

Abstract
We show that FRET between Pacific Blue (PB) and Alexa488 (A488) covalently attached to a DNA scaffold can be reversibly controlled by photochromic switching of a spiropyran derivative. With the spiropyran in the closed spiro isomeric form, FRET occurs freely between PB and A488. UV-induced isomerization to the open merocyanine form shuts down the FRET process by efficient quenching of the PB excited state. The process is reversed by exposure to visible light, triggering the isomerization to the spiro isomer.
Multichromophore fluorescence resonance energy transfer (FRET) cascades on self-assembled systems using DNA as a structural platform has attracted a great deal of interest in the last decades.1−8 The unique recognition properties governed by the base-pairing allow for bottom-up structural addressability on a very precise level, making DNA an excellent scaffold for nanometer-scale devices upon which the chromophores/photoactive units can be spatially arranged with high precision. This approach has allowed for the realization of DNA architectures where the flow of excitation energy can be controlled in one-, two-, and three-dimensions.9−11 Typically, the FRET processes are regulated by the addition of molecular functionalities capable of switching energy-transfer processes on or off. The downside of these pioneering studies is the irreversible nature of the switching process. Once the functionality is introduced, there is no straightforward means to remove it from the DNA construct in order to reset the system to the initial form.
Gaining dynamic reversible control over the FRET processes by external stimuli is essential for any future nanophotonic application that requires repeated on–off switching (i.e., all applications apart from the “one-time use”). Photochromic molecules are very promising candidates to be used as switching units, since photoisomerization between the implicated isomers typically occurs reversibly.12 Furthermore, the all-photonic nature of the light used to trigger the isomerization reactions makes this approach totally waste-free, as opposed to the majority of the systems triggered by chemical inputs, e.g., where DNA is being used as the “fuel”.13 In this context, the steadily growing research field of DNA-based logic operations and computing14−20 is another area where reversible switching processes will be an undeniable precondition.21 Here, we present a novel approach of photochromic on–off switching of FRET between a Pacific Blue (PB) donor chromophore and an Alexa488 (A488) acceptor chromophore covalently attached to a 20-mer DNA double-helix. This study represents the first example where FRET-switching on a DNA scaffold is done reversibly. It should be noted that in the majority of the previously reported studies on FRET-switching on DNA scaffolds, the typical approach has been to switch on the energy transfer between a donor- and an acceptor dye by the addition of FRET mediators. This allows for FRET cascades over distances longer than 10 nm. The major objective of the herein presented study was not to achieve wire-like long-range FRET processes but instead to gain reversibility of the switching by shutting on-and-off FRET between the donor- and the acceptor dye by all-photonic means.
We base the approach on our previous studies on photocontrolled DNA-binding of spiropyran derivatives, where we found that UV-activation of the spiro isomer SP to the merocyanine forms MC and MCH+ is required for binding to occur.22−25 Scheme 1 shows the structures and the interconversion pathways for the amidine appended spiropyran derivative (1) used here.24 The closed spiro form 1SP is isomerized to the corresponding open merocyanine isomer 1MC by UV light exposure (254 nm used in this study, isomerization quantum yield = 0.02 ± 0.01). The reverse reaction is triggered by visible light (λ > 450 nm used in this study, isomerization quantum yield = 0.014 ± 0.007). In the dark, a thermal equilibrium is established with a 20/80 [SP]/[MC] ratio and a time constant of ∼6 h at RT. The corresponding photostationary distribution using 254 nm light is 30/70. Upon acidification, the phenolate oxygen of the merocyanine isomer is protonated, yielding 1MCH+ with a pKa value of 5.2. This species is known to bind to DNA through intercalation with a binding constant of 3.4 × 104 M–1, whereas 1MC binds ∼35 times weaker.241SP shows no detectable DNA binding. Upon DNA binding, the pKa value of 1MCH+ increases to 6.8. Hence, at pH 5.4 where these studies were conducted, 1MCH+ is the dominating DNA binder. A comprehensive characterization of the DNA-binding properties of 1 has recently been reported.24 For the absorption spectra of all relevant forms of 1, see Figure S2.
