

Pathological changes in the livers of human abusers of alcohol range from mild (steatosis) to moderate (steatohepatitis and early fibrosis) to advanced (late fibrosis and cirrhosis), and depend on both the daily dose and pattern of exposure.1 Although the progression of alcoholic liver disease (ALD) is well characterized, there is no universally accepted drug therapy to prevent or treat this disease in humans. Instead, clinical treatment focuses predominantly on alcohol abstinence, nutritional support, and treatment of decompensation.1 These gaps in our knowledge have been due, in part, to the lack of an animal model of ALD that develops pathology that more completely recapitulates the human disease.
Numerous species are used to study ALD, including baboons and mini-pigs. However, owing to ease and cost, the majority of research is performed in rodents. Further, the availability of genetically altered strains makes mice the de facto species of choice for ALD research. However, rodents have a strong aversion to the taste or smell of ethanol and therefore do not voluntarily consume large enough quantities of ethanol in drinking water to produce significant liver pathology.2 Alternate techniques of ad libitum ethanol delivery, in which ethanol is incorporated into a liquid diet,3 partially overcome a rodent’s natural aversion to alcohol and daily ethanol intake is sufficient to produce steatosis. Although liquid diet models are distinct improvements over drinking water models, liver pathology is still limited predominantly to steatosis.3 Another means to bypass aversion to ethanol is by enteral feeding by way of a surgically implanted intragastric tube,4 which allows the researcher to achieve blood alcohol counts (BACs) that are much higher than ad libitum alcohol feeding. The main advantage of enteral feeding is that the pathology is more severe than in ad libitum models and better mimics early ALD in humans.4 However, the technical demands of this protocol have limited the number of research groups that actively employ it.
Although hepatic steatosis is generally considered an asymptomatic disease state, it sensitizes the liver to injury caused by a second insult.5 For example, steatosis caused by ethanol exposure is well known to enhance liver pathology induced by bolus injection of the bacterial cell wall product, lipopolysaccharide (LPS).6 However, although circulating LPS levels are elevated in both humans and in experimental animals consuming ethanol,7,8 these levels are much lower than observed after bolus injection in these models. Therefore, the relevance of the alcohol:LPS “2-hit” model to human ALD has been questioned.9 LPS is not the only hit for which ethanol exposure enhances hepatotoxicity. Indeed, human studies have suggested that the risk of ALD increases in individuals who engage in binge episodes of drinking on top of heavy daily consumption.10 These data suggest that acute high-dose ethanol exposure itself can serve as the second “hit” on the background of chronic consumption.
Gao and colleagues have “reverse-translated” these clinical observations into a mouse model of acute bolus ethanol exposure after chronic ad libitum exposure (i.e., the “NIAAA model”11,12). This model employs 10 days ad libitum exposure, followed by an acute bolus gavage (Fig. 1); the latter dose regimen yields BACs of ~400 mg/dL,12 which is in the range that “professional” drinkers can attain.13 Whereas neither the chronic nor the acute regimens cause major hepatic changes by themselves, their combination synergistically induces inflammatory liver damage11,12; this model may also therefore represent human acute alcoholic hepatitis (AH).
Fig. 1.
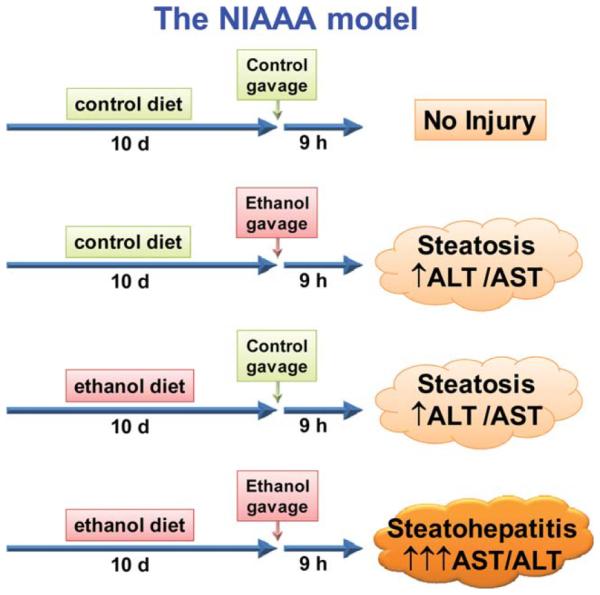
Schematic depiction of the NIAAA model. Ethanol or isocaloric maltose dextrin (control) liquid diet is fed ad libitum for 10 days. Mice are subsequently administered a bolus gavage of ethanol or isocaloric maltose-dextrin. Liver injury caused by ethanol diet or ethanol gavage alone is relatively minor and consists predominantly of steatosis. The combination of ethanol diet and ethanol gavage synergistically damages the liver, with active steatohepatitis. See Bertola et al.12 for methodological details.
