

Abstract
Glioblastoma (GBM) is a prototypical heterogeneous brain tumor refractory to conventional therapy. A small residual population of cells escapes surgery and chemoradiation, resulting in a typically fatal tumor recurrence ∼7 mo after diagnosis. Understanding the molecular architecture of this residual population is critical for the development of successful therapies. We used whole-genome sequencing and whole-exome sequencing of multiple sectors from primary and paired recurrent GBM tumors to reconstruct the genomic profile of residual, therapy resistant tumor initiating cells. We found that genetic alteration of the p53 pathway is a primary molecular event predictive of a high number of subclonal mutations in glioblastoma. The genomic road leading to recurrence is highly idiosyncratic but can be broadly classified into linear recurrences that share extensive genetic similarity with the primary tumor and can be directly traced to one of its specific sectors, and divergent recurrences that share few genetic alterations with the primary tumor and originate from cells that branched off early during tumorigenesis. Our study provides mechanistic insights into how genetic alterations in primary tumors impact the ensuing evolution of tumor cells and the emergence of subclonal heterogeneity.
The presence of multiple cancer cell clones within a single tumor has been explained as a Darwinian process in which different clones compete for limited resources, and the most phenotypically fit cells eventually prevail (Greaves and Maley 2012; Yates and Campbell 2012; Aparicio and Caldas 2013). It has been suggested that such heterogeneity allows a tumor to respond to local and systemic selective pressures, such as those exerted by therapeutic interventions (Nowak and Sigmund 2004; Greaves and Maley 2012; Bozic et al. 2013). For example, the presence of subclonal driver mutations in cancer cells was indicative of rapid disease progression in chronic lymphocytic leukemia (Landau et al. 2013). Using single-cell sequencing or massively parallel sequencing, clonal architectures ranging from complex polyclonal structures to monoclonal tumors have been described in cancer lineages such as those of the breast, kidney, and blood (Navin et al. 2011; Ding et al. 2012; Shah et al. 2012; Landau et al. 2013; Gerlinger et al. 2014). Distinct subclonal tumor cell populations relating to mosaic amplification of receptor tyrosine kinases were reported in glioblastoma (GBM), suggesting a similarly dynamic architecture for this disease (Snuderl et al. 2011; Nickel et al. 2012; Szerlip et al. 2012; Sottoriva et al. 2013).
GBM is the most common malignant brain tumor in adults (Van Meir et al. 2010; Dunn et al. 2012) and is standardly treated with surgical resection followed by concomitant radiotherapy and administration of the alkylating agent temozolomide (TMZ) (Stupp et al. 2005). Despite this aggressive treatment regimen, the median time to disease recurrence is 6.9 mo, with >90% of GBM tumors recurring at the original site (Wen and Kesari 2008). Therapy targeting the epidermal growth factor receptor variant III (EGFRvIII) led to an improved overall survival time among patients with GBM; however, 82% of these patients lost EGFRvIII expression when the tumor recurred, which suggests a competitive advantage for non-EGFRvIII expressing clones in these tumors (Sampson et al. 2010). Achieving a better understanding of the clonal structure of cancer cells is thus of vital importance and may inform the development of additional targeted therapies for rapidly lethal forms of cancer, such as GBM.
Here, we analyzed genomic profiles of 252 GBM samples from The Cancer Genome Atlas (TCGA) (Brennan et al. 2013), and 60 biopsies taken from 23 pairs of pre- and post-treatment GBMs, to understand (1) the intratumoral clonal compositions of primary GBM; and (2) how GBM responds to therapeutic intervention. Our results provide a molecular portrait of GBM recurrence.
Results
Sample characteristics and mutation calling
In this study, we performed an analysis of genomic data from 252 untreated GBM samples from The Cancer Genome Atlas (cohort I). To study tumor responses to treatment, we obtained a second cohort of tumor samples, for which we collected pairs of primary and first recurrent GBM samples from 21 patients and added pairs of secondary GBM and next disease occurrence samples from two patients (cohort II). Prior to disease recurrence, 21 patients in cohort II had received radiotherapy, and 17 of them had also received adjuvant TMZ. A variety of treatments, including carmustine and anti-inflammatory agents, were administered to the remaining patients in cohort II. An IDH1 R132 mutation was detected in two cases. The clinical data for cohort II is summarized in Supplemental Table 1.