Scheme 1. Isomerization Scheme for Spiropyran Derivative 1 Used in This Study.

Due to the dramatic differences in both the absorption spectra and the DNA-binding properties of the implicated isomers, the FRET process between a donor chromophore D and an acceptor chromophore A attached to a DNA scaffold could be affected by light-induced isomerization of 1. PB and A488 is an established D–A pair for FRET, with an R0 value of 52 Å (see below). It is obvious from Figure S1 that the absorption of DNA-bound 1MCH+ displays a substantial overlap with the PB emission. The R0 value for this D–A pair was determined to 38 Å. On the contrary, the absorption of 1SP does not overlap with the absorption of PB nor does it show any specific binding to DNA, which implies that 1SP cannot quench the excited state of PB by FRET.
PB and A488 were covalently linked to complementary 20-mer sequences, referred to as OPB and OA488, respectively (see Scheme 2). Using our experimentally determined values for the molar absorption coefficients and fluorescence quantum yields in aqueous solution for PB in OPB (17,700 M–1 cm–1 at 405 nm and 0.84) and A488 in OA488 (72,500 M–1 cm–1 at 495 nm and 0.81) hybridized to the respective unlabeled complementary strand, the R0 value for this D–A pair was determined to be 52 Å, assuming a κ2 value of 2/3. The relevant normalized spectra are shown in in Figure S1. Ideally, uninterrupted FRET will occur between PB and A488 in the OPB-OA488 double-strand. We hypothesized that the addition of 1SP to the sample should not interfere with the FRET process for the reasons described above. UV exposure isomerizes 1SP to 1MC, and subsequent protonation yields 1MCH+ that is expected to bind to the OPB-OA488 double-strand. Given a sufficiently high binding density, FRET from PB to DNA-bound 1MCH+ will be much more efficient than the corresponding process between PB and A488, and the latter process is efficiently shut off. Finally, exposure to visible light will trigger the formation of 1SP, which will dissociate from DNA, and switch on the FRET between PB and A488. This scenario is schematically depicted in Scheme 2, together with the sequences of the OPB and the OA488 oligonucleotides.
Scheme 2. Schematic Representation of the Reversible on–off Switching of FRET (top) and Sequences of the Dye-Labeled 20-Mer Oligonucleotides OPB and OA488 (bottom).
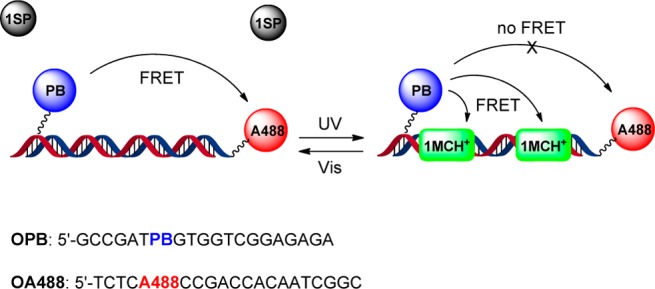
PB and A488 are both on T-bases. For the conjugate structures including the linkers, see the SI.
Figure 1 shows the emission intensities from OPB (blue line) and OA488 (red line) hybridized with the respective unlabeled complementary strand upon excitation at 410 nm ([OPB] = 1.2 μM, [OA488] = 1 μM). It is obvious that the 410 nm light directly excites a fraction of the A488 acceptor dye. The corresponding spectrum of the OPB-OA488 double-strand is shown in black and clearly shows quenched emission from PB and sensitized emission from A488. The efficiency of the FRET process is estimated to 78%, judging by the PB quenching and taking into account the 20% excess of the OPB strand (see Supporting Information, SI, for details).