The results of the current study indicate that neutrophil chemoattraction plays a key role in the observed liver damage in this model.14 Monocytes/ macrophages appear to play a more dominant role in most rodent models of AH/ALD, whereas human AH has a strong neutrophil component.15 Here, the production of neutrophil chemoattractants (e.g., monocyte chemoattractant protein-1 [MCP-1], and macrophage inflammatory proteins [MIPs]) was robustly induced by the chronic/binge paradigm. There was also a strong recruitment of neutrophils, the damaging role of which was validated with depletion experiments (anti-Ly6G antibodies). The authors demonstrated that E-selectin was induced to a much greater extent than other adhesion molecules (e.g., intercellular cell adhesion molecule-1 [ICAM-1] and vascular cell adhesion molecule-1 [VCAM-1]) that are involved in the rolling, sticking, and/or extravasation of neutrophils. Importantly, they demonstrated that E-selectin-deficient mice were almost completely protected against neutrophil recruitment and liver damage in this model. The authors were careful and thorough of their characterization of the damaging role of neutrophils and E-selectin in this work. The authors also took it one step further and demonstrated that E-selectin expression is induced in human AH patients and correlates with indices of neutrophil recruitment. Indeed, a major strength of this study is that the authors translated their novel benchtop findings into clinical samples, which makes a cohesive and convincing case. Taken together, these data make a strong and thorough case for a critical role of E-selectin-mediated neutrophil recruitment and damage in AH. Interestingly, this protein is not induced at later stages of the human disease (e.g., cirrhosis), which suggests that it might be selectively pathogenic in early phase ALD.
Although this model shows promise as a new paradigm for AH/ALD, there are several points that remain to be addressed. First, although the pathology in the NIAAA model appears to better represent the hepatic injury found in AH, the characterization of this pathology is incomplete. For example, Mallory- Denk bodies are characteristic pathologic changes found in livers from AH patients.15 Although the NIAAA model appears to produce necroinflammatory foci,14 whether or not these contain Mallory-Denk bodies has not been characterized. Second, no study as yet has demonstrated any fibrotic changes in the NIAAA model, although the authors claim that it is feasible.12 It would be interesting to determine if a more prolonged version of this model will indeed cause the appearance of fibrotic changes in the liver; this would be a great improvement over employing surrogate models of hepatic fibrosis (e.g., bile duct ligation and carbon tetrachloride [CCl4]). Related to this point is that liver pathology in AH/ALD is only a small part of a complex clinical picture. There are a host of effects associated with AH/ALD liver that are the major causes of clinical complications and mortality in AH/ALD.1 Aspects important to human AH/ ALD diagnosis and prognosis (e.g., prothrombin time, bilirubin) have not yet been characterized in this model. It would be very interesting to see if the NIAAA model induces any changes in the mice that are reflective of these clinical aspects of AH/ALD. If so, this model could then be used to screen for new drugs that target not only liver injury, but also the major causes of death. Therefore, additional studies will be required for the final verdict on whether or not this is indeed a new and improved rodent model of AH/ALD.
Abbreviations
- AH
alcoholic hepatitis
- ALD
alcoholic liver disease
- BAC
blood alcohol count
- LPS
lipopolysaccharide
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Bergheim I, McClain CJ, Arteel GE. Treatment of alcoholic liver disease. Dig Dis. 2005;2:3–275. doi: 10.1159/000090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Best CH, Hartroft WS, Lucas CC, Ridout JH. Liver damage produced by feeding alcohol or sugar and its prevention by choline. Br Med J. 1949;2:1001–1006. doi: 10.1136/bmj.2.4635.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieber CS, Jones DP, DeCarli LM. Effects of prolonged ethanol intake: Production of fatty liver despite adequate diets. J Clin Invest. 1965;44:1009–1021. doi: 10.1172/JCI105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsukamoto H, Reiderberger RD, French SW, Largman C. Long-term cannulation model for blood sampling and intragastric infusion in the rat. Am J Physiol. 1984;247:R595–R599. doi: 10.1152/ajpregu.1984.247.3.R595. [DOI] [PubMed] [Google Scholar]
- 5.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;1:14–842. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 6.Deaciuc IV, Nikolova-Karakashian M, Fortunato F, Lee EY, Hill DB, McClain CJ. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol Clin Exp Res. 2000;2:4–1557. [PubMed] [Google Scholar]
- 7.Fukui H, Brauner B, Bode JC, Bode C. Plasma endotoxin concentrations in patients with alcoholic and non-alcoholic liver disease: reevaluation with an improved chromogenic assay. J Hepatol. 1991;1:2–162. doi: 10.1016/0168-8278(91)90933-3. [DOI] [PubMed] [Google Scholar]
- 8.Nanji AA, Khettry U, Sadrzadeh SMH. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver disease. Proc Soc Exp Biol Med. 1994;2:05–243. doi: 10.3181/00379727-205-43703. [DOI] [PubMed] [Google Scholar]
- 9.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- 10.Mathurin P, Deltenre P. Effect of binge drinking on the liver: an alarming public health issue? Gut. 2009;5:8–613. doi: 10.1136/gut.2007.145573. [DOI] [PubMed] [Google Scholar]
- 11.Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. HEPATOLOGY. 2010;5:2–1291. doi: 10.1002/hep.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urso T, Gavaler JS, Van Thiel DH. Blood ethanol levels in sober alcohol users seen in an emergency room. Life Sci. 1981;2:8–1053. doi: 10.1016/0024-3205(81)90752-9. [DOI] [PubMed] [Google Scholar]
- 14.Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synerg istically induces neutrophil infiltration and liver injury: a critical role for E-selectin. HEPATOLOGY. 2013;5:8–1814. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arteel G, Marsano L, Mendez C, Bentley F, McClain CJ. Advances in alcoholic liver disease. Best Pract Res Clin Gastroenterol. 2003;1:7–625. doi: 10.1016/s1521-6918(03)00053-2. [DOI] [PubMed] [Google Scholar]