Integrative analysis identifies clonal and subclonal mutations
To investigate the clonal architecture of GBM, we classified somatic mutations into clonal and subclonal categories by integrating variant allele fraction, DNA copy number, genotype, and tumor purity (Methods). We used PyClone, a Bayesian clustering method that simultaneously estimates the distribution of the cellular frequency for each mutation (Roth et al. 2014). After correcting for tumor cell content, the cellular frequency of each mutation was used to infer the mutation clonality. By definition, clonal mutations occur early in or before malignant transformation and thus are present in all tumor cells. In contrast, subclonal mutations occur later in tumor expansion and are present in only a subset of tumor cells. Of 17,636 somatic single nucleotide variations (sSNVs) detected in cohort I (Brennan et al. 2013), we found 67.9% to be clonal (n = 11,973, median across samples; 68.3% ± 19.3%) and 29.8% (n = 5249) to be subclonal. A minor fraction of mutations could not be classified (2.3%). To validate our classification approach, we presumed that clonal mutations were present in all tumor compartments and thus should be spatially ubiquitous. We sequenced the exomes of two nonoverlapping sectors from 13 tumors in cohort II, classified detected mutations as clonal or subclonal, and compared both types of sSNVs with spatial distribution. The percentage of ubiquitous mutations (present in both tumor sectors) per sample ranged from 43.1% to 74.7% (59.3.5% ± 10%) (Supplemental Fig. 1A), and the majority of ubiquitous mutations (85.6%) were classified as clonal. In contrast, 41.2% of the private mutations were classified as clonal (Fig. 1A; Supplemental Table 2), which was a significant difference (P = 2.2 × 10−16, χ2 test). In a single recurrent tumor sample that had three biopsies available, we found a similar percentage of ubiquitous mutations (45.3%) (Supplemental Fig. 1B). Separating patients into discrete age groups by intervals of 10 yr, we found a significant linear correlation between clonal mutations and age (P = 3.69 × 10−7, multiple group analysis of variance) (Fig. 1B), suggesting that the majority of clonal mutations were acquired before gliomagenesis and accumulated over the life span of the patient at time of diagnosis. This observation was corroborated by the dominant presence of C > T transitions, reminiscent of germline substitutions, in the clonal mutation spectrum (Supplemental Fig. 2A; Nik-Zainal et al. 2012; Alexandrov et al. 2013; Tomasetti et al. 2013). We did not observe a correlation between age and subclonal mutations (P = 0.62). We evaluated the clonal status of significantly mutated GBM genes and recurrent cancer genes from the cancer gene census (nonsilent mutation frequency greater than three in the cohort) (Supplemental Fig. 2B). Very few genes were entirely clonal or subclonal (Andor et al. 2014). However, the majority (90.5%) of TP53 and PIK3CA/PIK3R1 mutations were clonal, suggesting a founder role for these events in GBM. In contrast, receptor tyrosine kinases such as EGFR, PDGFRA, and AKT pathway regulator PTEN were more evenly distributed between the clonal and subclonal mutation groups across cohort I, suggesting heterogeneous deregulation timing.
Figure 1.
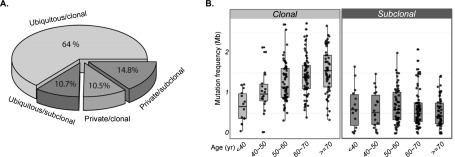
Inference of mutational clonality and association with patient age. (A) Validation of clonal and subclonal classifications using multisector sequencing. Mutations found in all nonoverlapping tumor sectors from the same tumor were defined as ubiquitous mutations, otherwise as private. Ubiquitous and private mutations were further subdivided into clonal and subclonal mutations, resulting in four categories (ubiquitous/clonal, ubiquitous/subclonal, private/clonal, and private/subclonal). (B) The clonal mutation frequency correlates with age across 221 primary GBMs with available age information (left panel), whereas the subclonal mutation frequency does not (right panel). The patients were separated into discrete age groups by intervals of 10 yr.
Subclonal mutations are associated with p53 pathway alterations
To identify genetic aberrations that are associated with subclonal progression, we juxtaposed somatic mutations, DNA copy number alterations, and clonal compositions in cohort I. Interestingly, deregulation of the p53 pathway, in particular TP53 somatic mutations (n = 78) and MDM2 amplifications (n = 21), was strongly associated with an increased fraction of subclonal mutations (P = 6 × 10−5, Wilcoxon rank-sum test) (Fig. 2A), and this association was independent of age (Methods). G-CIMP positive tumors, a subclass of GBM characterized by a hypermethylator phenotype, mutations in isocitrate dehydrogenase I (IDH1), and mutations in TP53 (Noushmehr et al. 2010), accordingly showed a high proportion of subclonal mutations. Since subclonal mutations were associated with unfavorable outcome in chronic lymphocytic leukemia (Landau et al. 2013), we compared the event-free survival of patients represented in our data set whose primary tumor had a dominance of clonal mutations to patients with tumors in which high fractions of subclonal mutations were detected. Interestingly, the subclonal tumor group showed significantly longer event-free survival than the clonal tumor group (P = 0.025, log-rank test), but only when limiting the analysis to patients younger than 55 yr of age at time of diagnosis (Fig. 2B; Supplemental Fig. 3). TP53 mutation status alone did not correlate with outcome (data not shown). The increased fraction of subclonal mutations in GBM harboring p53 pathway alterations may be indicative of an elevated tolerance to DNA damage or apoptosis suppression (Offer et al. 2002; Kojima et al. 2005; Pant et al. 2013).
Figure 2.
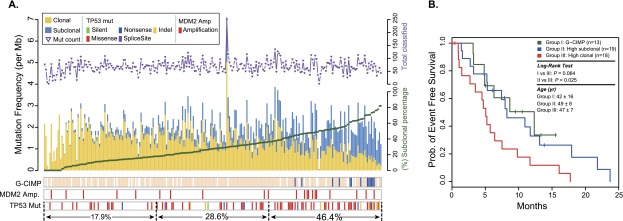
Association of subclonal fraction with TP53 mutation and event-free survival. (A) Clonal (yellow) and subclonal (blue) mutation frequencies and their association with G-CIMP methylation phenotype (blue) and p53 pathway-related alterations. The total number of sSNVs per patient is indicated in the top panel. (B) Patients with non-G-CIMP GBM harboring a higher subclonal mutation frequency (top 30% of non-G-CIMPs; Group II; blue) exhibited a longer event-free survival time compared to non-G-CIMP patients, whose tumor cell mutations had higher clonal frequency (bottom 30% of non-G-CIMPs; Group III; red). G-CIMP cases are grouped separately into Group I (dark green) and show event-free survival similar to that of Group II. Only patients younger than 55 yr of age were included in Groups II and III, and all G-CIMP cases were included in Group I.
