

Abstract
The availability of genetically tractable organisms with simple genomes is critical for the rapid, systems-level understanding of basic biological processes. Mycoplasma bacteria, with the smallest known genomes among free-living cellular organisms, are ideal models for this purpose, but the natural versions of these cells have genome complexities still too great to offer a comprehensive view of a fundamental life form. Here we describe an efficient method for reducing genomes from these organisms by identifying individually deletable regions using transposon mutagenesis and progressively clustering deleted genomic segments using meiotic recombination between the bacterial genomes harbored in yeast. Mycoplasmal genomes subjected to this process and transplanted into recipient cells yielded two mycoplasma strains. The first simultaneously lacked eight singly deletable regions of the genome, representing a total of 91 genes and ∼10% of the original genome. The second strain lacked seven of the eight regions, representing 84 genes. Growth assay data revealed an absence of genetic interactions among the 91 genes under tested conditions. Despite predicted effects of the deletions on sugar metabolism and the proteome, growth rates were unaffected by the gene deletions in the seven-deletion strain. These results support the feasibility of using single-gene disruption data to design and construct viable genomes lacking multiple genes, paving the way toward genome minimization. The progressive clustering method is expected to be effective for the reorganization of any mega-sized DNA molecules cloned in yeast, facilitating the construction of designer genomes in microbes as well as genomic fragments for genetic engineering of higher eukaryotes.
Complexities of natural biological systems make it difficult to understand and define precisely the roles of individual genes and their integrated functions. The use of model organisms with a relatively small number of genes enables the isolation of core biological processes from their complex regulatory networks for extensive characterization. However, even the simplest natural microbes contain many genes of unknown function, as well as genes that can be singly or simultaneously deleted without any noticeable effect on growth rate in a laboratory setting (Hutchison et al. 1999; Glass et al. 2006; Posfai et al. 2006). Ill-defined genes and those mediating functional redundancies both compound the challenge of understanding even the simplest life forms.
Toward generating a minimal cell where every gene is essential for the axenic viability of the organism, we are pursuing strategies to reduce the 1-Mb genome of Mycoplasma mycoides JCVI-syn1.0 (Gibson et al. 2010). Because we can (1) introduce this genome into yeast and maintain it as a plasmid (Benders et al. 2010; Karas et al. 2013a); and (2) “transplant” the genome from yeast into mycoplasma recipient cells (Lartigue et al. 2009), genetic tools in yeast are available for reducing this bacterial genome. Several systems offer advanced tools for bacterial genome engineering. Here we further exploit distinctive features of yeast for this purpose.
Methods for serially replacing genomic regions with selectable markers are limited by the number of available markers. One effective approach is to reuse the same marker after precise and scarless marker excision (Storici et al. 2001). We have previously used a self-excising marker (Noskov et al. 2010) six times in yeast to generate a JCVI-syn1.0 genome lacking all six restriction systems (JCVI-syn1.0 ∆1-6) (Karas et al. 2013a). Despite the advantages of scarless engineering, sequential procedures are time-consuming. When applied to poorly characterized genes with the potential to interact with other genes, some paths for multigene knockout may lead to dead ends that result from synergistic mutant phenotypes. When a dead end is reached, sequentially returning to a previous genome in an effort to find a detour to a viable higher-order multimutant may be prohibitively time-consuming.
An alternative approach to multigene engineering, available in yeast, is to prepare a set of single mutants and combine the deletions into a single strain via cycles of mating and meiotic recombination (Fig. 1A; Pinel et al. 2011; Suzuki et al. 2011, 2012). With a green fluorescent protein (GFP) reporter gene inserted in each deletion locus, the enrichment of higher-order yeast deletion strains in the meiotic population can be accomplished using flow cytometry. Here we apply this method to the JCVI-syn1.0 ∆1-6 exogenous, bacterial genome harbored in yeast to nonsequentially assemble deletions for genes predicted to be individually deletable based on biological knowledge or transposon-mediated disruption data. The functional identification of simultaneously deletable regions is expected to accelerate the effort to construct a minimal genome.
Figure 1.
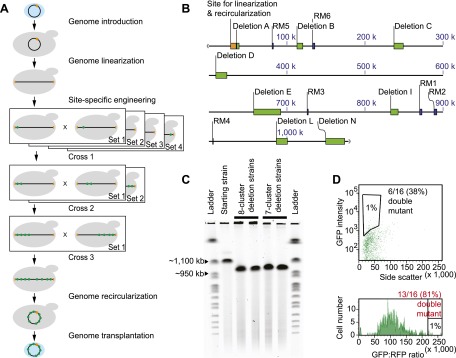
Progressive clustering of deleted genomic segments. (A) Scheme of the method. Light blue oval represents a bacterial cell. Black ring or horizontal line denotes a bacterial genome, with the orange box indicating the yeast vector used as a site for linearization and recircularization. Gray shape denotes a yeast cell. Green dot in the genome indicates a deletion replaced with a GFP marker. (B) Map of deleted regions. Orange box indicates the yeast vector sequence used for genome linearization and recircularization. Green boxes indicate regions deleted in multimutant mycoplasma strains. Blue boxes denote restriction modification (RM) systems that are also deleted in the strains. (C) Pulsed-gel electrophoresis result for deleted genomes. The starting strain was the JCVI-syn1.0 ∆1–6 strain (1062 kb). Two strains were analyzed for each design of simultaneous deletion (962 kb for eight-deletion or 974 kb for seven-deletion genome). Ladder is a set of yeast chromosomes (New England BioLabs). (D) GFP-RFP ratio sorting result. Standard sorting was compared with sorting based on a GFP-RFP ratio (Methods).
Results
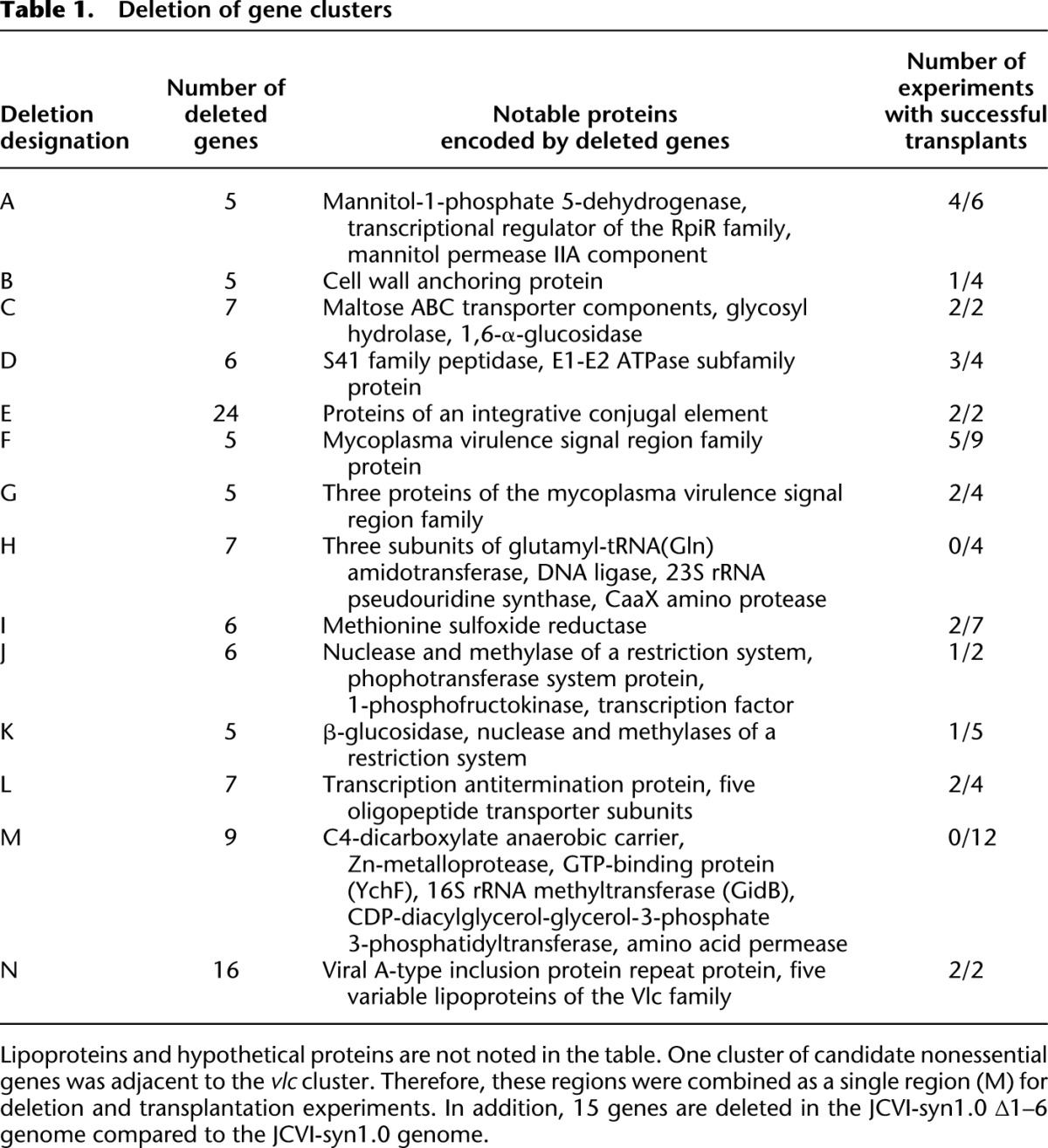
To select regions to delete in addition to those containing the restriction systems, we first considered genes that were unlikely to be required for growth in laboratory conditions. These genes included variable lipoproteins of large colony (vlc) genes presumably involved in evasion from the immune system (Wise et al. 2006) and genes within an integrative conjugal element (ICE) acquired by horizontal gene transfer (Dordet Frisoni et al. 2013). We found that a JCVI-syn1.0 genome lacking a cluster of five vlc genes or a cluster of 24 genes, including 22 genes of an ICE element (Table 1; Supplemental Table S1), when engineered in yeast and transplanted into mycoplasma recipient cells produced viable colonies.
