

Abstract
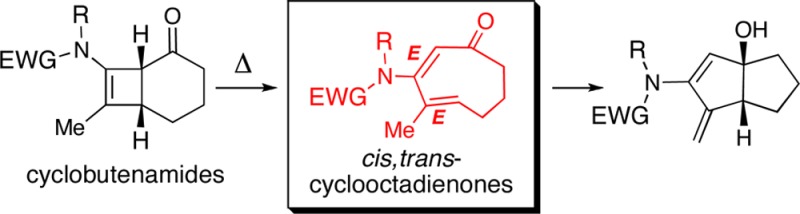
Electrocyclic ring opening of 4,6-fused cyclobutenamides 1 under thermal conditions leads to cis,trans-cyclooctadienones 2-E,E as transient intermediates, en route to 5,5-bicyclic products 3. Theoretical calculations predict that 4,5-fused cyclobutenamides should likewise undergo thermal ring opening, giving cis,trans-cycloheptadienones, but in this case conversion to 5,4-bicyclic products is thermodynamically disfavored, and these cyclobutenamides instead rearrange to vinyl cyclopentenones.
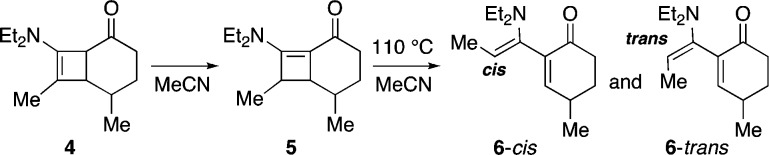
Small trans-cycloalkenes have unique reactivities and structural features, including inherent planar chirality, which have been the focus of many elegant experimental and seminal theoretical studies,1−6 and have recently attracted increasing interest relating to their applications in bioconjugate chemistry.1d Accommodating a trans olefin within a ring size of 7, 8, or 9 engenders significant ring strain, which may be alleviated if the ring contains heteroatoms such as N, O, or S but is exacerbated when the ring contains additional sp2-hybridized atoms.3−5 We report here the discovery of a new entry point into the chemistry of all-carbon trans-cycloalkenones, uncovered during our studies of cyclobutenamides 1 (Scheme 1).7−9 Under thermal conditions, 4,6-fused cyclobutenamides 1 undergo rearrangement to products whose structures are consistent with the intermediacy of the cyclooctadienone 2-E,E, containing one cis and one trans olefin, derived from cyclobutene ring opening. Small-ring trans-cycloalkenes are expected to be metastable intermediates that should undergo facile inter- or intramolecular transformations, and in the case of 2-E,E, this is manifested in a transannular cyclization leading to the 5,5-bicyclic product 3. This outcome is surprising as it is in direct contrast to previous studies of the related 4,6-fused cyclobutenamine 4 (Scheme 2), which showed no evidence for cycloalkadienone formation. Thermal rearrangement of 4 gave instead the dienes 6-cis and 6-trans.9,10 The sole difference is the amino group in 4 versus the amide in 1. Here we report our experimental and theoretical studies of the generation and fate of cis,trans-cyclooctadienones 2 from 4,6-fused cyclobutenamides 1 and the contrasting behavior of the corresponding 4,5-fused cyclobutenamides.
Scheme 1. Ring-Fused Cyclobutenamides as Potential Precursors to Small Cis,Trans-Cycloalkadienones.

Scheme 2. Ficini’s Thermal Rearrangement of a Fused Cyclobutenamine10.
Our experiments on the thermal rearrangements of cyclobutenamides 1 are summarized in Table 1. When heated at 100–120 °C in toluene, 4,6-fused cyclobutenamides 1 rearranged to the synthetically useful pentalanes 3 in 45–62% yield. The choice of appropriate conditions was crucial: no reaction took place either in DMF or chloroform (entries 1 and 2), or in the presence of base or 4 Å molecular sieves (entries 4 and 5). Yields were slightly higher when the reaction was conducted at 120 °C than at 100 °C (e.g., entry 3 vs 6) and were higher when lower concentrations of cyclobutenamide were used (e.g., entries 7, 8, 10), but in no case could the reaction be driven to completion.
Table 1. Thermal Rearrangements of 4,6-Fused Cyclobutenamides 1.