Figure 1.
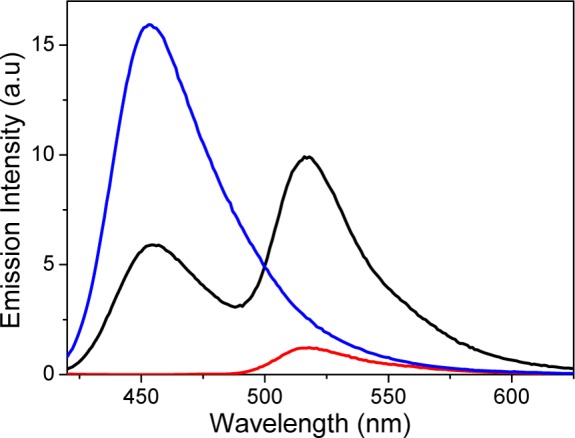
Fluorescence emission intensities (excitation at 410 nm) for OPB (blue line) and OA488 (red line) hybridized with the respective unlabeled complementary strand. Also shown is the emission spectra of the OPB-OA488 double-strand (black line). [OPB] = 1.2 μM and [OA488] = 1 μM. The same concentrations were used also for the corresponding unlabeled single-strands.
Given the nine base-pair separation between PB and A488 in the duplex, the distance between the chromophores is around 30.6 Å. With an R0 value of 52 Å, a FRET efficiency of 0.96 is expected from the Förster formulation. The observed discrepancy is likely a result of a κ2 value ≠ 2/3, well in line with the short linkers between the PB/A488 dyes and the oligonucleotides, hindering free rotation (see SI).
Time-resolved measurements using the single photon counting (SPC) technique clearly shows that the PB excited state is quenched in the OPB-OA488 double-strand. The lifetime of PB in the OPB strand hybridized with the unlabeled complementary strand is 3.4 ns, whereas the corresponding lifetime in the OPB-OA488 double-strand is 0.62 ns (see Figures S5 and S6). This implies a FRET efficiency of 82%, which is in excellent agreement with the 78% efficiency estimated from the steady-state measurements described above.
The effect of adding the dissolved form of 1, 1SP (25 μM), to the OPB-OA488 double-strand is shown in Figure 2. It is obvious that the emission intensity of both PB and A488 decreases significantly (green line vs black line). We assign this effect to a weak static quenching complex between 1SP and PB, as no short-lived component was observed in the time-resolved SPC fluorescence decay (data not shown). We can exclude residual amounts of 1MCH+, formed either thermally or from incomplete isomerization to 1SP in the stock solution, as the origin of the quenching (see Figures S3 and S4 for emission intensity variations with time and with different total concentrations of 1). The red line shows the emission spectrum after 2 min. 254 nm UV exposure, triggering the isomerization to the DNA-binding form 1MCH+. The substantially decreased emission intensity from both PB and A488 clearly shows that PB is further quenched and A488 less sensitized. Both these observations are in favor of the notion that DNA-bound 1MCH+ efficiently interrupts the FRET process between the dyes. The emission observed at wavelengths longer than 650 nm originates from 1MC.26 This emission does not overlap with the absorption spectra of PB or A488 and thus does not interfere with the intended function.
Figure 2.
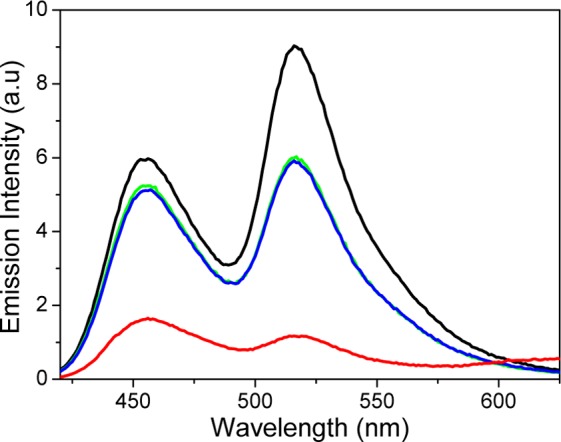
Emission intensities (excitation at 410 nm) for the OPB-A488 double-strand before (black line) and after (green line) the addition of 25 μM 1SP. The red line shows the emission intensity after 2 min. 254 nm UV irradiation to isomerize 1SP to the DNA-binding forms 1MC and 1MCH+, whereas the blue line is the emission after subsequent visible light irradiation (5 min, λ > 450 nm) to trigger the reverse isomerization to 1SP.