Table 1.
Deletion of gene clusters
To select additional regions for deletion, we carried out transposon-mediated mutagenesis (Hutchison et al. 1999; Glass et al. 2006) and identified singly nonessential genes. Genomic sequences upstream of Tn4001tet transposons from transformed cells were amplified using inverse PCR (Ochman et al. 1988) and sequenced using Roche 454 amplicon sequencing. Of the nonredundant reads, 4582 were mapped to the internal region of genes, where accidental expression of sequences upstream or downstream from the transposon was unlikely to restore gene activity (Methods; Hutchison et al. 1999). Rather than Sanger sequencing used in our previous studies in other mycoplasma species, we used next-generation sequencing. The advantage for this approach is that the standard procedure for library preparation replaces the isolation of mycoplasma transformants (Glass et al. 2006) or the cloning of sequences adjacent to transposons in Escherichia coli (Hutchison et al. 1999), which is needed for separating samples for analysis. Because the tetracycline resistance marker in the Tn4001tet transposon (Dybvig et al. 2000) is present in the JCVI-syn1.0 genome, we used wild-type M. mycoides (GM12) not containing this marker only for the transposon bombardment experiment. The two genomes are essentially identical with only small intended and unintended differences (Lartigue et al. 2009; Gibson et al. 2010). Therefore, insertion sites in the GM12 genome can be mapped onto the JCVI-syn1.0 genome.
We considered 368 genes in which the transposon was inserted at five or more positions as candidate nonessential genes for this study (Supplemental Table S2; Supplemental Fig. S1). As expected, these nonessential genes included 11 of 14 genes of the restriction systems (positive controls), three of five vlc genes, and seven of 22 genes of the ICE element. On the other hand, ribosomal protein genes, which we previously identified to be essential using other mycoplasma species (Hutchison et al. 1999; Glass et al. 2006), were underrepresented with only three of 22 rpl genes and zero of 20 rps genes included in the list. The candidate nonessential genes are distributed among various functional categories (Supplemental Fig. S2). Notably, none of the 41 RNA genes are present among the nonessential genes.
To efficiently reduce the genome by progressive clustering of deletions, we selected 14 regions where five or more contiguous genes were scored as nonessential (Table 1; Supplemental Table S1). When genomes individually lacking one region were transplanted from yeast into mycoplasma recipient cells, we obtained viable organisms for 12 of the 14 regions (Table 1).
Using yeast mating and meiotic recombination as described below, we combined within yeast the deletions of the first eight regions found to be dispensable in a mycoplasma genome (Fig. 1B; Supplemental Fig. S1; Supplemental Table S3). We also made the genomes containing seven deletions after finding that deleting a region containing the genes MMSYN1_0840–MMSYN1_0846 resulted in slow growth of mycoplasma cells (see below).
Because recombination between circular genomes in yeast can generate a dicentric plasmid that can be damaged during cell division (Oertel and Mayer 1984), our first step was to linearize the circular JCVI-syn1.0 ∆1-6 genome (Fig. 1A). This was accomplished by integrating two telomeric fragments into the circular molecule (Methods; Sugiyama et al. 2005). This method was previously used to split endogenous chromosomes, but not to linearize a circular exogenous DNA entity.
Using the linear genome, we generated each of the eight deletions in a separate strain and marked each with a GFP cassette (Methods; Suzuki et al. 2011, 2012). We then introduced sets of haploid- and diploid-specific selection markers into these strains (Suzuki et al. 2011, 2012) and carried out three rounds of mating and meiosis. In each round, we isolated strains and genotyped the mycoplasma genomes using PCR (Methods). We “exponentially” assembled the deletions with the maximal number of deletions in the genotyped set of genomes increasing from one, to two, to four, and to eight (Supplemental Table S4).
The generated linear genomes containing eight or seven deletions were circularized via homologous recombination in yeast using a DNA fragment with homology with either end of the linear genome (Methods; Cocchia et al. 2000). When the circularized genomes were transplanted into recipient cells, viable mycoplasma cells were generated.
To confirm genomic deletions in these mycoplasma cells, we analyzed DNA samples isolated from two eight-deletion strains and two seven-deletion strains. PCR amplification of the upstream and downstream junctions between the GFP cassette and the genome produced bands of the expected size for each of the deletions for all strains tested. In addition, the absence of sequences within the deleted regions was confirmed by PCR. We then showed that the generated genomes had the expected size using pulsed-field gel electrophoresis (Fig. 1C; Supplemental Fig. S3). Finally, we determined the sequences of the genomes for one eight-deletion strain and one seven-deletion strain and found that no mutation was introduced during the whole reduction process.
When growth rates were determined using a method based on DNA quantification (Methods), the eight-deletion strain was slow-growing with a doubling time of 113 ± 5 min (n = 3) (Supplemental Table S5). However, it was not slower than one of the single mutant strains with a doubling time of 138 ± 2 min, suggesting that deletions of the eight tested gene clusters do not have any synergistic deleterious effect on cell growth. When the one “slow” deletion was excluded from the construction, the doubling time of the seven-deletion strain was 63 ± 2 min, which is comparable to the doubling times of the JCVI-syn1.0 and JCVI-syn1.0 ∆1–6 strains (63 ± 4 min and 59 ± 2 min, respectively) and to those of single mutants carrying one of the seven deletions (68.3–76 min) (Supplemental Table S5).