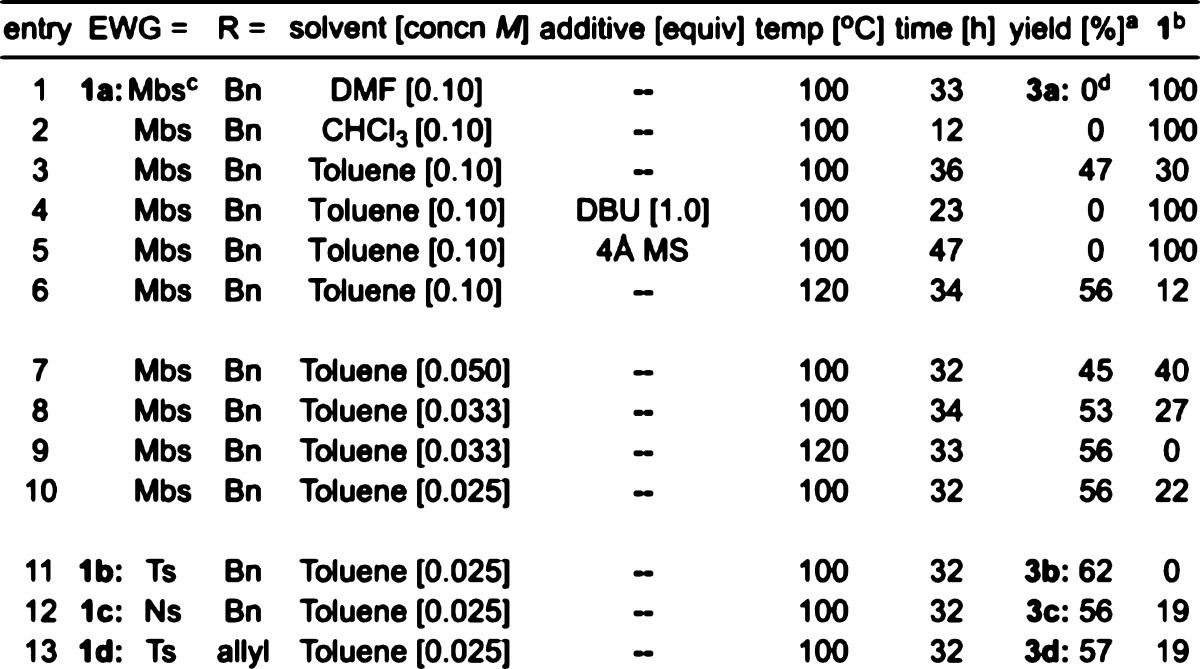
All are isolated yields.
Recovered starting 1.
Mbs = para-methoxy-benzene-sulfonyl; Ns = para-nitro-benzene-sulfonyl.
Complete recovery of 1.
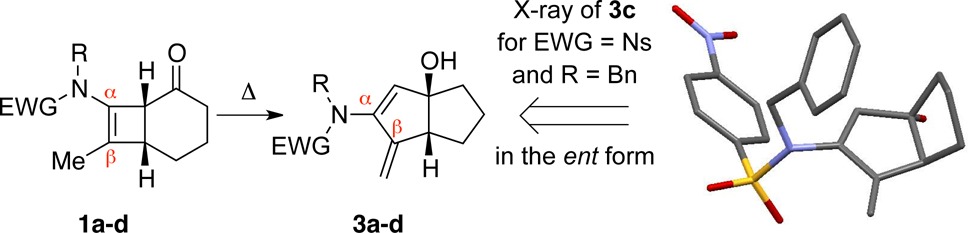
The scope and generality of this fascinating cascade are accentuated in Figure 1. Variation of substituents on the nitrogen atom and/or the β-carbon of the starting cyclobutenamides led to the pentalanes 3d (Table 1) and 3e–h (Figure 1). The initially formed hydroxypentalanes could undergo syn-dehydration to fulvenes, which may be isolated (3e′ and 3f′) or trapped by an intramolecular [4 + 2] cycloaddition onto a tethered alkene to give tetracycles (3h″ and 3g″). These last two examples reveal the synthetic potential of this rearrangement for the rapid assembly of structural complexity. It is also noteworthy that, in the case of 3e/3e′, only the Z exocyclic olefin was found.
Figure 1.