p53 pathway status affects mutational burden in recurrent tumor
To further examine the effect of TP53 mutations on tumor evolution, we performed exome sequencing and DNA copy number analysis on 23 pairs of first GBM and matched recurrent tumors in cohort II. In line with a recent report (Johnson et al. 2014), we found that the majority of primary GBM mutations could also be detected in the tumor after disease relapse (median overlap 67%; range 18%–85%) (Supplemental Table 3). Five recurrent tumors showed a very high number of clonal and/or subclonal sSNVs and were considered hypermutated (12–131 mutations per Mb). Despite the high overall population frequency of genetic alterations in TP53 (27.9%), EGFR (57.4%), and CDKN2A (57.8%) (Brennan et al. 2013), copy number alterations and sSNVs in these and other GBM driver genes were frequently present at time of diagnosis but absent in the tumor recurrence, mutated at a difference base, or deleted/amplified with different copy number breakpoints (Fig. 3A). It is therefore unlikely that any of these genes can be unambiguously flagged as an initiating mutation of IDH1 wild-type glioblastoma, and their frequent alterations can be best explained by intratumoral evolutionary pressures resulting in convergent evolutionary events. For example, we observed focal amplification of the Rb pathway regulator CDK4 in three primary GBM tumors but not in two of the three corresponding recurrent tumors (Fig. 3A). Interestingly, both cases had acquired a homozygous deletion of upstream Rb pathway regulator CDKN2A, suggesting a compensatory effect to maintain the deregulation of the Rb pathway. We speculate that one common pathway of tumor initiation starts with loss of 10q and gain of Chromosome 7, which predispose to deactivation of PTEN and activating mutations/amplifications in major oncogenes.
Figure 3.

Spectrum of somatic mutations in primary and recurrent GBM. (A) The number of mutations detected in exome sequencing (top panel), clinical characteristics (second panel), EGFRvIII aberrations (third panel), copy number alterations (fourth panel), and somatic exonic mutations (bottom panel). Only frequently amplified, deleted, and mutated GBM genes are included in the copy number and mutation panels. ω (mutation of a different nucleotide in the same gene); Ω (DNA copy number alteration defined by different breakpoints). Clonal status for one sample with an extreme number of mutations could not be reliably classified and is shown in gray. (B) Comparison of subclonal (left panel) and clonal (right panel) mutation frequencies between primary or secondary GBM and matching recurrent tumors. Patients treated with TMZ are indicated with a brown triangle. (C) Association of TP53 mutation with subclonal mutational burden. Changes in clonal (left panel) and subclonal (right panel) mutation frequency are indicated as yellow dots for GBM harboring wild-type TP53 and purple triangles for tumors with mutated TP53 in both primary and recurrent tumors. The green diamond represents a patient who acquired a TP53 mutation at GBM recurrence; the blue diamond represents a case where a TP53 mutation was observed in the primary but not the matching tumor recurrence. A P-value was obtained by performing a Wilcoxon rank-sum test comparing subclonal mutation frequency change between TP53 wild-type and mutant groups.
Mutations in TP53 were detected in seven primary GBMs, and in two cases, the DNA binding domain mutation was no longer detected in the recurrent tumor (mutant locus coverage 279× and 46× in the respective recurrent samples). In one case, the original TP53 mutation was replaced with a different variant. One recurrent tumor acquired a TP53 mutation that was not found in the matching primary, an observation confirmed by ultradeep sequencing of the diagnostic tumor sample (average mutant locus coverage 1100×) (Supplemental Fig. 4). We compared the mutation frequencies (mutations per megabase, adjusted for coverage) at diagnosis and after disease relapse, and found disparate mutagenesis trends between TP53 mutant tumors and TP53 wild-type tumors (Fig. 3B). When compared to the matching primary tumor, TP53 mutant recurrent GBM showed an increase in subclonal mutation frequency, whereas GBM with wild-type TP53 did not, and the difference was statistically significant (P = 0.0015; Wilcoxon rank-sum test) (Fig. 3C). The clonal mutation frequency was unaffected (P = 0.23; Wilcoxon rank-sum test). Tumor purity, estimated by allele-specific copy number analysis of tumors (ASCAT) (Van Loo et al. 2010), did not confound the results (Supplemental Fig. 5). This increase in subclonal mutation frequency in TP53 mutant but not TP53 wild-type tumors after disease relapse further exposed the association between TP53 mutation and subclonal tumor progression.
Whole-genome sequencing inferred tumor evolution
To comprehensively cover GBM tumor heterogeneity and disease progression, we performed whole-genome sequencing on 10 primary-recurrence pairs from cohort II (Supplemental Table 1). We identified a median number of 4224 mutations (range from 2063 to ∼142,984) in the genome-wide sequencing data from 20 tumors (Supplemental Table 4). One recurrent tumor had an extremely high number (142,984) of mutations, and this sample was among the five recurrent tumors with hypermutated exomes (Fig. 3A).