We included a flow-cytometric enrichment step in each round of the progressive clustering process, but it was ineffective (Supplemental Table S4). The likely reason is that multiple copies of the genome can coexist in yeast (Popov et al. 1997). For example, cells containing one copy of the mycoplasma genome with four GFP-marked deletions appear equivalent to cells containing two duplicated copies of the genome with two deletions. To see if this problem could be circumvented in future experiments, we introduced one copy of a red fluorescent protein (RFP) (Shaner et al. 2004) as an internal metric of mycoplasma genome copy number and used the GFP-to-RFP ratio to sort cells (Methods). When we used PCR to genotype the “greenest” 1% within the meiotic progeny of a diploid strain doubly heterozygous for two deletions, B and I (Table 1; Supplemental Table S1), 81% of the cells sorted based on the GFP-to-RFP ratio (n = 16) and only 38% of the cells sorted based on GFP alone (n = 16) had a genome containing both deletions (P = 0.03) (Fig. 1D).
Discussion
Despite the normal growth rate in the generated seven-deletion strain, significant changes in its metabolic potential and nonessential intracellular processes may have occurred. For example, capabilities for mannitol metabolism and signaling are expected to be compromised in this strain due to the deletion of cluster MMSYN1_0019-0021. The deleted gene MMSYN1_0021 is predicted to encode mannitol permease. MMSYN1_0019 encodes mannitol-1-phosphate 5-dehydrogenase, which is involved in the conversion of mannitol phosphate to fructose 6-phosphate in the glycolysis metabolic pathway. The deleted gene MMSYN1_0020 encodes a transcriptional regulator of the RpiR family, which may regulate sugar metabolism by repressing or activating downstream genes in response to levels of different sugars (Sørensen and Hove-Jensen 1996). The dispensability of mannitol metabolism reflects the laboratory culture condition with abundant glucose.
Similarly, a whole cluster of genes involved in maltose metabolism is deleted in both strains. Among the deleted genes, MMSYN1_0182–0184 are predicted to encode components of the maltose ABC transporter. The gene product for MMSYN1_0180 is annotated as putative lipoprotein, but a BLAST search with this sequence finds matches to several sugar ABC transporter proteins, including a periplasmic maltose transporter from Streptobacillus moniliformis (Nolan et al. 2009). A search with MMSYN1_0185 finds maltose phosphorylase from other mycoplasmas. This enzyme converts maltose and phosphate to D-glucose and β-D-glucose 1-phosphate. The MMSYN1_0186 gene is predicted to encode a carbohydrate-active enzyme of the GH13 family. A BLAST search shows matches with numerous 1,6-α-glucosidases from Streptococcus pneumoniae and other species, rather than 1,4-α-glucosidases needed for cleaving maltose, raising the possibility that isomaltosaccharides or dextrans with α-1,6 linkages are relevant to mycoplasma survival in its natural habitat.
The MMSYN1_0241 gene encoding a peptidase of the S41 family is also deleted in both strains. A previous study of M. mycoides showed that disruption of this gene affects the concentration of 61 of 221 proteins tested (Allam et al. 2012). Deletion of the gene for methionine sulfoxide reductase (MMSYN1_0699) with a role in reversing oxidized methionine is also expected to affect the functions of multiple proteins (Ezraty et al. 2005). It is interesting to investigate how the organism adapts to pleiotropic changes while maintaining its growth rate.
To examine the extent of redundancy within the genome, we performed a search for duplicated genes (Methods). Besides the transposase genes associated with mobile elements, ribosomal RNA genes, and tRNA genes, there were only six pairs of candidate duplicated genes (Supplemental Table S6) at the cutoff we chose. Only one of these genes, MMSYN1_0560, was deleted in our strain. In addition, some genes deleted in our strains have undeleted counterpart genes with the same annotations. For example, the deleted gene MMSYN1_0241 and the undeleted genes MMSYN1_0112 and MMSYN1_0292 are all predicted to encode S41 peptidase. The deleted gene MMSYN1_0246 and the undeleted gene MMSYN1_0879 both have the same annotation for putative E1-E2 ATPase. The deleted genes MMSYN1_0841–0846 are predicted to encode subunits of an oligopeptide ABC transporter, but there is an undeleted set (MMSYN1_0165–0168) for another oligopeptide ABC transporter. Whether the generated strains remain viable due to the activity of undeleted homologs will be a subject of future studies.
Alteration of multiple sequences throughout a genome is often required for generating microbial strains capable of addressing complex questions in genomics and systems biology. However, the necessary tools for genetic engineering have been developed for relatively few organisms. Establishing the strategy of cloning and engineering bacterial genomes in yeast and using powerful genetic tools developed in this host alleviate the need for techniques such as genome editing mediated by CRISPR (clustered regularly interspaced short palindromic repeat) in numerous bacterial systems. Genome synthesis is arguably the most powerful genome engineering technique because the resulting sequence is not constrained by the native DNA sequence in the organism. Moreover, genome synthesis is commercially available as a service, contributing to the streamlining of the design-build-test cycle for microbial strain development. Because yeast is used for the assembly of DNA fragments during the genome synthesis process (Gibson et al. 2010), complementary genome manipulation tools developed in yeast further promote the synthetic genomics approach. The method of recombining bacterial genomes in yeast can be used to progressively cluster not only deletions, as described in this paper, but also any combinations of deletions, insertions, and substitutions including synthesized fragments introduced into the genome. Our method can also be used to recombine two finished synthetic genomes to exchange modular parts or to combine intermediate synthetic genomic segments when regions of homology are introduced into these segments.
We expect the progressive clustering method to become applicable to genome engineering in an increasing number of organisms. Cloning of a whole genome in yeast has been demonstrated for genomes from several bacteria (Gibson et al. 2008, 2010; Lartigue et al. 2009; Benders et al. 2010; Karas et al. 2011, 2013a; Tagwerker et al. 2012). This approach has also been expanded to eukaryotic chromosomes (Karas et al. 2013b). The procedure for transferring bacterial genomes into yeast has been further facilitated by our discovery of genome transfer within a mixture of bacterial cells and yeast spheroplasts (Karas et al. 2013a). Meiotic recombination, linearization, and recircularization in yeast are applicable to any of these megasized DNA clones. Genome transplantation should become available for an increasing number of organisms. In addition, there will be technologies to transfer genomes in pieces, as demonstrated in E. coli (Krishnakumar et al. 2014) and Bacillus subtilis (Itaya et al. 2005), and these can be used to reconstitute the engineered genome.
It is theoretically possible that the accumulation of GFP cassettes would result in toxicity for the engineered organisms due to overproduction of GFP. Our approach includes two layers of protection to address this possibility. First, because engineering based on GFP occurs within yeast, a promoter that is inactive in mycoplasma (or other prokaryotes) can be used in the GFP construct. In fact, the generated mycoplasma strains were not fluorescent. Second, to avoid any impairment of yeast during genome construction, we placed the GFP gene under the control of a titratable tetracycline-responsive promoter (Suzuki et al. 2011). If a deleterious effect is observed, the inducer concentration can be lowered to modulate GFP expression. We have not observed any toxicity associated with the deletion marker in yeast (Suzuki et al. 2011) or in mycoplasma, but it is expected that having too many copies leads to genome instability due to unwanted homologous recombination. In such a case, a GFP count reset may be needed. We have not yet devised a method to remove the GFP cassettes from the generated genome. However, methods for scarless engineering, including whole genome synthesis (Gibson et al. 2010), can be applied to constructing genomes based on information from our approach on the identities of the genes that can be simultaneously deleted.
The CRISPR method has been widely adapted for genome editing in various organisms and may be considered a competing technology, but the lack of examples in mycoplasma makes comparison difficult. Plasmids stably maintained in M. mycoides have not been established, so methods involving purified Cas9 protein or expressing the nuclease from a transient plasmid or a construct integrated in the genome may need to be devised. The efficiency of homologous recombination required for CRISPR-mediated editing is also uncertain, especially in organisms with reduced genomes lacking some of the genes for DNA repair enzymes. In contrast, progressive clustering is unaffected by the progress in genome reduction because the genome is engineered in yeast using the yeast machinery for recombination, and the engineered genome is always introduced into the same bacterial recipient cell.