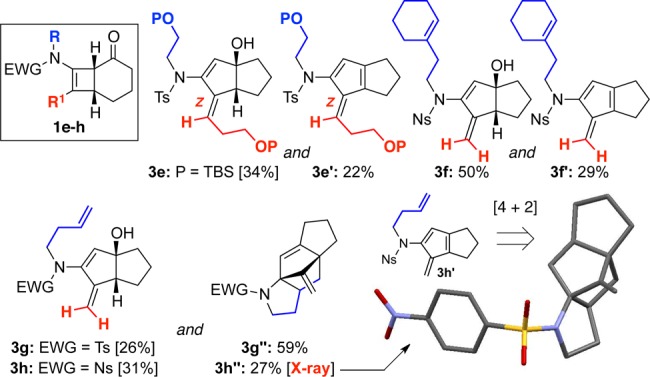
Structurally diverse products accessed through thermal rearrangements of cyclobutenamides 1.
Having uncovered this novel cascade reaction, we were intrigued by its mechanism and distinct contrast to Ficini’s observations on the 4,6-fused cyclobutenamines 4.10 We propose that the rearrangement of 1 to 3 follows the mechanism shown in Scheme 3. The details of the mechanism were established with the aid of density functional theory calculations,11 performed at the M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) level of theory (Figure 2).12 The 4π electrocyclic ring opening of 1 predictably13 favors the conrotatory mode in which the cyclohexanone alkyl group rotates outward and the carbonyl rotates inward (direction a) leading to 2-E,E. This intermediate lies 9.4 kcal/mol above 1. The computed torquoselectivity (ΔΔG‡) is 9.8 kcal/mol.
Scheme 3. Proposed Mechanism for Thermal Rearrangement of 1 to 3.

Figure 2.
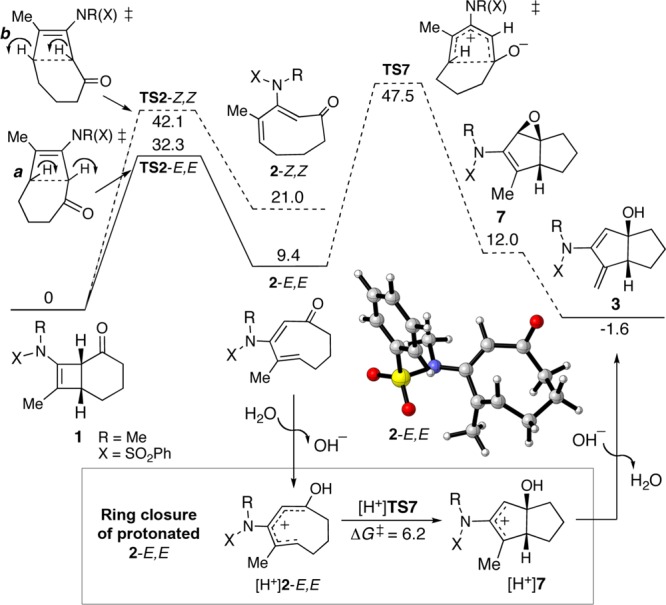
Free energy profile for thermal rearrangement of 1 to 3 in toluene, calculated at the M06-2X/6-311+G(d,p)//B3LYP/6-31G(d) level of theory with SMD solvent corrections. ΔG in kcal/mol.
The E,E isomer of 2 is not only kinetically favored but also the only isomer possessing an appropriate geometry for cyclization in the 5,5 mode to give 3 (in 2-Z,Z, the carbonyl group and diene terminus are too far apart to form a bond). Ring closure of 2-E,E resembles a Nazarov cyclization (cf. resonance structure 2′-E,E, Scheme 3). Starting from the neutral cyclooctadienone, 5,5 ring closure leading to epoxide 7 (the result of barrierless collapse of a 5,5-bicyclic zwitterion) has a very high barrier (TS7, 47.5 kcal/mol), too high to be feasible at 100–120 °C. However, protonation of the carbonyl group (Figure 2 inset) lowers the energy of the Nazarov TS significantly, such that it lies only 6.2 kcal/mol above protonated 2-E,E.14,15 Based on this result, together with the lack of reaction in the presence of 4 Å molecular sieves (Table 1, entry 5), we propose that the cyclization step is catalyzed by adventitious water acting as an acid catalyst.15 Unfortunately, the yield of 3 could not be improved by deliberate addition of an acid catalyst; for example, addition of camphorsulfonic acid (CSA, 0.4 equiv) brought about severe decomposition.16
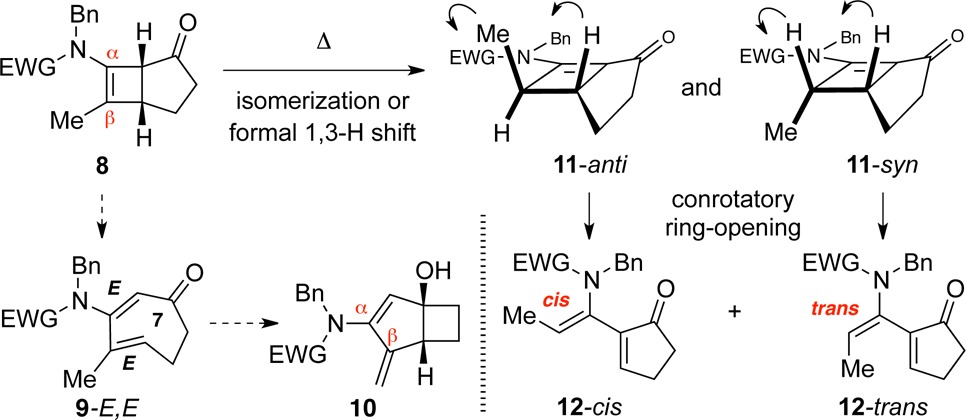
The 4,5-fused cyclobutenamides 8 behaved differently from 1, instead echoing the behavior of 4 observed by Ficini (Scheme 2).10 As shown in Table 2, no products were identified that could be traced to the cis,trans-cycloheptadienone intermediate 9-E,E. Instead, 4,5-fused cyclobutenamides 8 rearranged to amido-dienes 12 as cis/trans mixtures.17 The rearrangements involving the 4,5-fused series required much higher temperatures (195 °C) than those in the 4,6-fused series (100–120 °C).
Table 2. Thermal Rearrangements of 4,5-Fused Cyclobutenamides 8.
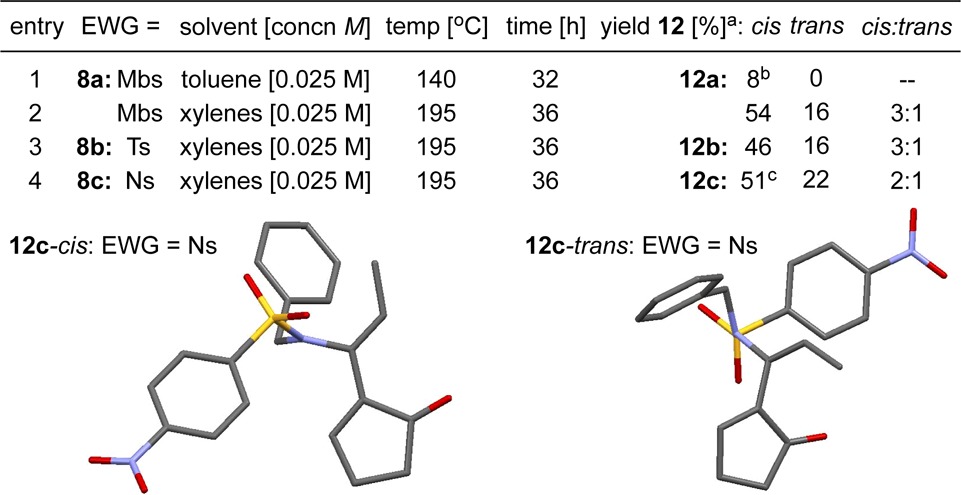
All are isolated yields.
72% recovered 8a.
16% recovered 8c.
The rearrangement of 8 to 12 is proposed to commence with formation of the conjugated cyclobutenone 11 (syn/anti), as previously observed by Ficini10c for 4 (Scheme 2). 4π electrocyclic ring opening of 11 gives 12. Each isomer of 11 undergoes ring opening with complete torquoselectivity, avoiding unfavorable inward rotation by the cyclopentanone alkyl group that would lead to a highly strained trans-cyclopentenone. Thus, 12-cis is derived from 11-anti and 12-trans from 11-syn.