SPC measurements were undertaken in order to quantify the FRET efficiency between PB and DNA-bound 1MCH+. The results show that the PB excited state is too efficiently quenched for the lifetime to be resolved in the measurements, i.e., a lifetime significantly shorter than the instrumental response function of around 40 ps. The R0 value for PB and DNA-bound 1MCH+ is 38 Å, which implies that binding of 1MCH+ within a four base-pair distance from PB will quench the lifetime to around 7 ps or shorter.27 Given the concentrations of 1MCH+ and DNA binding sites used in this experiment, virtually 100% of the OPB-OA488 double-strands are expected to display such a binding density. The residual emission from PB and A488 observed in the steady-state measurements (Figure 2) is thus explained by the 20% unhybridized OPB single-strands and directly excited A488 in the OPB-OA488 double-strand, respectively.28 Hence, we can conclude that UV-induced isomerization of 1SP to the 1MCH+ form totally shuts off the FRET process between PB and A488 on the DNA scaffold. Furthermore, exposure to visible light (5 min, λ > 450 nm) triggers the isomerization back to 1SP which restores the emission intensities of both PB and A488 to the initial values (blue line in Figure 2). Thus, the process is indeed fully reversible. The repeated operation of the switching cycle is shown in Figure S7, and it is seen that the system degrades significantly after 10 UV–vis cycles. What ultimately limits the fatigue resistance of the system seems to be photocleavage of the linker between the PB/A488 dyes and the DNA oligonucleotides (see SI for details).
A few things should be commented on. Photochromic FRET switching in covalently linked molecular dyads and triads, etc., has been previously reported.29−35 In these studies, the FRET switching relies on the ability of only the UV generated colored isomeric form of the photoswitch to act as a FRET acceptor to the covalently attached donor chromophore, thereby switching the donor fluorescence emission on and off in concert with the isomerization processes. While this switching scheme certainly is interesting per se, it comes with several limitations when contrasted to multichromophoric/photochromic constructs on, e.g., DNA scaffolds: (i) With few exceptions,36,37 the colored isomeric forms of photoswitches from the most frequently used photochromic families (spiropyrans, azobenzenes, diarylethenes, fulgimides, etc.) display very low fluorescence quantum yields. Hence, fluorescence switching is only observed for the donor chromophore, which implies that energy transfer cascades and other higher order processes are very hard to achieve. (ii) Many applications require switch-to-switch communication. With increasing level of sophistication, larger numbers of molecular switching units and chromophores need to be incorporated into the constructs. Relying on covalently linked supramolecules implies that the complexity of the synthetic chemistry rapidly grows. This is why a self-assembly approach is preferable. (iii) Finally, it is worth emphasizing that the FRET efficiency between PB and A488 in this study does not necessarily have to be switched between the two extremes. Instead it can be tuned continuously to intermediate values by varying the UV–vis exposure times to achieve variations in the binding density of 1MCH+, resulting in a “transistor-like” FRET response. This is not possible for covalently linked dyads, where the FRET process between the donor and the photochromic acceptor can only be swithed in a binary fashion.
In summary, we have presented a novel method to control the flow of excitation energy from a donor to an acceptor on a DNA scaffold using all-photonic switching of a DNA-binding photochromic spiropyran derivative. Due to the all-photonic nature of the switching stimuli, the operation leaves no chemical waste behind. The process is totally reversible, but the repeated cycling is ultimately restricted by the limited resistance to photofatigue.
