

Abstract
How do biochemical signaling pathways generate biological specificity? This question is fundamental to modern biology, and its enigma has been accentuated by the discovery that most proteins in signaling networks serve multifunctional roles. An answer to this question may lie in analyzing network properties rather than individual traits of proteins in order to elucidate design principles of biochemical networks that enable biological decision-making. We discuss how this is achieved in the MST2/Hippo-Raf-1 signaling network with the help of mathematical modeling and model-based analysis, which showed that competing protein interactions with affinities controlled by dynamic protein modifications can function as Boolean computing devices that determine cell fate decisions. In addition, we discuss areas of interest for future research and highlight how systems approaches would be of benefit.
Keywords: affinity mediated phosphorylation, apoptosis, cell fate decision, competing protein interaction, mathematical modeling, proliferation, signaling switches, systems analysis
Introduction
Normal development and tissue-size homeostasis at the embryonic as well as adult level rely crucially on a fine balance between cell proliferation and apoptosis.1,2 Failure to maintain this balance leading to either exceeding proliferation or apoptosis could tip the cell toward aberrant growth or tissue loss that underlie serious pathologies, such as cancer or neurodegenerative diseases. Although much has been learned about the molecular mechanisms regulating cell proliferation and cell death in isolation, our understanding of how these opposite outcomes are coordinated at the mechanistic level remains patchy.
Originally identified in the fruit fly Drosophila melanogaster through genetic screenings for growth suppressors, the MST2/Hippo signaling pathway has emerged as an important pathway for the regulation of growth, apoptosis and proliferation in mammalian cells.3 The core pathway in Drosophila was defined as Hippo-Warts-Yorkie, where the Hippo kinase activates the Warts kinase, which phosphorylates and inhibits the transcription factor Yorkie.4,5 The individual components of this pathway are well conserved in mammals, with Hippo corresponding to MST1/2, Warts to LATS1/2, and Yorkie to YAP1/2. However, the upstream activators and downstream effectors have diverged substantially.5-7 For instance, in mammalian cells RASSF1A, a protein of the RASSF tumor suppressor family, activates MST1/2 to promote apoptosis, while the single RASSF homolog in the fly suppresses Hippo activity. Even more striking is the divergence of the pathway structure. There is robust evidence that in mammalian cells YAP can be regulated independently of MST and LATS,810 and that both MST and LATS have substrates outside of the classic Hippo pathway that was defined by genetic studies in Drosophila.11-16 These findings highlight that a modern concept of the Hippo/MST pathway needs to take organismal differences into account.
Having such a central function in the control of growth, apoptosis and the cytoskeleton it is no surprise that the Hippo/MST2/Hippo pathway is embedded in a network of crosstalk with other pathways, e.g. the Wnt, Notch, TGFβ, PI3K/Akt and the Raf/ERK pathway (reviewed in refs.4,17-19) Here, we will discuss the crosstalk between the Raf/ERK and Hippo/MST2 pathway and its role in the regulation of transformation and apoptosis in mammalian cells.
MST2/Hippo crosstalks with Raf-1 signaling through a complex network of PPIs modulated by phosphorylation
Crosstalk between the MST2/Hippo pathway and the Raf/ERK pathway occurs at several levels as summarized in Figure 1.6,20-24 At the heart of the crosstalk are dynamic changes in PPIs between the kinases MST2 and Raf-1 and their respective upstream activators RASSF1A and Ras. In un-stimulated conditions, Raf-1 binds to the SARAH domain of MST2 and interferes with its recruitment by the scaffold protein RASSF1A. This prevents RASSF1A-mediated MST2 dimerization and auto-phosphorylation on Thr180 which are both required for full activation of MST2. In addition, MST2 kinase activity is also suppressed due to dephosphorylation by a phosphatase associated with Raf-1.3,6 Interestingly, the inhibition of MST2 by Raf-1 does not require Raf kinase activity but only relies on binding. In fact, there seems to be an inverse relationship between Raf specific catalytic activity and MST2 binding and inhibition. A-Raf, which possesses barely measureable kinase activity binds MST2 strongly, while B-Raf, which has the highest kinase activity, only weakly interacts with MST2.25 This differential ability of Raf isoforms to regulate MST2 likely is the reason why this interaction was not picked up in Drosophila genetic screens. Drosophila has a single Raf ortholog that is most closely related to B-Raf, and hence not expected to interact with MST2. In mammalian cells MST2 can be released from its inhibitory complex with Raf-1 by RASSF1A, leading to MST2 activation and subsequent binding to its substrate LATS1. Depending on the input stimulus LATS1 can triggers apoptosis by inducing the formation of a YAP1-p73 transcriptional complex22 or by stabilizing the p53 tumor suppressor protein20 (Fig. 1). These pro-apoptotic pathways are obliterated by the frequent loss of RASSF1A expression in human cancers.26,27 In addition, MST2 and Ras compete for binding to Raf-1. Binding of activated Ras initiates the Raf-1 activation process.28 The main binding site of MST2 in Raf-1 overlaps with the RBD causing MST2 to interfere with Ras binding and Raf-1 activation.