Why does 8 rearrange to 12, rather than to 10? Theoretical calculations (Figure 3a) in fact predict that the cis,trans-cycloheptadienone 9-E,E, which would be the precursor of 10, can be accessed from 8 with a barrier of 33.1 kcal/mol (TS9). This value is only 0.8 kcal/mol higher than the barrier for formation of 2-E,E from 1. It is therefore likely that some 9-E,E is formed transiently during the course of the reaction at 195 °C. The transient formation of 9-E,E is not reflected in the final product distribution, however, because the transannular cyclization product 10 is higher in energy than the starting cyclobutenamide (ΔG = 10.4 kcal/mol).18 Given the thermodynamic driving force against the formation of 10, the cis,trans-cycloheptadienone 9-E,E reverts to 8 which rearranges by the alternative pathway shown in Figure 3b to 12. Conversion of 8 to the α,β-unsaturated intermediate 11 is likely to be acid catalyzed.19 Formation of 12-cis and 12-trans from 8 has ΔG ≈ −12 kcal/mol.
Figure 3.
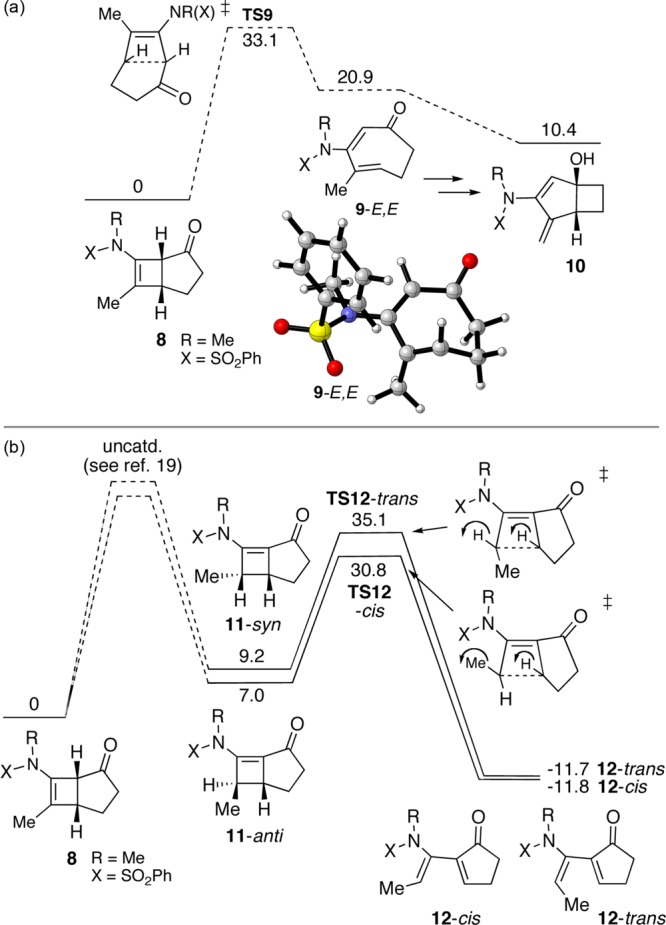
Free energy profiles for thermal rearrangements of 8 to (a) 10 and (b) 12 in toluene.
We have documented here the thermal rearrangements of 4,6-fused and 4,5-fused cyclobutenamides 1 and 8 and the discovery of contrasting mechanistic pathways. Theory predicts that 1 and 8 undergo electrocyclic ring opening to cis,trans-cyclooctadienone 2-E,E and cis,trans-cycloheptadienone 9-E,E, respectively. For 2-E,E, Nazarov-like ring closure leads to 5,5-bicyclic amido-dienes 3, but for 9-E,E, Nazarov cyclization is thermodynamically disfavored and an alternative rearrangement leads to monocyclic amido-dienes 12. The differing behavior between 1 and Ficini’s 4,6-fused cyclobutenamines 4 likely reflects the lower basicity of 1, which inhibits the isomerization to a conjugated cyclobutenamide (cf. 5) that would trigger rearrangement to monocyclic amido-dienes. Further synthetic and mechanistic studies and development of an asymmetric variant are underway.
Acknowledgments
We thank the NIH (GM-66055 to R.P.H.), NSF (CHE-0548209 to K.N.H.), and Australian Research Council (FT120100632 to E.H.K.) for generous financial support, and the NCI NF (Australia) and University of Queensland Research Computing Centre for computer resources. R.C.J. thanks Oregon State University for a 2013–2014 Graduate Internationalization Grant.
