using a 3130 genetic analyzer (Applied Biosystems) (Watanabe et al. 2011). All sequences were searched for matches with HBV and pCR-XL-TOPO sequences using Geneious 5.5.8 (Biomatters) sequence analysis and assembly software and the BLAST program available on the UCSC Genome Browser (http://genome.ucsc.edu/).
Analysis of HBV DNA integration site sequences using NGS
Agilent’s SureSelect target enrichment system is a highly efficient hybrid selection technique for optimizing NGS. We used this system and 12,391 custom baits covering the DNA sequences of HBV genotypes A to J and PLC/PRF/5 HBV sequences and optimized experiments for a GS FLX Titanium system (Roche). PLC/PRF/5, HepG2.2.15, and four paired tumor and nontumor samples (sample nos. 7, 8, 9, and 10 in Fig. 1B) were analyzed.
DNA methylation analysis of the integrated HBV genome as well as the adjacent human genome
DNA methylation was analyzed using bisulfite pyrosequencing (Oishi et al. 2012). The pyrosequencing reactions were performed using the PyroMark Q24 and/or PyroMark Q24 advanced (Qiagen). PLC/PRF/5, HepG2.2.15, and four paired tumor and nontumor samples (sample nos. 7, 8, 9, and 10 in Fig. 1B) were analyzed. Primers for methylation analysis of integration sites in PLC/PRF/5 are shown in Supplemental Table 1.
DNA methylation analysis of orthologous loci
Methylation levels of orthologous loci in HepG2.2.15 cells and in PBLs of a healthy volunteer at the same (empty) target sites of PLC/PRF/5 cells were analyzed using bisulfite pyrosequencing. Similarly, methylation levels of orthologous loci in PLC/PRF/5 cells and in PBLs at the same (empty) target sites of HepG2.2.15 cells were analyzed.
Allele-specific DNA methylation analysis of the integrated HBV genome as well as the adjacent human genome
Allele-specific DNA methylation was analyzed as described previously (Yamada and Ito 2011). The pyrosequencing reactions were performed using the PyroMark Q24 and/or PyroMark Q24 advanced (Qiagen).
Correlation analysis between the methylation pattern of the integrated HBV DNA and that of the human genome
DNA fragments, including 200 bp of the integrated HBV DNA and 200 bp of the human genome around the boundary, were analyzed for average methylation, GC content, and repetitive sequences in cell lines and clinical samples. RepeatMasker was used to identify repetitive elements in genomic sequences (AFA Smit, R Hubley, P Green, unpubl.). The Spearman correlation coefficient was used to assess correlations between the average methylation of the HBV DNA and that of the human genome. Correlations between GC content or repetitive sequences in the HBV DNA and the human genome were analyzed by using the Spearman correlation coefficient for continuous variables, and P < 0.05 was considered significant. All statistical analyses were performed using PRISM software for Windows, version 4 (GraphPad Prism).
Chromatin structure at the integrated HBV site
Using Bander software (Cheung et al. 2001; Furey and Haussler 2003), we analyzed the chromatin structure at the integrated HBV site in PLC/PRF/5 and HepG2.2.15.
Data access
All raw sequence data from this study have been submitted to the DDBJ Japanese Genotype-phenotype Archive (JGA; http://trace.ddbj.nig.ac.jp/jga/index_e.html) under accession number JGAS00000000015. Array data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE59405.
Supplementary Material
Acknowledgments
We thank Drs. K. Watashi and T. Wakita for cell lines and Drs. T. Takayama, K. Takasaki, and S. Kawasaki for clinical samples. We also thank Drs. N. Matsumoto and N. Yamada-Ohkawa, as well as other members of the laboratory, for advice and suggestions. A part of the data used for this research was originally obtained by a research project of Hiroyuki Yamamoto and Yoshiyuki Watanabe led by Professor Fumio Itoh and is available at the website of the NBDC/JST (http://biosciencedbc.jp/en/). This work was supported in part by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (JSPS KAKENHI grant no. 23590964 to H. Yotsuyanagi).
Author contributions: Y.W. conceived the study, designed and performed the experiments, analyzed the data, and wrote the manuscript. H. Yamamoto designed the experiments, analyzed the data and wrote the manuscript. R.O. performed the experiments and analyzed the data. M.T. provided intellectual support. M.Y., N.K., S.T., and A.S. provided clinical samples. H. Yotsuyanagi designed the experiments, analyzed the data and wrote the manuscript. K.K. provided intellectual support. F.I. supervised all aspects of the study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.175240.114.