Figure 1.
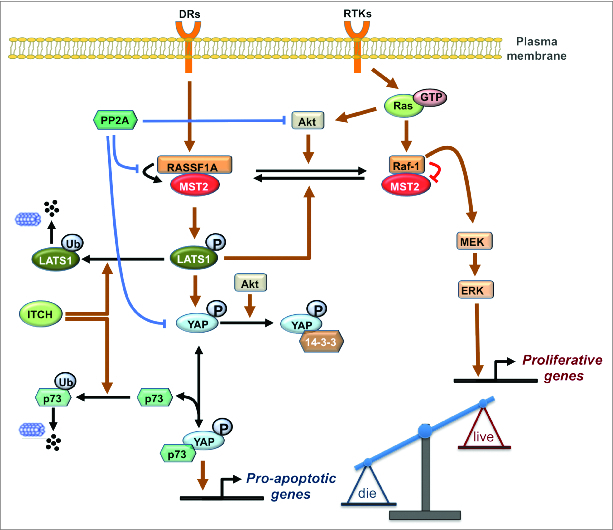
The integrated MST2/Hippo-Raf-1 signaling network schematic diagram. Normal and blunt arrows represent positive and negative regulations, respectively. DRs: Death Receptor, RTKs: Receptor Tyrosine Kinases.
The complexity of this network of competing protein interactions indicated that the resulting pathway behavior will be equally complex. Therefore, we employed a systems approach combining mathematical modeling and experimentation to investigate the mechanistic details of the crosstalk between the MST2 and Raf-1 pathways elucidating a surprising dynamic switch and feedback loop that orchestrate the activities of these pathways.24 At the heart of these switches are the competing PPIs described above and phosphorylations that change the affinity of the binding partners. Simple competitions between 2 proteins (A,B) for binding to a third protein (C) generate smooth transitions between the abundance of the respective complexes where AC declines and BC increases proportionally to the concentration of B. However, when combined with changes in affinity these transitions can become switch-like.24 The relevant affinity changes are caused by phosphorylations of Raf-1 and MST2. Akt phosphorylates MST2 and promotes its binding to Raf-1, thereby inhibiting MST2 activation.23,24 Conversely, in resting cells Raf-1 is phosphorylated on Ser259, which inhibits its kinase activity toward MEK and is dephosphorylated during the Raf-1 activation process.29 Interestingly, pSer259 enhances MST2 binding, thus diverting Raf-1 from activating MEK to inhibiting MST2. Ser259 has been identified previously as an important phosphorylation site for PKA.30,31 However, PKA inhibition did not affect the basal levels of pSer259 suggesting the existence of another Ser259 kinase. We identified LATS1 as kinase that maintains basal levels of pSer259, thereby constituting a feedback loop that inhibits both the MST2 as well as the ERK pathway.24
Analyzing molecular switches in the MST2-Raf-1 network by mathematical modeling
The operation of several concurrent competing protein interactions coupled with dynamic changes in their binding affinities regulated by different phosphorylations bring highly non-linear dynamics to the network, which challenge an intuitive analysis of network behavior. To systematically explore and understand the emergent properties of this integrated circuitry, we developed a mathematical model which allowed us to analyze the salient features of the crosstalk in a unified and quantitative framework. For this purpose we constructed a number of mathematical models that capture the network at different levels of abstraction.24 In the most coarse-grained model, the network was simplified to its essentials containing only the MST2 and Raf-1 PPI and reversible phosphorylation cycles (Fig. 2A). Two fine-grained models contained the relevant known components of both pathways, incorporating all observed protein interactions, phosphorylation reactions and feedback loops. One used Michaelis-Menten kinetics which emphasizes the role of enzymatic reactions, and another mass-action, which explicitly includes the role of PPIs in the reactions. The employment of models having different levels of detail enabled us to flexibly zoom-in and –out the network structure, which facilitates not only numerical simulations but also analytical analysis. In combination, these in silico analyses allowed us to untangle the network complexity and identify the key conditions that characterize the network behavior. Importantly, model predictions helped to articulate novel hypotheses and design appropriate experiments to test them.