We utilized the large number of mutations detected in each tumor to investigate the architecture of disease evolution by analyzing the distribution of purity-scaled variant allele fractions (PS-VAFs) determined using the SciClone method (Miller et al. 2014). SciClone is a variational Bayesian algorithm to infer genetic composition of subclones from the variant allele frequencies of somatic mutations. To avoid potential confounding effects from DNA dosage, loss of heterozygosity (LOH), and sequencing depth, we limited the analysis to mutations with at least 60× coverage and located in copy-neutral and LOH-free regions. Theoretically, heterozygous mutations with 0.5 PS-VAF are clonal mutations present in all tumor cells.
Analysis of PS-VAFs from paired primary and recurrent tumors revealed at least two evident patterns of GBM relapse. The ancestral cell of origin pattern was demonstrated by cases TCGA-06-0152 (Fig. 4A) and TCGA-14-1034 (Supplemental Fig. 6A). In this pattern, primary and recurrent tumors shared a cluster of clonal mutations close to PS-VAF 0.5; whereas at the same time, a large cluster of clonal mutations in the primary tumor disappeared in the matching recurrent tumor. The limited overlap of clonal mutations implicated a common ancestral cell that gave rise to both primary and recurrent tumors. The lack in overlapping subclonal mutations and the wide range of PS-VAFs from 0 to 0.5 in primary and recurrent specific mutations suggested that the two subsequent disease instances evolved independently (Fig. 5A).
Figure 4.

Comprehensive comparison of mutations in four pairs of primary and recurrent GBM. Each row in the upper panels of A–D represents exonic mutations detected in sectors of the primary tumor (P.1, P.2) and exonic mutations detected in sectors of the matching recurrent GBM (R.1, R.2, R.3). Mutations are color-coded for cancer cell fractions (tumor purity-corrected cellular frequencies), in which a cancer cell fraction of 1.0 indicates that a mutation was present in all tumor cells. The columns represent the same chromosomal nucleotide position in all samples displayed. The bottom panels of A–D show scatter plots of the copy-neutral and non-LOH localizing mutations detected in whole-genome sequencing data of the primary and recurrent tumor. The x-axis and y-axis indicate purity-scaled variant allele fractions. Variant allele frequency distribution plots for individual tumors are also displayed at the bottom for primary tumor and at the right side for its matching recurrent tumor.
Figure 5.

Schematic illustration of the patterns of tumor recurrence. (A) In the ancestral cell origin model, therapeutic interventions removed all dominant disease clones from the primary tumor but not refractory ancestral cells. The ancestral cell accumulates new mutations and proliferates to become the recurrent tumor. In this model, mutations shared by primary and recurrent tumors were presumably only from the ancestral cell, and the two subsequent tumors were thus more divergent. (B) In the clonal evolution model, the treatment removed most of the primary tumor cells, but cells from the major primary disease clones survived and continued to grow to result in a recurrent tumor. During this process, additional mutations were accumulated and therefore detected in the recurrent tumor, and all primary tumor clonal mutations are retained in the recurrence.
A different pattern was observed in cases TCGA-06-0125 (Fig. 4B), TCGA-06-0190, TCGA-06-0221, and TCGA-14-1402 (Supplemental Fig. 6B,C,D). In these samples, a high degree of overlap between clonal mutations in the primary and the recurrent tumor was observed, as well as a general absence of primary-specific clonal mutations, which would argue against the evolution of the recurrent tumor from an ancestral cell. For these cases, it appeared more likely that the recurrent tumors developed from the residual primary disease, and specific subclones were retained and subsequently expanded at tumor relapse. We note that this residual-based recurrence was dynamic. For instance, in the case TCGA-06-0221 (Supplemental Fig. 6C), a primary subclone at 0.1 PS-VAF underwent clonal expansion, resulting in a cluster of mutations at 0.5 PS-VAF in the relapse (Fig. 5B).
Two cases, TCGA-06-0210 and TCGA-06-0211, could not be classified into either model (Fig. 4C,D). The presence and expansion of subclonal mutations of the primary tumors at recurrence suggested residual disease development, but the presence of clonal primary-only mutations suggested the ancestral cell model. We speculate that the two models might both be operating simultaneously in these tumors. In the remaining two pairs, either the primary tumor (TCGA-19-1389) or the recurrent tumor (TCGA-06-0171) was found to be monoclonal, which coincided with predicted tumor purity levels of 0.31 and 0.2, respectively; and we expect that the relative lack of tumor cells interfered with prediction of evolutionary patterns (Supplemental Fig. 6E,F).
To address the possibility that the lack of overlap between subsequent tumors and subclones was due to intratumoral heterogeneity, we analyzed exome sequencing data from a second, and in one case a third, sector of seven primary and six recurrent cohort II tumors. On average, 20 additional exonic mutations were detected in the second primary tumor biopsy, which represented a 25% increase on the total number of mutations found in the primary tumor. However, the additionally recovered mutations were in majority unique to the second primary sample (median: 17 of 20) and not detected in the tumor recurrence (Supplemental Table 5). Analysis of a second recurrent tumor biopsy did not alter this result but confirmed that the majority of “second biopsy” mutations were not conserved between time points. We therefore suggest that intratumoral heterogeneity did not explain the large number of mutations that were uniquely detected in primary and recurrence.