Supporting Information Available
Experimental procedures, compound characterizations, NMR spectra, X-ray data, computational methods and data, supporting mechanistic discussion, and a complete citation for ref (11). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Visiting PhD student from Department of Chemistry, Oregon State University, 153 Gilbert Hall, Corvallis, Oregon 97331, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Reviews on planar chirality and trans-cycloalkenes:; a Eliel E. L.; Wilen S. H.; Mander L. N. In Stereochemistry of Organic Compounds; Wiley: New York, 1994; pp 1172–1175. [Google Scholar]; b Nakazaki M.; Yamamoto K.; Naemura K. Top. Curr. Chem. 1984, 125, 1. [Google Scholar]; c Marshall J. A. Acc. Chem. Res. 1980, 13, 213. [Google Scholar]; d Selvaraj R.; Fox J. M. Curr. Opin. Chem. Biol. 2013, 17, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For earlier studies on trans-cycloalkenes, see:; a Marshall J. A.; Konicek T. R.; Flynn K. E. J. Am. Chem. Soc. 1980, 102, 3287. [Google Scholar]; b Cope A. C.; Moore W. R.; Bach R. D.; Winkler H. J. S. J. Am. Chem. Soc. 1970, 92, 1243. [Google Scholar]; c Binsch G.; Roberts J. D. J. Am. Chem. Soc. 1965, 87, 5157. [Google Scholar]
- For recent leading examples, see:; a Tomooka K.; Ezawa T.; Inoue H.; Uehara K.; Igawa K. J. Am. Chem. Soc. 2011, 133, 1754. [DOI] [PubMed] [Google Scholar]; b Tomooka K.; Inoue H.; Igawa K. Chem. Lett. 2011, 40, 591. [Google Scholar]; c Royzen M.; Yap G. P. A.; Fox J. M. J. Am. Chem. Soc. 2008, 130, 3760. [DOI] [PubMed] [Google Scholar]; d Taylor M. T.; Blackman M. L.; Dmitrenko O.; Fox J. M. J. Am. Chem. Soc. 2011, 133, 9646. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Royzen M.; Taylor M. T.; DeAngelis A.; Fox J. M. Chem. Sci. 2011, 2, 2162. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Seitchik J. L.; Peeler J. C.; Taylor M. T.; Blackman M. L.; Rhoads T. W.; Cooley R. B.; Refakis C.; Fox J. M.; Mehl R. A. J. Am. Chem. Soc. 2012, 134, 2898. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Herth M. M.; Andersen V. L.; Lehel S.; Madsen J.; Knudsen G. M.; Kristensen J. L. Chem. Commun. 2013, 49, 3805. [DOI] [PubMed] [Google Scholar]
- For examples containing N, O, or S linkages, see:; a Tomooka K.; Uehara K.; Nishikawa R.; Suzuki M.; Igawa K. J. Am. Chem. Soc. 2010, 132, 9232. [DOI] [PubMed] [Google Scholar]; b Tomooka K.; Suzuki M.; Shimada M.; Ni R.; Uehara K. Org. Lett. 2011, 13, 4926. [DOI] [PubMed] [Google Scholar]; c Tomooka K.; Komine N.; Fujiki D.; Nakai T.; Yanagitsuru S.-i. J. Am. Chem. Soc. 2005, 127, 12182. [DOI] [PubMed] [Google Scholar]; d Uehara K.; Tomooka K. Chem. Lett. 2009, 38, 1028. [Google Scholar]
- For seminal studies of 1,5-cis,trans-cyclooctadiene, see:; a Cope A. C.; Howell C. F.; Knowles A. J. Am. Chem. Soc. 1962, 84, 3190. [Google Scholar]; b Whitesides G. M.; Goe G. L.; Cope A. C. J. Am. Chem. Soc. 1969, 91, 2608. [Google Scholar]
- For experimental and theoretical studies on the electrocyclic ring opening of cis-bicyclo[4.2.0]oct-7-ene to cis,trans-1,3-cyclooctadiene, and subsequent isomerization to cis,cis-1,3-cyclooctadiene, see:; a Silva López C.; Nieto Faza O.; de Lera Á. R. Chem.—Eur. J. 2007, 13, 5009. [DOI] [PubMed] [Google Scholar]; b Silva López C.; Nieto Faza O.; de Lera Á. R. Org. Lett. 2006, 8, 2055. [DOI] [PubMed] [Google Scholar]; c Baldwin J. E.; Gallagher S. S.; Leber P. A.; Raghavan A. S.; Shukla R. J. Org. Chem. 2004, 69, 7212. [DOI] [PubMed] [Google Scholar]; d Baldwin J. E.; Gallagher S. S.; Leber P. A.; Raghavan A. Org. Lett. 2004, 6, 1457. [DOI] [PubMed] [Google Scholar]; e Bramham J.; Samuel C. J. J. Chem. Soc., Chem. Commun. 1989, 29. [Google Scholar]; f Bloomfield J. J.; McConaghy J. S. Jr.; Hortmann A. G. Tetrahedron Lett. 1969, 10, 3723. [Google Scholar]