CRISPR has been demonstrated in yeast (DiCarlo et al. 2013), but its use for multisite engineering of mycoplasma or other genomes may be hampered by the instability of nonchromosomal DNA, as observed for engineering with meganucleases (Noskov et al. 2010). On the other hand, our analysis indicates that the genomes are stable during progressive clustering. Each round of CRISPR engineering takes only a few days, but screening of correctly engineered genomes and confirmation of correct engineering would add extra days. Multisite CRISPR engineering on a mycoplasma genome harbored in yeast is expected to be more difficult than single-site engineering. One round of the progressive clustering process requires 12 d (Methods), and the number of deletions exponentially increases in each round. With eight deletions generated in 36 d, the average is 4.5 d per deletion after yeast strains, each carrying a linear genome with a single deletion, are prepared. This rate is comparable to that of a sequential, single-gene CRISPR approach.
A minimal cell containing only genes that are essential for life is expected to provide opportunities for understanding how molecular machines interact to form a functioning organism. It can also serve as a simple and predictable platform for studying exogenous genes or for engineering an exceptionally efficient microbe designed to carry out specific tasks. Starting with the M. mycoides genome, one of the smallest among free-living organisms, we have shown that 10% reduction of the genome does not result in any noticeable change in growth rate or viability in a rich medium. As perhaps expected for a genome that has naturally undergone dramatic genome reduction, we did not observe any synergistic growth defective phenotype that can result from simultaneous deletion of redundant or functionally equivalent genes. These two findings suggest the feasibility of minimizing a genome by simultaneously deleting genes that are singly dispensable.
Methods
Strains and culture conditions
The Mycoplasma mycoides subspecies capri strains GM12 (DaMassa et al. 1983), JCVI-syn1.0 (Gibson et al. 2010), and JCVI-syn1.0 ∆1–6 (Karas et al. 2013a) were used. GM12 was used only for transposon bombardment because it is similar to JCVI-syn1.0, and it does not have a tetracycline resistance gene (Lartigue et al. 2009; Gibson et al. 2010). JCVI-syn1.0 ∆1–6 lacks genes of the restriction systems. It is derived from JCVI-syn1.0. These strains were cultured at 37°C in SP-4 medium (Tully et al. 1977) containing 17% fetal bovine serum (Life Technologies). Five micrograms per milliliter of tetracycline was also added when culturing the JCVI-syn1.0 and JCVI-syn1.0 ∆1–6 strains unless otherwise indicated or when cells transformed with a transposon containing a tetracycline resistance marker (see below) were selected in the transposon bombardment experiment using the GM12 strain. Mycoplasma capricolum subspecies capricolum (NCTC 10154) was used as a recipient and cultured as previously described (Lartigue et al. 2009) for transplanting engineered JCVI-syn1.0 and JCVI-syn1.0 ∆1–6 genomes. For engineering the mycoplasma genomes, the Saccharomyces cerevisiae strains BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Brachmann et al. 1998); RY0146 (MATα lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 can1Δ∷GMToolkit-a); and RY0148 (MATα lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 can1Δ∷GMToolkit-α) (Suzuki et al. 2011, 2012) were used. These strains and strains derived from these strains were cultured at 30°C in rich YPDA or GNA (Brachmann et al. 1998) medium or in synthetic complete (SC) medium lacking relevant auxotrophic components, or at room temperature (∼23°C) and then at 30°C in sporulation medium (Brachmann et al. 1998). GMToolkit-a contains CMVpr-rtTA KanMX4 STE2pr-Sp-his5. GMToolkit-α contains CMVpr-rtTA NatMX4 STE3pr-LEU2 (pr denotes a promoter). KanMX4 (Wach et al. 1994) and NatMX4 (Goldstein and McCusker 1999) enable cells to grow in the presence of 200 μg mL−1 G418 (Sigma-Aldrich) and 100 μg mL−1 nourseothricin (Nat; Werner BioAgents), respectively, in YPD. These markers are used to select diploids in the sexual cycling scheme of the progressive clustering method (Supplemental Fig. S4). STE2pr∷Sp-his5 and STE3pr∷LEU2 are specifically expressed in a-haploids and α-haploids, respectively. The his5 gene from Schizosaccharomyces pombe (Sp) complements his3Δ1. The LEU2 gene complements leu2Δ0. These markers are used to select haploids (Suzuki et al. 2011, 2012). The JCVI-syn1.0 and JCVI-syn1.0 ∆1–6 genomes are marked with the yeast marker HIS3. The linearized forms of the JCVI-syn1.0 ∆1–6 genome were marked with MET15. Deletions in these genomes were first generated with a KanMX4 marker (Wach et al. 1994); and for the Green Monster process, these were converted to a cassette containing a GFP reporter gene and URA3 (Suzuki et al. 2011, 2012).
Transposon mutagenesis
The pIVT-1 plasmid containing the Tn4001 transposon bearing the tetracycline-resistance gene tetM (Dybvig et al. 2000) was methylated using a Mycoplasma mycoides extract as previously described (Lartigue et al. 2009) to avoid the restriction of the introduced plasmid DNA in the host organism. Transformation of wild-type M. mycoides subspecies capri GM12 was accomplished using a previously described method (King and Dybvig 1991; Lartigue et al. 2003). More than 2000 colonies selected on SP-4 (Tully et al. 1977) agar plates containing 17% fetal bovine serum and 5 μg/mL of tetracycline were pooled and harvested. Genomic DNA (gDNA) from the pooled sample was prepared using Wizard Genomic DNA Purification Kit (Promega). To identify the transposon insertion sites, an inverse PCR approach (Ochman et al. 1988) was used. One microgram of gDNA was digested using SspI. To promote self-circularization of SspI-digested fragments as opposed to generation of tandem multimers, the reaction was diluted to 5 ng/µL DNA and treated with T4 ligase. There are 2615 SspI sites in the GM12 genome. The average distance between adjacent sites is ∼400 bp. SspI also digests Tn4001, but not within the region flanked by the annealing sites for the primers AR_Tn_Jct and AR_Tn_SspI_Jct (all primers described in Supplemental Table S7). These primers were used in PCR to capture the genomic sequences preceding the first SspI fragment of Tn4001 along with the transposon junction from the pool (Supplemental Fig. S5). The PCR products were used to make a library, which was then sequenced using a 454 GS FLX sequencer (Roche).
Of the 756,568 reads obtained, 4924 reads were (1) not redundant with another read (Gomez-Alvarez et al. 2009); (2) longer than 100 nucleotides; (3) mapped to the Tn4001 transposon sequence using the Basic Local Alignment Search Tool algorithm (Altschul et al. 1990); (4) mapped to the JCVI-syn1.0 genome (GenBank accession number CP002027); and (5) characterized as having the matched sequences to Tn4001 and JCVI-syn1.0 not separated by more than 13 nucleotides within the read. Transposon insertions downstream from the ninth nucleotide and upstream of the 80% point along the length of the coding or structural region of an annotated gene in the JCVI-syn1.0 genome are assumed to be disruptive to the gene function (Hutchison et al. 1999). Of the 4924 reads, 4582 were in the disruptive category, with the Tn4001 insertions defining 675 potentially nonessential genes. Two hundred forty-two genes were not found to have any disruptive insertion (Supplemental Table S2).
Some reads were probably derived from transient insertions of Tn4001 into essential genes. Transient insertions were possible because the transposase encoded in Tn4001 enabled secondary or recursive transposition. These reads could not be eliminated from data based on the low abundance of the identical reads, because replication of reads is a known artefact during 454 sequencing that precludes the direct correlation of read counts and abundances of the original DNA materials (Gomez-Alvarez et al. 2009). Nevertheless, unique reads representing different Tn4001 insertions within a gene should be more numerous for nonessential genes than for essential genes, because cells that contain a transposon in a nonessential gene propagate and have a higher chance of being sampled. Therefore, genes that were hit five times or more were considered to be candidate nonessential genes for this study (Supplemental Table S2; Supplemental Fig. S1).