Evolutionary phylogenetic trees constructed from exome mutation profiles showed that primary and recurrent tumor sectors grouped in separate branches, with varying separation structures (Fig. 6). In one sample, TCGA-06-0125, the tree suggested that the tumor cells seeding the recurrent tumor were derived from primary tumor sector 2, which is consistent with the observations made in our analysis of the whole-genome sequencing data. Similarly, in TCGA-06-0211, the structure suggested that the tumor cells responsible for disease relapse were predominantly found in sector 1, where sectors 2 and 3 branched off linearly at a later time point.
Figure 6.

Phylogenetic trees based on multisector sequencing of primary and recurrent GBM. Phylogenetic trees were constructed using mutations from exome sequencing data of all biopsies of individual tumors. The length of branches in the tree proportionally represents the number of mutations, and putative cancer genes are labeled.
Therapy-induced hypermutation is a subclonal process
We observed temozolomide treatment induced hypermutation, previously described in secondary GBM (Hunter et al. 2006; Johnson et al. 2014), in the recurrent tumor of five patients. The majority of mutations were classified as subclonal, and this was confirmed by the analysis of two independently sequenced biopsies from one hypermutated recurrent tumor, in which we observed 2429 and 5980 somatic point mutations, respectively, but only 163 shared mutations between the two biopsies (Supplemental Table 2). Multiple DNA mismatch repair genes, including MSH2, MSH6, MLH1, MLH3, and POLD3, were found to be mutated in this specific tumor; however, none was shared by the two independently sequenced biopsies, suggesting that mechanisms not related to these genes and resulting in hypermutation could exist. Although the number of samples was too small to identify genes associated with hypermutator phenotype at statistical significance, we noted that the receptor tyrosine kinases EGFR and ALK were among the eight genes mutated in all six hypermutator samples (Supplemental Table 6). MGMT was methylated in all five primary tumors and in three of the five recurrent tumors, whereas MGMT status was not available for the remaining three recurrent tumors.
Following their second surgery to remove a GBM tumor, the five patients had respective lengths of survival of 35, 64, 107, 191, and 245 d, relative to a median survival of approximately 8 mo after surgery upon disease progression (Wen and Kesari 2008). The clinical outcome of the hypermutators illustrated the lethality of GBM, but the limited number of cases is unable to confirm that a high level of mutation is associated with relatively aggressive disease progression at statistically significant levels.
Discussion
As indicated by the adjective “multiforme,” the histopathological features of glioblastoma show a high degree of intratumor heterogeneity. Genomic studies have further illustrated this disease characteristic by demonstrating local variation in the amplification patterns of receptor tyrosine kinases (Snuderl et al. 2011; Szerlip et al. 2012; Sottoriva et al. 2013; Francis et al. 2014) as well as a wide landscape of somatic alterations, expression subtypes, and epigenetic differences across GBM (Noushmehr et al. 2010; Verhaak et al. 2010; Brennan et al. 2013; Zheng et al. 2013). Here, we expand our knowledge of GBM by evaluating heterogeneity in a large number of primary tumor samples from TCGA as well as through a comparison of pre- and post-treatment GBM tumor pairs.
We developed a computational approach to infer the cellular frequency of sSNVs and to classify mutations as clonal or subclonal, which we then validated using multisector sequencing (Gerlinger et al. 2012). Corroborating the findings from previous reports (Nik-Zainal et al. 2012; Alexandrov et al. 2013; Tomasetti et al. 2013), we note that the number of clonal but not subclonal mutations found at the time of diagnosis increased with the age of the patient, and that clonal mutations reflected the signature of germline substitutions. Over the lifetime of the patient, the cancer cell of origin may thus have been subjected to mutational processes before acquiring the necessary alterations to become tumorigenic, and this observation could be used to increase the specificity of algorithms aimed at identifying cancer-contributory genes (Lawrence et al. 2013). Mutation of TP53 has been related to an increased frequency of double-strand breaks and chromothripsis in medulloblastoma (Rausch et al. 2012) and tumor progression from low-grade to high-grade glioma (Ishii et al. 1999; Fulci et al. 2002). We extended our knowledge of the damaging effects of TP53 deactivation by identifying a significant correlation with increased subclonal mutation frequency. The causality of this association could not be determined in our analysis, but we speculate that the apoptosis-negating properties of TP53 DNA binding domain mutations result in an increased tolerance for acquiring and sustaining single nucleotide variants. GBM as a cancer type is uniquely characterized by the general absence of metastasis and a perceived lack of self-seeding by circulating tumor cells (Kim et al. 2009). Intratumor heterogeneity is therefore likely dictated by local competitive advantages resulting from the proliferation of enhanced genomic alterations, increased vascularity, or the morphological aspects of tumor growth such as the ability to mix with primary tissues or expand into the ventricles (González-García et al. 2002). Through longitudinal comparisons of tumor samples before and after treatment, we found that TP53-mutated tumors showed a further increase in clonal complexity at time of relapse, whereas TP53 wild-type recurrences appeared to have gone through an evolutionary bottleneck, which resulted in relatively monoclonal recurrent tumors. A high rate of complexity may provide the tumor more routes to escape therapeutic challenges, yet our data also suggested that increased frequency of subclonal mutations was related to a relatively favorable event-free survival. Although further research is needed, we suggest two explanations for this paradox. First, the longer interval between tumor occurrences may be a reflection of a slower developing disease, which allows more time for subclones to proliferate to detectable levels. Alternatively, the absence of a dominant aggressive clone may indicate a reduced rate of tumor growth as a result of the larger number of cells competing for space in the cranial cavity. A number of different genetically engineered mouse models of GBM have been developed by manipulating tumor suppressors such as Pten, Trp53, and Rb1, and it would be of interest to evaluate whether the association between p53 pathway alterations and subclonal tumor progression is captured in these models. Importantly, such experimental systems may be used to determine whether higher levels of intratumoral heterogeneity are associated with decreased efficacy of targeted therapies, which one might predict as more variety may mean more routes to resistance. Focusing on the general patterns of disease recurrence, we found a general instability of genomic alterations after GBM recurrence relative to GBM primary tumor samples, which prevented the identification of founding mutational events. The size of our cohort II (n = 23) may not be sufficient to detect more subtle disease pathway associations at the levels of statistical significance, and this analysis was further complicated by the variety of therapies provided to the patients, ranging from the current standard of care for primary GBM (concomitant radiotherapy and temozolomide, termed the Stupp protocol) (Stupp et al. 2005) to pre-2005 treatment modalities.