Acknowledgments
This work was financed by the Swedish Research Council VR (grant 622-2010-280), the European Research Council (ERC FP7/2007-2013 grant no. 203952), and the BBSRC (sLOLA grant BB/J001694/1 “extending the boundaries of nucleic acid chemistry”). The authors thank Melina Gilbert Gatty for assistance with the SPC measurements and Niklas Bosaeus for running the electrophoresis gels.
Supporting Information Available
Experimental section, oligonucleotide synthesis, labeling, purification, and characterization, spectral properties of the dyes and spiropyran derivative 1, SPC decays, and repeated operation of the switching cycle. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Su W.; Bonnard V.; Burley G. A. Chem.—Eur. J. 2011, 17, 7982–7991. [DOI] [PubMed] [Google Scholar]
- Teo Y. N.; Kool E. T. Chem. Rev. 2012, 112, 4221–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeneman K.; Prasuhn D. E.; Blanco-Canosa J. B.; Dawson P. E.; Melinger J. S.; Ancona M.; Stewart M. H.; Susumu K.; Huston A.; Medintz I. L. J. Am. Chem. Soc. 2010, 132, 18177–18190. [DOI] [PubMed] [Google Scholar]
- Hannestad J. K.; Sandin P.; Albinsson B. J. Am. Chem. Soc. 2008, 130, 15889–15895. [DOI] [PubMed] [Google Scholar]
- Kawahara S.; Uchimaru T.; Murata S. Chem. Commun. 1999, 563–564. [Google Scholar]
- Su W.; Schuster M.; Bagshaw C. R.; Rant U.; Burley G. A. Angew. Chem., Int. Ed. 2011, 50, 2712–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilemann M.; Tinnefeld P.; Mosteiro G. S.; Garcia-Parajo M.; Van Hulst N. F.; Sauer M. J. Am. Chem. Soc. 2004, 126, 6514–6515. [DOI] [PubMed] [Google Scholar]
- Spillmann C. M.; Buckhout-White S.; Oh E.; Goldman E. R.; Ancona M. G.; Medintz I. L. Chem. Commun. 2014, 50, 7246–7249. [DOI] [PubMed] [Google Scholar]
- Hannestad J. K.; Gerrard S. R.; Brown T.; Albinsson B. Small 2011, 7, 3178–3185. [DOI] [PubMed] [Google Scholar]
- Stein I. H.; Steinhauer C.; Tinnefeld P. J. Am. Chem. Soc. 2011, 133, 4193–4195. [DOI] [PubMed] [Google Scholar]
- Özhalici-Ünal H.; Armitage B. A. ACS Nano 2009, 3, 425–433. [DOI] [PubMed] [Google Scholar]
- Irie M. Chem. Rev. 2000, 100, 1685–1816. [DOI] [PubMed] [Google Scholar]
- Graugnarg E.; Kellis D. L.; Bui H.; Barnes S.; Kuang W.; Lee J.; Hughes W. L.; Knowlton W. B.; Yurke B. Nano Lett. 2012, 12, 2117–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpashchikov D. M.; Stojanovic M. N. J. Am. Chem. Soc. 2005, 127, 11348–11351. [DOI] [PubMed] [Google Scholar]
- Seelig G.; Soloveichik D.; Zhang D. Y.; Winfree E. Science 2006, 314, 1585–1588. [DOI] [PubMed] [Google Scholar]
- Frezza B. M.; Cockroft S. L.; Ghadiri M. R. J. Am. Chem. Soc. 2007, 129, 14875–14879. [DOI] [PubMed] [Google Scholar]
- Lake A.; Shang S.; Kolpashchikov D. M. Angew. Chem., Int. Ed. 2010, 49, 4459–4462. [DOI] [PubMed] [Google Scholar]