Introduction of mycoplasma genomes into yeast
For testing whether deletion of a single gene cluster is tolerated by mycoplasma, the JCVI-syn1.0 genome was used. For generating a multideletion genome, the JCVI-syn1.0 ∆1–6 genome was used, because additional genes were already deleted in this genome. The JCVI-syn1.0 genome was purified from the synthetic cell (Gibson et al. 2010) and introduced into the yeast strain BY4741 as described (Benders et al. 2010). The JCVI-syn1.0 ∆1–6 (Karas et al. 2013a) genome was transferred from mycoplasma to BY4741 using direct genome transfer (Karas et al. 2013a) with a detailed protocol published elsewhere (Karas et al. 2014) to make the yeast strain SY141. These genomes are marked with the HIS3 marker for maintenance in yeast.
Construction of KanMX4 deletion cassettes
Three rounds of PCR with Phusion polymerase (New England BioLabs) were used to generate mycoplasma gene targeting constructs containing a KanMX4 transformation marker (Wach et al. 1994). In the first round, the primers Myc_del_7_F and Myc_del_7_R1, as well as the pFA-kanMX4 plasmid (Wach et al. 1994) were used. In the second round, the gene- or region-specific primer MMSYN1_(gene number)_F0 paired with the primer Myc_del_7_R2 and the product from the first reaction purified using the PCR purification kit (Qiagen) as a template were used. In the third round, the region-specific primers MMSYN1_(gene number)_F and MMSYN1_(gene number)_R were used with the product of the second round pin-transferred from the previous reaction. These cassettes contain DNA barcodes (Giaever et al. 2002) that enable en masse characterization of generated strains (Hillenmeyer et al. 2008).
Construction of single-deletion genomes based on the JCVI-syn1.0 genome
To see if deletion of each cluster results in viable organisms, the JCVI-syn1.0 genome was used. A standard transformation procedure in yeast (Woods and Gietz 2001) followed by selection on a YPDA plate supplemented with G418 was used to introduce each of the generated KanMX4-marked deletion cassettes into the JCVI-syn1.0 genome. Correct integration of the cassettes was confirmed using colony PCR with the primer that anneals to the genome upstream of the deleted region paired with the primer TEFterm_seq. For the positive clones, colony PCR with the downstream primer paired with the primer TEFpr_seq was also performed. Because mycoplasma genomes can reside in yeast as a multicopy entity, unaltered genomes need to be eliminated before a functional test is conducted for the yeast clone. For this purpose, yeast clones with a correctly made deletion were cultured overnight in a liquid medium containing G418. These cultures were plated to make isolated colonies. The resulting colonies were analyzed by colony PCR to confirm the absence of a band representing a wild-type sequence within the targeted region using the GTIF and GTIR primers. Lysate of a yeast strain containing the JCVI-syn1.0 genome served as a positive control for the PCR. The integrity of the engineered genome was confirmed using multiplex PCR (Qiagen) using six amplicons scattered throughout the genome.
Mycoplasma genome transplantation
M. mycoides genomic DNA was purified from yeast and transplanted into M. capricolum recipient cells as previously described (Lartigue et al. 2009). The recipient cells were cultured at 30°C, rather than 37°C, before transplantation to increase the transplantation efficiency.
Construction of a genome carrying a MET15 marker
A version of JCVI-syn1.0 marked with MET15, JCVI-syn1.0 (MET15), was constructed and used for making plasmids that served as templates for producing linearization fragments below. For this construction, two rounds of PCR were used to generate a targeting fragment that had a MET15 gene between sequences that were homologous to the sequences flanking the HIS3 marker in JCVI-syn1.0. The first reaction contained the primers MET15_syn1_1F and MET15_syn1_1R, as well as the genomic DNA of the yeast strain sMmyc235 (Gibson et al. 2010) derived from the strain VL6-48 (Met+) as a template. The second reaction contained the primers MET15_syn1_2F and MET15_syn1_2R, as well as the purified product of the first reaction as a template. Met+ transformant colonies were streaked out on a fresh SC-Met plate, and cells from the resulting patches were lysed and analyzed for the presence of the junction downstream from MET15 using PCR with the primers MET15_syn1_Chk_R and MET15_seq_F1. Twelve of the 60 patches analyzed were positive for this PCR. Ten of the 12 positive patches were also positive for PCR with the primers MET15_syn1_Chk_F and MET15_seq_R1, indicating that the upstream junction was also correct.
Linearization of mycoplasma genomes in yeast
A method for constructing telomeres within a chromosome using PCR fragments termed chromosome splitting (Sugiyama et al. 2005) was used to linearize the JCVI-syn1.0 ∆1–6 genome in yeast. Only the JCVI-syn1.0 ∆1–6 genome was linearized in this study and used for removing multiple gene clusters. The fragment for one telomeric end was marked with MET15 (Supplemental Fig. S6). The one for the other end was not marked. These fragments were simultaneously introduced into yeast and into the yeast vector region of the JCVI-syn1.0 ∆1–6 genome to open the genome while either completely or partially deleting the HIS3 marker in the vector region. The benefit of only partially deleting HIS3 is that a DNA fragment not containing the complete HIS3 sequence can be later used to recircularize the linearized genome via homologous recombination (see below). The chance of selecting strains with a correctly recircularized genome would increase with such a fragment because a mistargeted truncated HIS3 fragment is unlikely to provide the transformants with the ability to grow in the absence of added histidine (Supplemental Fig. S6). For generating the PCR fragments with a telomeric end, convenient plasmids (pYO018 and pYO019) containing MET15 and parts of JCVI-syn1.0 were used as templates. These plasmids vary in the length of the sequence that can be used as a template for generating the unmarked PCR fragment. pYO018 only contains a sequence of the vector region up to the HIS3 boundary, whereas the sequence in pYO019 extends into HIS3.
The preparation of these plasmids involves the following three-step process. A PCR fragment containing MET15 and 360 bp of the JCVI-syn1.0 vector region was generated using the primers Lin(syn1)_3F_Long and Lin(syn1)_3R, as well as the template JCVI-syn1.0 (MET15) genomic DNA, digested with NotI and SalI, and cloned into pBluescript SK+ digested with NotI and XhoI, to generate the plasmid pYO014. To introduce a spacer sequence to be used as a primer annealing site for later PCR analysis or to separate the MET15 marker from the telomere, which may be affected by transcriptional silencing in the final linearized genome, a short fragment was amplified from the genomic DNA of the cyanobacterium Synechococcus elongatus PCC 7942 (Noskov et al. 2012) using the primers Lin(syn1)_2F_Linker and Lin(syn1)_2R_Linker, digested with NotI and SacI, and cloned into pYO014 digested with NotI and SacI to generate the plasmid pYO017. A PCR fragment containing 1000 bp of the JCVI-syn1.0 vector region made with the primers Lin(syn1)_1F and Lin(syn1)_1Ra and the template JCVI-syn1.0 genomic DNA was digested with BamHI and SacI and cloned into pYO017 digested with BamHI and SacI to generate pYO018. A PCR fragment containing 1374 bp of the JCVI-syn1.0 vector region including part of HIS3 made with the primers Lin(syn1)_1F and Lin(syn1)_1Rb and the same template was digested with BamHI and SacI, and cloned into pYO017 digested with BamHI and SacI, to generate pYO019.
The primers LC_DnCirF and CA_syn1_1R were used with the template plasmid pYO018 or pYO019 to generate a fragment containing 320 bp (with pYO018) or 694 bp (with pYO019) of homology with the JCVI-syn1.0 ∆1–6 genome, 42 bp of spacer sequence, and the (C4A2)6 sequence that initiates the development of a telomere (Sugiyama et al. 2005). The primers CA_syn1_2F and CA_syn1_2R were used with the template pYO018 to generate a fragment containing the (C4A2)6 repeat, 59 bp of spacer sequence, MET15, and 347 bp of homology with JCVI-syn1.0 ∆1–6. These PCR products were purified using the QIAquick PCR purification kit (Qiagen). The unmarked fragment and the MET15-marked fragment were mixed in a >30:1 ratio and simultaneously introduced into a BY4741 strain carrying the JCVI-syn1.0 ∆1–6 genome. Met+ transformants were selected on a SC-Met medium containing agar.