We combined whole-genome sequencing and exome sequencing of multiple tumor biopsies to capture the genealogy of GBM recurrence at sufficient detail to infer the phylogeny of tumor subclones and to trace their pattern of disease recurrence. The existence of glioma stem cells has been a subject of much debate and has been suggested to relate to radio- and chemotherapy resistance (Bao et al. 2006), and this model may explain the patterns of private and shared mutations observed in the two GBMs where the recurrence appeared to have evolved from an ancestral cell (Gilbertson and Rich 2007). In contrast, we also identified four cases in which all clonal primary tumor mutations could be detected in the relapse, suggesting a clonal evolution model. Finally, the exact pattern of the four remaining cases could not be derived, suggesting either extensive intratumoral heterogeneity and/or lack of granularity due to tumor purity.
In conclusion, this study has provided a better understanding of intratumor heterogeneity and disease progression in GBM. Our dissection of the temporal sequence of mutations revealed mutational forces acting on the cancer genome and resulted in new insights into the patterns and dynamics of tumor evolution. Although the therapeutic opportunities for GBM remain limited, continuing efforts to detail the mechanisms of disease relapse will contribute to our ability to provide curative treatment for this lethal disease.
Methods
Whole exome DNA sequencing and targeted resequencing
DNA of multisector and additional non-TCGA primary and recurrent paired tumor samples in cohort II was extracted by the University of Texas MD Anderson Cancer Center DNA Analysis Facility following standard protocols. All specimens were obtained from patients with appropriate consent from the relevant institutional review board. Samples were then multiplexed and sequenced on Illumina HiSeq 2000 by the Sequencing and Microarray Facility at an average target exome coverage of 100× using 76-bp paired-end reads. Targeted resequencing of selected mutations for validation was performed by PCR using a microfluidic device (Fluidigm Corp.). PCR primers with Fluidigm-compatible tails were designed to flank the sites of interest and produce amplicons of 200 bp ± 20 bp. Oligonucleotides containing Illumina adaptor sequences were mixed with 20–50 ng of each DNA sample along with a sample-specific molecular barcode and a sequence complementary to the primer tails. This mixture was used as the PCR template for each sample amplified on the Fluidigm access array. PCR was performed on the Fluidigm access array according to the manufacturer’s instructions. Barcoded libraries were recovered for each sample in a single collection well on the Fluidigm access array, quantified using PicoGreen dsDNA Quantitation Reagent (Invitrogen), and concentrations were normalized for use across libraries. Libraries were loaded on the Illumina MiSeq instrument and sequenced using paired-end 150-bp sequencing reads.
Whole genome DNA sequencing
Library construction and sequencing alignment (using the Human Genome Reference Consortium build 37) were performed by the Broad Institute as previously described (Brennan et al. 2013). All whole-genome sequencing BAM files are available through The Cancer Genomics Hub (https://cghub.ucsc.edu/).
Exome sequence data processing and mutation calling
Exome sequencing reads were aligned to the human reference genome (hg19) using Burrows-Wheeler Aligner 0.7.2 (Li and Durbin 2009). BAM files were subjected to duplication marking, indel realignment, and recalibration using Picard version 1.7 (http://broadinstitute.github.io/picard/) and GATK version 2.4.9 (McKenna et al. 2010). On average, 70% of the bases on exonic regions were covered with at least 20 reads of sequence. We then applied MuTect algorithm (version 1.14) (Cibulskis et al. 2013) to identify somatic single nucleotide variations (sSNVs) from tumor and patient-matched normal blood samples. In this manuscript, we limited our analysis to the mutations whose variant allele fractions were at least 0.05. All sSNVs were annotated by ANNOVAR (Wang et al. 2010) and Oncotator (http://www.broadinstitute.org/oncotator). Only mutations in exon regions were included in subsequent analyses. For primary samples from cohort I, sSNVs were publically available and downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov/docs/publications/gbm_2013/). Mutation frequency was calculated by dividing the number of somatic mutations by the number of bases that were properly covered for mutation calling.
Validation of somatic single nucleotide variants
To validate our somatic single nucleotide mutation calls, we performed targeted resequencing at high coverage (>1100×). We randomly selected 239 unique bases, which had been found to be mutated in primary, recurrence, or both, and sequenced these in both primary and recurrences. These sites corresponded to 367 sSNVs and 101 wild-type nucleotides. In total, 343 of 367 mutations called from the exome sequencing data were detected in the high coverage data, resulting in a true positive validation rate of 93.5%. Evidence for a low allelic fraction somatic mutation was observed in nine of 101 wild-type nucleotides. The variant allelic fractions (VAFs), i.e., the number of reads harboring the variant allele divided by all reads covering to that base, of exome and validation sequencing were highly correlated (Pearson correlation = 0.93) (Supplemental Fig. 4).