- Orbach R.; Remacle F.; Levine R. D.; Willner I. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 21228–21233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu F.; Ren J.; Qu X. Adv. Mater. 2014, 26, 5742–5757. [DOI] [PubMed] [Google Scholar]
- Orbach R.; Wang F.; Lioubashevski O.; Levine R. D.; Remacle F.; Willner I. Chem. Sci. 2014, 5, 3381–3387. [Google Scholar]
- Nishimura T.; Ogura Y.; Tanida J.. Appl. Phys. Express 2013, 6, 015201. [Google Scholar]
- Andersson J.; Li S. M.; Lincoln P.; Andréasson J. J. Am. Chem. Soc. 2008, 130, 11836–11837. [DOI] [PubMed] [Google Scholar]
- Hammarson M.; Andersson J.; Li S. M.; Lincoln P.; Andréasson J. Chem. Commun. 2010, 46, 7130–7132. [DOI] [PubMed] [Google Scholar]
- Hammarson M.; Nilsson J. R.; Li S. M.; Lincoln P.; Andréasson J. Chem.—Eur. J. 2014, 20, 15855–15862. [DOI] [PubMed] [Google Scholar]
- Hammarson M.; Nilsson J. R.; Li S. M.; Beke-Somfai T.; Andréasson J. J. Phys. Chem. B 2013, 117, 13561–13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1MCH+ is a photoacid, i.e., it deprotonates in the excited state to emit as 1MC.
- At very high binding densities, photoinduced electron-transfer reactions between PB and 1MCH+ cannot be excluded. The thermodynamic driving force of this process (if any) is not known due to the absence of redox data for the involved species.
- The residual emission from PB is somewhat lower than what is expected from the 20% excess of single-stranded OPB. This is accounted for by the experimental observation that 1MCH+ is able to quench PB also in the OPB single-strand, but to a much less extent (data not shown). Likewise, the residual emission intensity from A488 is also lower than what is expected from unquenched emission of the directly excited fraction. This is accounted for by FRET from A488 to small amounts of DNA-bound 1MC.
- Bälter M.; Li S. M.; Nilsson J. R.; Andréasson J.; Pischel U. J. Am. Chem. Soc. 2013, 135, 10230–10233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz S. A.; Giordano L.; Jovin T. M.; Jares-Erijman E. A. Nano Lett. 2012, 12, 3537–3544. [DOI] [PubMed] [Google Scholar]
- Deniz E.; Tomasulo M.; DeFazio R. A.; Watson B. D.; Raymo F. M. Phys. Chem. Chem. Phys. 2010, 12, 11630–11634. [DOI] [PubMed] [Google Scholar]
- Raymo F. M.; Tomasulo M. Chem. Soc. Rev. 2005, 34, 327–336. [DOI] [PubMed] [Google Scholar]
- Fukaminato T.; Sasaki T.; Kawai T.; Tamai N.; Irie M. J. Am. Chem. Soc. 2004, 126, 14843–14849. [DOI] [PubMed] [Google Scholar]
- Keirstead A. E.; Bridgewater J. W.; Terazono Y.; Kodis G.; Straight S.; Liddell P. A.; Moore A. L.; Moore T. A.; Gust D. J. Am. Chem. Soc. 2010, 132, 6588–6595. [DOI] [PubMed] [Google Scholar]
- Norsten T. B.; Branda N. R. J. Am. Chem. Soc. 2001, 123, 1784–1785. [DOI] [PubMed] [Google Scholar]
- Uno K.; Niikura H.; Morimoto M.; Ishibashi Y.; Miyasaka H.; Irie M. J. Am. Chem. Soc. 2011, 133, 13558–13564. [DOI] [PubMed] [Google Scholar]
- Jeong Y.-C.; Yang S. I.; Ahn K.-H.; Kim E. Chem. Commun. 2005, 2503–2505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.