When the correct integration of the unmarked fragment was analyzed using colony PCR with the primers LC_UpF3 and LC_UpR, five of 20 colonies and one of 14 colonies were positive for the experimental designs using pYO018 and pYO019, respectively. One yeast clone from each design was analyzed using pulsed-field gel electrophoresis. Genomic DNA was purified using CHEF Yeast Genomic DNA Plug Kit (Bio-Rad) and digested using the restriction enzymes NotI, FseI, RsrII, and PspXI within plugs (New England BioLabs). This treatment results in the degradation of the yeast DNA into small pieces, but in only a single cut in the JCVI-syn1.0 ∆1–6 genome through the action of PspXI. Two fragments of the expected sizes (292 kb and 785 kb) were observed for the design with pYO019, consistent with the successful linearization. However, there were no distinct bands for the design with pYO018. When the correct integration of the MET15-containing PCR fragment was analyzed using colony PCR with the primers Lin_Chk_MET_F and LC_DnR2, the clone with the pYO018 design was negative, suggesting that the MET15 fragment was integrated into a random location in a yeast chromosome in this strain, and the JCVI-syn1.0 ∆1–6 genome was lost during propagation. The clone with the pYO019 design was positive for this PCR and used for the subsequent study.
Construction of the ProMonster strains (yeast strains carrying single-deletion genomes) for deletion assembly
To generate multideletion genomes, each of the selected clusters (A, B, C, D, E, I, L, and N) was deleted in the linearized JCVI-syn1.0 ∆1–6 genome to establish single-deletion genomes in yeast. The procedures for making the KanMX4 deletions, confirming the presence of deletion junctions, confirming the absence of the wild-type sequences, and confirming the integrity of the generated genomes were identical to those described for the JCVI-syn1.0 genome above.
The KanMX4-marked deletions in the linearized JCVI-syn1.0 ∆1–6 genome were converted to GFP-marked deletions using a restriction fragment derived from the plasmid pYOGM057 as previously described (Suzuki et al. 2012). Correct integration was confirmed with colony PCR using the upstream primer paired with the primer URA3_seq_2 and the downstream primer paired with the primer GFP_GT_R. To confirm the loss of the unaltered genome still containing the KanMX4 marker, the growth of the yeast strain was tested on YPDA medium supplemented with G418.
The strains containing the GFP cassette were crossed with a strain containing GMToolkit-a or GMToolkit-α (Suzuki et al. 2011, 2012). The mated diploids were selected based on the presence of markers introduced from the two haploid strains, by sequentially culturing the cells in YPDA supplemented with G418 (resistance provided by KanMX4 in GMToolkit-a) or nourseothricin (resistance provided by NatMX4 in GMToolkit-α) and in SC-Ura (GFP cassette in the JCVI-syn1.0 ∆1–6 genome). After sporulation, haploids termed the ProMonster strains were selected in SC-His-Ura-Met (a-haploids) or SC-Leu-Ura-Met (α-haploids). The integrity of the genome was confirmed using multiplex PCR as described above.
Assembly of multiple deletions into single genomes using the Green Monster process
The ProMonster strains were cultured in 100 µL of SC-His (for growing a-haploids) or SC-Leu (for growing α-haploids) containing 1 mg L−1 (one 100th of the standard amount) of methionine (SC1%M-H or SC1%M-L), in an attempt to select for cells containing a single copy of the mycoplasma genome. The a-haploids and the α-haploids were mated in 10 µL of YPDA initially at the optical density of 1 by combining appropriate and roughly equal numbers of cells based on OD reading using a NanoDrop instrument (Thermo Scientific) in a tube, centrifuging the cells, resuspending the cells in 10 µL of YPDA, centrifuging again, and incubating the tube for 8 h at 30°C in a damp box (Suzuki et al. 2011, 2012). Cells from this culture were streaked out on a YPDA plate containing G418 and nourseothricin and incubated for 2 d at 30°C. The colonies were confirmed to contain both genomes by PCR-amplifying one of the deletion junction(s) from each of the two genomes. Seventy-five percent to 100% of the diploid colonies tested had both parental genomes. Positive colonies were cultured for 5 h in 100 µL of 2×YPDA containing G418 and nourseothricin. This was rinsed in 500 µL of sporulation medium three times (Suzuki et al. 2011, 2012) and resuspended in 1 mL of a sporulation medium containing 5 mg L−1 histidine, 5 mg L−1 leucine, 5 mg L−1 lysine, and 1.25 mg L−1 uracil. After culturing at room temperature for 1 d and at 30°C for 2 d, the spores were processed as described (Suzuki et al. 2011, 2012). The spores were germinated in 100 µL of SC1%Met-His and SC1%Met-Leu to select for a-haploids and α-haploids, respectively, for 16 h. These cultures were combined with 100 µL of SC1%Met-His or -Leu medium containing 20 µg mL−1 doxycycline (10 µg mL−1 final) to induce GFP expression for 2 d. After flow cytometry, the cells were plated on SC-Met-His or SC-Met-Leu plates. The colonies were genotyped (Supplemental Table S4), and selected strains were used for the next round of the Green Monster process. The cycles of the Green Monster process were repeated a total of three times to generate genomes containing eight or seven GFP-marked deletions.
Circularization of mycoplasma genomes in yeast
To construct a plasmid that contains a fragment used for recircularizing the genome, a PCR fragment generated using the primers Cir_b_(syn1)_1F and Cir_b_(syn1)_1R, as well as the template JCVI-syn1.0 genomic DNA, was digested with SacI and SalI and cloned into pBluescript SK+ digested with SacI and SalI. This plasmid (pYO016) was digested using BglII and purified using QIAquick PCR Purification Kit (Qiagen). This sample was introduced into yeast using a lithium acetate method (Woods and Gietz 2001). Transformants were analyzed using colony PCR using the primers Recir_chk_F and Recir_chk_R to see if genomes in the transformants were circular. Twelve of 18 (∼70%) colonies tested were positive for this PCR. Genome integrity was confirmed for six of six strains tested using multiplex PCR as described above. Two recircularized genomes for each of the eight- and seven-deletion designs were transplanted using the method described above.
Whole genome sequencing
The genomes of the mycoplasma strains were sequenced as previously described (Karas et al. 2013a). One eight-deletion strain and one seven-deletion strain were sequenced at 689- and 1147-fold coverage, respectively. Only two single-nucleotide polymorphisms were categorized as probable (Karas et al. 2013a) when obtained sequences were compared to the reference sequence of JCVI-syn1.0 (Gibson et al. 2010). However, these polymorphisms were present in the starting strain for this study JCVI-syn1.0 ∆1–6 (Karas et al. 2013a), indicating that no mutation was introduced during the sexual recombination procedure.
Mycoplasma growth assay
Growth rates were determined by quantifying the increase in cell-associated DNA during logarithmic phase in liquid cultures statically grown at 37°C in SP-4 medium containing 17% fetal bovine serum. At selected times, samples from a set of replicate aliquots made from an initial culture were transferred to ice to arrest growth. After final collection, a 0.4-mL cushion of 0.5 M sucrose (in 10 mM Tris-HCl, pH 7.5) was placed under each of the 0.80-mL samples in graduated microfuge tubes. Cells were sedimented by centrifugation at 16,000g for 10 min. Medium was aspirated to adjust the volume of the remaining clear cushion to 100 µL without disturbing a cell pellet. Cells were lysed by trituration after adding sodium dodecyl sulfate (SDS) to a final concentration of 0.1% (w/v) and TE buffer to 1× (10 mM Tris-HCl, pH 7.5, 1 mM ethylenediaminetetraacetic acid) in 150 µL. The lysate was then diluted to 0.01% SDS in TE buffer. To quantify DNA, equal volumes (80 µL) of each lysate and Quant-iT PicoGreen reagent (Life Technologies) were mixed for all strains and all time points using individual wells of opaque black 96-well plates (Costar, catalog no. 3915) and incubated at room temperature for 5 min. Fluorescence was measured using a FlexStation 3 fluorimeter (Molecular Devices) with excitation at 488 nm and detection of emission at 525 nm. Relative fluorescence units (RFUs) were plotted as log2(RFU) against time. Regression values (R2) were calculated from exponential growth curves, and doubling times were determined from the formula ln2/exponential rate. Standard deviations were calculated from replicate samples taken at each time point.