Copy number and LOH analysis
For cohort II, Affymetrix SNP 6.0 arrays were preprocessed by either aroma.affymetrix (Bengtsson et al. 2009) or CopyNumberInferencePipeline in GenePattern (http://genepattern.broadinstitute.org) and segmented by the circular binary segmentation algorithm (Olshen et al. 2004). Tumor and matched normal samples were used in pairs to obtain somatic alterations. The GISTIC2 algorithm (Mermel et al. 2011) was applied to the segmented copy number profiles for significant aberrations and sample-specific events. For cohort I, GISTIC2 outputs were downloaded from the Broad GDAC Firehose website (https://confluence.broadinstitute.org/display/GDAC/Home). In both cohorts I and II, the loss of heterozygosity (LOH) levels were determined by the paired parent-specific circular binary segmentation method (PSCBS, v0.28.1) (Olshen et al. 2011) to raw Affymetrix SNP 6.0 CEL files from paired tumor and normal samples.
Inference of cellular frequency distributions of sSNVs
We defined cellular frequency of a mutation as the fraction of cells harboring the mutation. To estimate cellular frequency in individual tumors, we integrated reference and variant allele counts, LOH, and copy number alterations using the PyClone algorithm (version 0.2) (Shah et al. 2012). In brief, PyClone implements a Dirichlet process clustering model that simultaneously estimates the distribution of the cellular frequency for each mutation. Copy number levels at somatic mutation sites were inferred by thresholded copy number profiles determined by GISTIC 2.0 on a single sample basis. We considered “−2” as homozygous deletion, “−1” as hemizygous deletion, “0” as copy number neutral, “1” as low level gain, and “2” as high level gain. LOH status was inferred from the PSCBS LOH file. PyClone was performed using default settings. The outputs were a pairwise mutation co-occurrence matrix and cellular frequency value distribution per sSNV estimated from Markov-chain Monte Carlo (MCMC) sampling. The median value of the MCMC sampling-derived distribution was used as a representative cellular frequency for each mutation.
Inferring cancer cell fraction and clonal status of sSNVs
When corrected for tumor cell content, the cellular frequency of each mutation can be used to infer the cancer cell fraction of the mutation. We assumed a group of mutations were shared by all cancer cells in a biopsy, and their cellular frequencies reflected the relative abundance of cancer cells (cellularity) in the admixture of infiltrating normal cells. Then, the cancer cell abundance in each sample can be used as a scale factor for rescaling cellular frequencies to the cancer cell fractions of the mutations in the sample. To infer cancer cell abundance, we performed hierarchical clustering of the pairwise mutation co-occurrence matrix and then selected a group of at least 10 mutations with the highest median cellular frequency at a given tree cutoff threshold. Multiple grouping thresholds were applied, and we manually inspected the results to select the most reasonable one. Euclidean distances were used in hierarchical clustering. The distribution of rescaled cancer cell fraction per mutation in a tumor was used to compute the clonal probability of the mutation. We defined a mutation to be clonal if the probability of the mutation being present in >95% of the cancer cells was more than 0.5, or subclonal otherwise. The cutoff of 0.5 was selected based on the empirical distribution of clonal probabilities for our data set.
Tumor purity estimation
In cohort II, tumor purities and ploidy were estimated by the ASCAT algorithm (v 2.1) using Affymetrix SNP 6.0 array data (Van Loo et al. 2010).
Survival analysis
We carried out a survival analysis using the “Survival” package in R version 3.1.0 (R Core Team 2014). Clinical information for cohort I was downloaded from TCGA data portal (Brennan et al. 2013). An event was considered to be either disease progression or death. Survival curves were estimated by the Kaplan-Meier method. The statistical significance of survival differences was calculated using the log-rank test in the “Survival” package.
Statistical analysis
All statistical analysis was performed with R version 3.0.1 (R Core Team 2014).
Data access
SNP6 array and sequencing data from this study have been submitted to the European Genome-phenome Archive (EGA; http://www.ebi.ac.uk/ega/) under accession number EGAS00001001033.
Competing interest statement
M.M. is a founder of and equity holder in Foundation Medicine.