References
- Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. 2013. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers WA, Blanchon L, Pradhan S, de Launoit Y, Kouzarides T, Fuks F. 2007. Viral oncoproteins target the DNA methyltransferases. Oncogene 26: 1650–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Nowak N, Jang W, Kirsch IR, Zhao S, Chen XN, Furey TS, Kim UJ, Kuo WL, Olivier M, et al. 2001. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature 409: 953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding D, Lou X, Hua D, Yu W, Li L, Wang J, Gao F, Zhao N, Ren G, Li L, et al. 2012. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next-generation sequencing-based approach. PLoS Genet 8: e1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerfler W. 2008. In pursuit of the first recognized epigenetic signal-DNA methylation: a 1976 to 2008 synopsis. Epigenetics 3: 125–133. [DOI] [PubMed] [Google Scholar]
- Doerfler W, Remus R, Müller K, Heller H, Hohlweg U, Schubbert R. 2001. The fate of foreign DNA in mammalian cells and organisms. Dev Biol 106: 89–97. [PubMed] [Google Scholar]
- Durbin R, Eddy S, Krogh A, Mitchison G. 1998. Biological sequence analysis. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Fernandez AF, Rosales C, Lopez-Nieva P, Graña O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A, Fraga MF, et al. 2009. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res 19: 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al. 2012. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 44: 760–764. [DOI] [PubMed] [Google Scholar]
- Furey TS, Haussler D. 2003. Integration of the cytogenetic map with the draft human genome sequence. Hum Mol Genet 12: 1037–1044. [DOI] [PubMed] [Google Scholar]
- Gatza ML, Chandhasin C, Ducu RI, Marriott SJ. 2005. Impact of transforming viruses on cellular mutagenesis, genome stability, and cellular transformation. Environ Mol Mutagen 45: 304–325. [DOI] [PubMed] [Google Scholar]
- Hilleman MR. 2004. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc Natl Acad Sci 101: 14560–14566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi S, Suzuki H, Niinuma T, Shimizu H, Nojima M, Iwaki H, Nobuoka T, Nishida T, Miyazaki Y, Takamaru H, et al. 2010. A novel correlation between LINE-1 hypomethylation and the malignancy of gastrointestinal stromal tumors. Clin Cancer Res 16: 5114–5123. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S, et al. 2012. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res 22: 593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, et al. 2013. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res 23: 1422–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28: 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CM, Koike K, Saito I, Miyamura T, Jay DG. 1991. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351: 317–320. [DOI] [PubMed] [Google Scholar]
- Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, Kurokawa K. 1994. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 19: 810–819. [PubMed] [Google Scholar]
- Lau C-C, Sun T, Ching AK, He M, Li JW, Wong AM, Co NN, Chan AW, Li PS, Lung RW, et al. 2014. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 25: 335–349. [DOI] [PubMed] [Google Scholar]
- Li S, Mao M. 2013. Next generation sequencing reveals genetic landscape of hepatocellular carcinomas. Cancer Lett 340: 247–253. [DOI] [PubMed] [Google Scholar]
- Lupberger J, Hildt E. 2007. Hepatitis B virus–induced oncogenesis. World J Gastroenterol 13: 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami M, Poussin K, Brechot C, Paterlini P. 1995. A novel PCR technique using Alu-specific primers to identify unknown flanking sequences from the human genome. Genomics 29: 403–408. [DOI] [PubMed] [Google Scholar]
- Mirabello L, Sun C, Ghosh A, Rodriguez AC, Schiffman M, Wentzensen N, Hildesheim A, Herrero R, Wacholder S, Lorincz A, et al. 2012. Methylation of human papillomavirus type 16 genome and risk of cervical precancer in a Costa Rican population. J Natl Cancer Inst 104: 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Minami M, Daimon Y, Okanoue T. 2004. Hepatitis B virus DNA in liver, serum, and peripheral blood mononuclear cells after the clearance of serum hepatitis B virus surface antigen. J Med Virol 72: 203–214. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Shibata T. 2013. Comprehensive genome sequencing of the liver cancer genome. Cancer Lett 340: 234–240. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Watanabe Y, Yoshida Y, Sato Y, Hiraishi T, Oikawa R, Maehata T, Suzuki H, Toyota M, Niwa H, et al. 2012. Hypermethylation of Sox17 gene is useful as a molecular diagnostic application in early gastric cancer. Tumour Biol 33: 383–393. [DOI] [PubMed] [Google Scholar]
- Orend G, Kuhlmann I, Doerfler W. 1991. Spreading of DNA methylation across integrated foreign (adenovirus type 12) genomes in mammalian cells. J Virol 65: 4301–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriml LM, Padilla-Nash HM, Coleman A, Moen P, Nash WG, Menninger J, Jones G, Ried T, Dean M. 1999. Tyramide signal amplification (TSA)-FISH applied to mapping PCR-labeled probes less than 1 kb in size. Biotechniques 27: 608–613. [DOI] [PubMed] [Google Scholar]
- Sells MA, Chen ML, Acs G. 1987. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci 84: 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speel EJ, Ramaekers FC, Hopman AH. 1997. Sensitive multicolor fluorescence in situ hybridization using catalyzed reporter deposition (CARD) amplification. J Histochem Cytochem 45: 1439–1446. [DOI] [PubMed] [Google Scholar]
- Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al. 2012. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 44: 765–769. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Nosaka K, Yasunaga J, Maeda M, Mueller N, Okayama A, Matsuoka M. 2005. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Q, Robertson KD. 2003. Stealth technology: how Epstein-Barr virus utilizes DNA methylation to cloak itself from immune detection. Clin Immunol 109: 53–63. [DOI] [PubMed] [Google Scholar]
- Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, et al. 2013. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 34: 787–798. [DOI] [PubMed] [Google Scholar]
- Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe-Toyota M, Baylin SB, Issa JP. 1999. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res 59: 2307–2312. [PubMed] [Google Scholar]
- Uozaki H, Fukayama M. 2008. Epstein-Barr virus and gastric carcinoma-viral carcinogenesis through epigenetic mechanisms. Int J Clin Exp Pathol 1: 198–216. [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Castoro RJ, Kim HS, North B, Oikawa R, Hiraishi T, Ahmed SS, Chung W, Cho MY, Toyota M, et al. 2011. Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. PLoS ONE 6: e23320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Ito T. 2011. Highly efficient PCR assay to discriminate allelic DNA methylation status using whole genome amplification. BMC Res Notes 4: 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.