Introduction of RFP internal standards
To generate a tdTomato (RFP) reporter gene, a tdTomato sequence was amplified from pRSET-B tdTomato (Shaner et al. 2004) using the primers JS_tdTomato_tetO2_F and JS_tdTomato_tetO2_R. The pCM251 vector (Bellí et al. 1998) containing a tetracycline inducible system was amplified using the primers tdTomato/mCherry_pCM251_F and tdTomato/mCherry_pCM251_R. These fragments were combined to make a plasmid using Gibson assembly (Gibson et al. 2009). To generate a version of the plasmid containing the ADH1 promoter rather than the tetO2 promoter, the ADH1 promoter was amplified from the BY4741 genomic DNA using the primers ADH1pr_F and ADH1pr_R. The vector backbone was amplified from the tetO2pr-tdTomato plasmid using the primers ADH1pr_Ins_F and ADH1pr_Ins_R. These fragments were combined using Gibson assembly.
To generate a robust hygromycin B resistance marker, the TDH3 promoter, the hph gene, the GAL7 terminator, and the pUC19 vector backbone were amplified from the yeast BY4741 genomic DNA, the plasmid pAG32 (Goldstein and McCusker 1999), the BY4741 genomic DNA, and the plasmid pUC19, respectively, and combined into a single plasmid using Gibson assembly. The primers used to amplify these fragments were TDH3pr_F paired with TDH3pr_R (TDH3pr), hph_F paired with hph_R (hph), GAL7term_F paired with GAL7term_R (GAL7), and pUC19_F paired with pUC19_R (pUC19).
To combine each of the two tdTomato reporter genes (one with the tetO2 promoter and the other with the ADH1 promoter) with the hygromycin B resistance marker, a fragment containing a tdTomato construct was amplified using the primers Gibson_RFP_into_HpH_F and Gibson_RFP_into_HpH_R. This was assembled with the vector fragment amplified from the hygromycin B marker plasmid using the primers HpH_Backbone_FWD and HpH_Backbone_REV.
A homologous sequence and a telomeric sequence for integration and genome linearization were attached to the resulting construct using PCR with the primers Telo_RFP_Hph_Prox and Telo_RFP_Hph_Dist. This fragment was introduced into the genome along with the MET15 telomeric fragment described above.
For the experiment for sorting based on GFP-to-RFP ratio, a version of the GFP reporter gene containing an improved translation initiation signal was used. To make this GFP construct, the plasmid pYOGM057 (Suzuki et al. 2012) was amplified using the primers Fix_Kozak_pYOGM057F and Fix_Kozak_pYOGM057R. The PCR fragment was self-ligated using Gibson assembly.
For using the tdTomato constructs in genome construction, the Green Monster process was performed as described above except that the following changes were made during flow cytometry. The tdTomato signal was detected using a 605/40-nm filter. The excitation wavelength was 488 nm. After excluding outliers using forward scatter and side scatter during flow cytometry (Suzuki et al. 2011, 2012), cells in the top 10% range in RFP intensity were included to generate a histogram of GFP-to-RFP ratio. Using this histogram, cells within the highest 1% range in GFP-to-RFP ratio were sorted. The result with the tetO2pr-driven tdTomato construct is shown in Figure 1D.
Search for redundant genes
Duplicated or paralogous genes were identified by all-by-all BLASTN (NCBI blast+ v. 2.2.24) (Camacho et al. 2009) of predicted gene and RNA nucleotide sequences for the JCVI-syn1.0 genome (GenBank accession number CP002027) at e-value below 1 × 10−12, filtered for reciprocal hits at or above 85% identity and with the gene length not differing by more than 20%.
Statistical analysis
In some experiments to enrich for yeast strains carrying a larger number of GFP-marked deletions in the genome using flow cytometry, sorted cells and unsorted cells were genotyped using PCR (the main text; Supplemental Table S4). One-tailed Mann-Whitney U-tests were used to evaluate the significance of differences in distributions of numbers of detected deletions between the samples. When sorting based on the GFP-RFP ratio was compared to sorting based on GFP alone, only the frequencies of double mutants were described. Fisher’s exact test was used to calculate the P-value. All statistical tests used α = 0.05.
Data access
The sequencing data from the transposon bombardment study and from the whole-genome sequencing of the engineered mycoplasma strains, as well as a control strain, have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP050098 and SRP044962, respectively.
Competing interest statement
The authors declare competing financial interests. J.C.V. is Chief Executive Officer and Co-Chief Scientific Officer of Synthetic Genomics, Inc. (SGI). H.O.S. is Co-Chief Scientific Officer and on the Board of Directors of SGI. C.A.H. is Chairman of the Scientific Advisory Board of SGI. D.G.G. is a Vice President at SGI. All four of these authors and the J. Craig Venter Institute hold SGI stock.
Supplementary Material
Acknowledgments
We thank Roger Tsien for sharing a tdTomato plasmid with us. The described work was supported by the US Defense Advanced Research Projects Agency contract N66001-12-C-4039 to Y.S. and the US Department of Energy cooperative agreement DE-FC02-02ER63453 to J.C.V. The research group was also supported by the US Department of Energy cooperative agreement DE-EE0006109 to Y.S. and the Synthetic Genomics, Inc. grant to H.O.S. and C.A.H.
Author contributions: Y.S., K.S.W., S.V., E.A.W., C.A.H., D.G.G., H.O.S., J.I.G., and J.C.V. designed the research. Y.S., N.A., M.K., V.N.N., K.S.W., B.J.K., J.S., M.G.M., T.J.H., N.J.E., A.R., G.M.G., R.A.R., R.C., and D.G.G. performed experiments. Y.S., M.K., K.S.W., B.J.K., M.G.M., T.J.H., N.J.E., A.R., R.A.R., and H.O.S. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.182477.114.