- For leading reviews on ynamide chemistry, see:; a Wang X.-N.; Yeom H.-S.; Fang L.-C.; He S.; Ma Z.-X.; Kedrowski B. L.; Hsung R. P. Acc. Chem. Res. 2014, 47, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]; b DeKorver K. A.; Li H.; Lohse A. G.; Hayashi R.; Lu Z.; Zhang Y.; Hsung R. P. Chem. Rev. 2010, 110, 5064. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Evano G.; Coste A.; Jouvin K. Angew. Chem., Int. Ed. 2010, 49, 2840. [DOI] [PubMed] [Google Scholar]
- a Li H.; Hsung R. P.; DeKorver K. A.; Wei Y. Org. Lett. 2010, 12, 3780. [DOI] [PMC free article] [PubMed] [Google Scholar]; Also see:; b Schotes C.; Mezzetti A. Angew. Chem., Int. Ed. 2011, 50, 3072. [DOI] [PubMed] [Google Scholar]
- Ficini J. Tetrahedron 1976, 32, 1449. [Google Scholar]
- a Ficini J.; Eman A.; Touzin A. M. Tetrahedron Lett. 1976, 17, 679. [Google Scholar]; b Ficini J.; Touzin A. M. Tetrahedron Lett. 1972, 13, 2093.and 2097. [Google Scholar]; c Ficini J.; Duréault A. Tetrahedron Lett. 1977, 18, 809. [Google Scholar]
- Frisch M. J., et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
- B3LYP:; a Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]; b Becke A. D. J. Chem. Phys. 1993, 98, 1372. [Google Scholar]; c Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]; d Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. J. Phys. Chem. 1994, 98, 11623. [Google Scholar]; M06-2X:; e Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]; SMD:; f Marenich A. V.; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378. [DOI] [PubMed] [Google Scholar]; 6-311+G(d,p) basis:; g Hehre W. J.; Ditchfeld R.; Pople J. A. J. Chem. Phys. 1972, 56, 2257. [Google Scholar]; h Hariharan P. C.; Pople J. A. Theor. Chim. Acta 1973, 28, 213. [Google Scholar]
- Dolbier W. R. Jr.; Koroniak H.; Houk K. N.; Sheu C. Acc. Chem. Res. 1996, 29, 471. [Google Scholar]
- In contrast, H-bonding catalysis by a water molecule lowers the barrier of the Nazarov cyclization by only 5.8 kcal/mol.
- O-Protonation of cyclobutenamide 1 (R = Me, X = SO2Ph) lowers the barrier for cyclobutene ring opening to 21.3 kcal/mol.
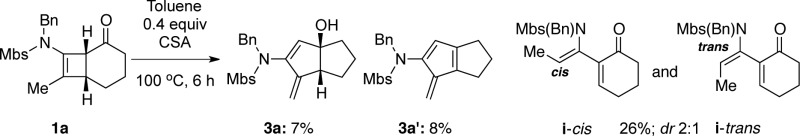
- When 1a was heated at 100 °C
in the presence of 0.4 equiv of CSA, we obtained a complex mixture
due to decomposition of 1a. However, we were able to
identify from the mixture all four products including those corresponding
[see i] to Ficini’s observations.
- Amido-dienes 12-cis and 12-trans interconvert under the reaction conditions but do not fully equilibrate during the time scale of the synthetic experiments. The cis/trans ratios of 12 in Table 2 do not reflect thermodynamic control but are determined by the rates of isomerization of 8 to 11-syn and 11-anti, which are rate-determining under thermal conditions.19
- It is noteworthy that, in the calculations on the 4,6 series (Figure 2), the final product 3 is only 1.6 kcal/mol lower in energy than the reactant 1. This likely explains our observation that these reactions could not be driven to completion in the laboratory.
- Isomerization of 8a–c to 11a–c (syn/anti) appears to be catalyzed by adventitious acid. In the presence of 0.4 equiv of CSA, amido-dienes 12a–c were obtained (85%–92% yield) after only 6 h, as compared with 36 h in the absence of CSA. Further discussion of the mechanism of conversion of 8 to 11 is provided in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.