Supplementary Material
Acknowledgments
We thank all members of TCGA GBM working group, Patricia S. Fox for assistance in survival analysis, and Lee Ann Chastain for assistance in manuscript editing. The results published here are in whole or part based upon data generated by The Cancer Genome Atlas project established by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) of the National Institutes of Health. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov. This work is supported by grants from the NCI, grant numbers P50 CA127001, P50 CA083639-12, P01 CA085878, R01 CA190121, Cancer Prevention & Research Institute of Texas (CPRIT) grant number R140606, and the University Cancer Foundation via the Institutional Research Grant program at the University of Texas MD Anderson Cancer Center to R.G.W.V.; grant number R01 CA163722 and TCGA contract number 28×S100 to E.G.V.M.; grant number CA143883 (MD Anderson Genome Data Analysis Center) to J.N.W.; and contract number HHSN261201000057C for A.E.S., M.L.C., and J.S.B. The MD Anderson Sequencing and Microarray Facility is funded by NCI Grant CA016672. This work is also supported by generous funding from the H.A. and Mary K. Chapman Foundation and the Michael and Susan Dell Foundation (honoring Lorraine Dell) to J.N.W. H.K. is supported in part by the Odyssey Program and the Theodore N. Law Endowment for Scientific Achievement at The University of Texas MD Anderson Cancer Center.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.180612.114.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andor N, Harness JV, Müller S, Mewes HW, Petritsch C. 2014. EXPANDS: expanding ploidy and allele frequency on nested subpopulations. Bioinformatics 30: 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio S, Caldas C. 2013. The implications of clonal genome evolution for cancer medicine. N Engl J Med 368: 842–851. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. 2006. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444: 756–760. [DOI] [PubMed] [Google Scholar]
- Bengtsson H, Wirapati P, Speed TP. 2009. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6. Bioinformatics 25: 2149–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT, et al. 2013. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2: e00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. 2012. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481: 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. 2012. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev 26: 756–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis JM, Zhang CZ, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, Homer H, Haidar S, Blumenstiel B, Pedamallu CS et al. 2014. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov 4: 956–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulci G, Ishii N, Maurici D, Gernert KM, Hainaut P, Kaur B, Van Meir EG. 2002. Initiation of human astrocytoma by clonal evolution of cells with progressive loss of p53 functions in a patient with a 283H TP53 germ-line mutation: evidence for a precursor lesion. Cancer Res 62: 2897–2905. [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. 2014. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46: 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson RJ, Rich JN. 2007. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7: 733–736. [DOI] [PubMed] [Google Scholar]
- González-García I, Solé RV, Costa J. 2002. Metapopulation dynamics and spatial heterogeneity in cancer. Proc Natl Acad Sci 99: 13085–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, Maley CC. 2012. Clonal evolution in cancer. Nature 481: 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, et al. 2006. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 66: 3987–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Tada M, Hamou MF, Janzer RC, Meagher-Villemure K, Wiestler OD, Tribolet N, Van Meir EG. 1999. Cells with TP53 mutations in low grade astrocytic tumors evolve clonally to malignancy and are an unfavorable prognostic factor. Oncogene 18: 5870–5878. [DOI] [PubMed] [Google Scholar]
- Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, et al. 2014. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343: 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massague J. 2009. Tumor self-seeding by circulating cancer cells. Cell 139: 1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev LT, et al. 2005. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood 106: 3150–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. 2013. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152: 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. 2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. 2011. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12: R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, Vij R, Tomasson MH, Graubert TA, Walter MJ, et al. 2014. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol 10: e1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel GC, Barnholtz-Sloan J, Gould MP, McMahon S, Cohen A, Adams MD, Guda K, Cohen M, Sloan AE, LaFramboise T. 2012. Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence. PLoS ONE 7: e35262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. 2012. The life history of 21 breast cancers. Cell 149: 994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. 2010. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MA, Sigmund K. 2004. Evolutionary dynamics of biological games. Science 303: 793–799. [DOI] [PubMed] [Google Scholar]
- Offer H, Erez N, Zurer I, Tang X, Milyavsky M, Goldfinger N, Rotter V. 2002. The onset of p53-dependent DNA repair or apoptosis is determined by the level of accumulated damaged DNA. Carcinogenesis 23: 1025–1032. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. 2004. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5: 557–572. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Bengtsson H, Neuvial P, Spellman PT, Olshen RA, Seshan VE. 2011. Parent-specific copy number in paired tumor-normal studies using circular binary segmentation. Bioinformatics 27: 2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant V, Xiong S, Jackson JG, Post SM, Abbas HA, Quintás-Cardama A, Hamir AN, Lozano G. 2013. The p53–Mdm2 feedback loop protects against DNA damage by inhibiting p53 activity but is dispensable for p53 stability, development, and longevity. Genes Dev 27: 1857–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team 2014. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/.
- Rausch T, Jones DT, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, Jäger N, Remke M, Shih D, Northcott PA, et al. 2012. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148: 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, Ha G, Aparicio S, Bouchard-Côté A, Shah SP. 2014. PyClone: statistical inference of clonal population structure in cancer. Nat Methods 11: 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE II, McLendon RE, Mitchell DA, et al. 2010. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 28: 4722–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. 2012. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486: 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, et al. 2011. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20: 810–817. [DOI] [PubMed] [Google Scholar]
- Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, Curtis C, Watts C, Tavare S. 2013. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci 110: 4009–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996. [DOI] [PubMed] [Google Scholar]
- Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, et al. 2012. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci 109: 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B, Parmigiani G. 2013. Half or more of the somatic mutations in cancers of self-renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci 110: 1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, et al. 2010. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci 107: 16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. 2010. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 60: 166–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. 2010. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PY, Kesari S. 2008. Malignant gliomas in adults. N Engl J Med 359: 492–507. [DOI] [PubMed] [Google Scholar]
- Yates LR, Campbell PJ. 2012. Evolution of the cancer genome. Nat Rev Genet 13: 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Fu J, Vegesna R, Mao Y, Heathcock LE, Torres-Garcia W, Ezhilarasan R, Wang S, McKenna A, Chin L, et al. 2013. A survey of intragenic breakpoints in glioblastoma identifies a distinct subset associated with poor survival. Genes Dev 27: 1462–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.