References
- Allam AB, Brown MB, Reyes L. 2012. Disruption of the S41 peptidase gene in Mycoplasma mycoides capri impacts proteome profile, H2O2 production, and sensitivity to heat shock. PLoS ONE 7: e51345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Bellí G, Garí E, Piedrafita L, Aldea M, Herrero E. 1998. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res 26: 942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benders GA, Noskov VN, Denisova EA, Lartigue C, Gibson DG, Assad-Garcia N, Chuang RY, Carrera W, Moodie M, Algire MA, et al. 2010. Cloning whole bacterial genomes in yeast. Nucleic Acids Res 38: 2558–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchia M, Kouprina N, Kim SJ, Larionov V, Schlessinger D, Nagaraja R. 2000. Recovery and potential utility of YACs as circular YACs/BACs. Nucleic Acids Res 28: e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaMassa AJ, Brooks DL, Adler HE. 1983. Caprine mycoplasmosis: widespread infection in goats with Mycoplasma mycoides subsp mycoides (large-colony type). Am J Vet Res 44: 322–325. [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41: 4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dordet Frisoni E, Marenda MS, Sagné E, Nouvel LX, Guérillot R, Glaser P, Blanchard A, Tardy F, Sirand-Pugnet P, Baranowski E, et al. 2013. ICEA of Mycoplasma agalactiae: a new family of self-transmissible integrative elements that confers conjugative properties to the recipient strain. Mol Microbiol 89: 1226–1239. [DOI] [PubMed] [Google Scholar]
- Dybvig K, French CT, Voelker LL. 2000. Construction and use of derivatives of transposon Tn4001 that function in Mycoplasma pulmonis and Mycoplasma arthritidis. J Bacteriol 182: 4343–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezraty B, Aussel L, Barras F.. 2005. Methionine sulfoxide reductases in prokaryotes. Biochim Biophys Acta 1703: 221–229. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, et al. 2008. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319: 1215–1220. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA, Benders GA, Montague MG, Ma L, Moodie MM, et al. 2010. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 329: 52–56. [DOI] [PubMed] [Google Scholar]
- Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, Maruf M, Hutchison CA, Smith HO, Venter JC. 2006. Essential genes of a minimal bacterium. Proc Natl Acad Sci 103: 425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15: 1541–1553. [DOI] [PubMed] [Google Scholar]
- Gomez-Alvarez V, Teal TK, Schmidt TM. 2009. Systematic artifacts in metagenomes from complex microbial communities. ISME J 3: 1314–1317. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, et al. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320: 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CA, Peterson SN, Gill SR, Cline RT, White O, Fraser CM, Smith HO, Craig Venter J. 1999. Global transposon mutagenesis and a minimal mycoplasma genome. Science 286: 2165–2169. [DOI] [PubMed] [Google Scholar]
- Itaya M, Tsuge K, Koizumi M, Fujita K. 2005. Combining two genomes in one cell: stable cloning of the Synechocystis PCC6803 genome in the Bacillus subtilis 168 genome. Proc Natl Acad Sci 102: 15971–15976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas BJ, Tagwerker C, Yonemoto IT, Hutchison CA III, Smith HO. 2011. Cloning the Acholeplasma laidlawii PG-8A genome in Saccharomyces cerevisiae as a yeast centromeric plasmid. ACS Synth Biol 1: 22–28. [DOI] [PubMed] [Google Scholar]
- Karas BJ, Jablanovic J, Sun L, Ma L, Goldgof GM, Stam J, Ramon A, Manary MJ, Winzeler EA, Venter JC, et al. 2013a. Direct transfer of whole genomes from bacteria to yeast. Nat Methods 10: 410–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas BJ, Molparia B, Jablanovic J, Hermann WJ, Lin YC, Dupont CL, Tagwerker C, Yonemoto IT, Noskov VN, Chuang RY, et al. 2013b. Assembly of eukaryotic algal chromosomes in yeast. J Biol Eng 7: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas BJ, Jablanovic J, Irvine E, Sun L, Ma L, Weyman PD, Gibson DG, Glass JI, Venter JC, Hutchison CA III, et al. 2014. Transferring whole genomes from bacteria to yeast spheroplasts using entire bacterial cells to reduce DNA shearing. Nat Protoc 9: 743–750. [DOI] [PubMed] [Google Scholar]
- King KW, Dybvig K. 1991. Plasmid transformation of Mycoplasma mycoides subspecies mycoides is promoted by high concentrations of polyethylene glycol. Plasmid 26: 108–115. [DOI] [PubMed] [Google Scholar]
- Krishnakumar R, Grose C, Haft DH, Zaveri J, Alperovich N, Gibson DG, Merryman C, Glass JI. 2014. Simultaneous non-contiguous deletions using large synthetic DNA and site-specific recombinases. Nucleic Acids Res 42: e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartigue C, Blanchard A, Renaudin J, Thiaucourt F, Sirand-Pugnet P. 2003. Host specificity of mollicutes oriC plasmids: functional analysis of replication origin. Nucleic Acids Res 31: 6610–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartigue C, Vashee S, Algire MA, Chuang RY, Benders GA, Ma L, Noskov VN, Denisova EA, Gibson DG, Assad-Garcia N, et al. 2009. Creating bacterial strains from genomes that have been cloned and engineered in yeast. Science 325: 1693–1696. [DOI] [PubMed] [Google Scholar]
- Nolan M, Gronow S, Lapidus A, Ivanova N, Copeland A, Lucas S, Del Rio TG, Chen F, Tice H, Pitluck S, et al. 2009. Complete genome sequence of Streptobacillus moniliformis type strain (9901). Stand Genomic Sci 1: 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov VN, Segall-Shapiro TH, Chuang RY. 2010. Tandem repeat coupled with endonuclease cleavage (TREC): a seamless modification tool for genome engineering in yeast. Nucleic Acids Res 38: 2570–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov VN, Karas BJ, Young L, Chuang RY, Gibson DG, Lin YC, Stam J, Yonemoto IT, Suzuki Y, Andrews-Pfannkoch C, et al. 2012. Assembly of large, high G+C bacterial DNA fragments in yeast. ACS Synth Biol 1: 267–273. [DOI] [PubMed] [Google Scholar]
- Ochman H, Gerber AS, Hartl DL. 1988. Genetic applications of an inverse polymerase chain reaction. Genetics 120: 621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel W, Mayer M. 1984. Structure and mitotic stability of minichromosomes originating in yeast cells transformed with tandem dimers of CEN11 plasmids. MGG Mol Gen Genet 195: 300–307. [DOI] [PubMed] [Google Scholar]
- Pinel D, D’Aoust F, del Cardayre SB, Bajwa PK, Lee H, Martin VJJ. 2011. Saccharomyces cerevisiae genome shuffling through recursive population mating leads to improved tolerance to spent sulfite liquor. Appl Environ Microbiol 77: 4736–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov AV, Bützler C, Brüggemann M. 1997. Yeast colony size reflects YAC copy number. Nucleic Acids Res 25: 2039–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posfai G, Plunkett G, Feher T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V, Stahl B, Sharma SS, de Arruda M, et al. 2006. Emergent properties of reduced-genome Escherichia coli. Science 312: 1044–1046. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572. [DOI] [PubMed] [Google Scholar]
- Sørensen KI, Hove-Jensen B. 1996. Ribose catabolism of Escherichia coli: characterization of the rpiB gene encoding ribose phosphate isomerase B and of the rpiR gene, which is involved in regulation of rpiB expression. J Bacteriol 178: 1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Lewis LK, Resnick MA. 2001. In vivo site-directed mutagenesis using oligonucleotides. Nat Biotechnol 19: 773–776. [DOI] [PubMed] [Google Scholar]
- Sugiyama M, Ikushima S, Nakazawa T, Kaneko Y, Harashima S. 2005. PCR-mediated repeated chromosome splitting in Saccharomyces cerevisiae. Biotechniques 38: 909. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, St Onge RP, Mani R, King OD, Heilbut A, Labunskyy VM, Chen W, Pham L, Zhang LV, Tong AHY, et al. 2011. Knocking out multigene redundancies via cycles of sexual assortment and fluorescence selection. Nat Methods 8: 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Stam J, Novotny M, Yachie N, Lasken RS, Roth FP. 2012. The green monster process for the generation of yeast strains carrying multiple gene deletions. J Vis Exp 15: e4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagwerker C, Dupont CL, Karas BJ, Ma L, Chuang RY, Benders GA, Ramon A, Novotny M, Montague MG, Venepally P, et al. 2012. Sequence analysis of a complete 1.66 Mb Prochlorococcus marinus MED4 genome cloned in yeast. Nucleic Acids Res 40: 10375–10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully JG, Whitcomb RF, Clark HF, Williamson DL. 1977. Pathogenic mycoplasmas: cultivation and vertebrate pathogenicity of a new spiroplasma. Science 195: 892–894. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Pöhlmann R, Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10: 1793–1808. [DOI] [PubMed] [Google Scholar]
- Wise KS, Foecking MF, Röske K, Lee YJ, Lee YM, Madan A, Calcutt MJ. 2006. Distinctive repertoire of contingency genes conferring mutation-based phase variation and combinatorial expression of surface lipoproteins in Mycoplasma capricolum subsp. capricolum of the Mycoplasma mycoides phylogenetic cluster. J Bacteriol 188: 4926–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods RA, Gietz RD. 2001. High-efficiency transformation of plasmid DNA into yeast. Methods Mol Biol 177: 85–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.