Figure 2.
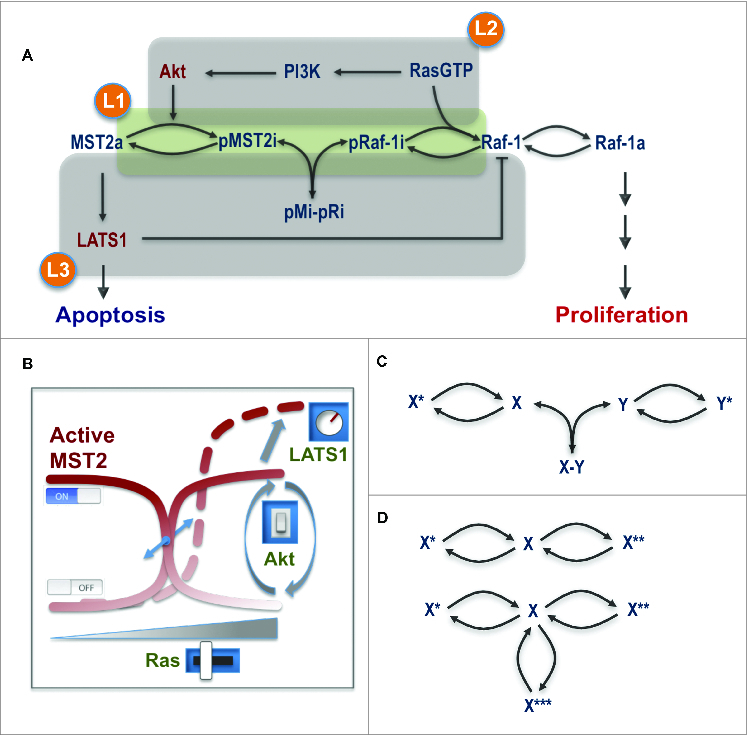
Main layers of regulation controlling the MST2-Raf-1 network behavior. (A) The core interaction scheme with overlapping regulatory layers. (B) The level and degree of the switches in the network being modulated by Ras, Akt and LATS1. (C) The switch-generating motif in the MST2-Raf-1 network. (D) The switch-generating motif by a single protein. X*, X** and X*** represent different forms of the unmodified protein X.
Model analysis revealed non-intuitive dynamic properties of the MST2-Raf-1 network.24 Most remarkably, model simulations followed by experimental verification predicted and confirmed the occurrence of sharp switches between the activities of the Raf-1 and MST2 pathways. Notably, a graded increase in Ras activity led to sharp OFF-ON switches in MST2 and Raf-1 activities. Interestingly, the model predicted that while Raf-1 switches on regardless of Akt activity, MST2 activity is strongly Akt sensitive being ON at low and OFF at high Akt activities (Fig. 2B). This unexpected prediction was confirmed experimentally using Ras mutants that induced different levels of Akt activation. Likewise, increasing serum stimulation switched Raf-1 on but switched off MST2 activities, probably due to a high Akt activation induced by serum. Ras, thus in principle, could sharply activate ERK and MST2 triggering both cell proliferation and apoptosis in cells where Akt is weakly activated by Ras. In contrast, the pro-apoptotic role of Ras is shut down in cells where it activates Akt strongly.
Our model further showed that the LATS1 to Raf-1 feedback phosphorylation affects both arms of the MST2-Raf-1 network. Stronger feedback strength attenuates MST2 activity as expected, but unexpectedly desensitizes Raf-1 and ERK activation. Weakening the feedback by LATS1 knockdown increased both the amplitude and degree of the serum growth factor-induced Raf-1 activation switch. In addition, while graded Akt activation unequivocally switches off MST2 activity independent of the LATS1 feedback, the model interestingly suggested that Akt could switch from a Raf-1 inhibiting role to a Raf-1 activating role at high LATS1 feedback strength. Taken together, the molecular module comprising the core proteins Ras, Akt, Raf-1, MST2, RASSF1A and LATS1 constitutes a highly integrated and delicate signaling apparatus, which can “digitally” coordinate opposing pathway outcomes in a switch-like manner. In this “digital” machinery, Ras acts like a switch inducer while Akt serves to direct the switches' state (ON or OFF) and LATS1 via its feedback on Raf-1 functions as a tuner for the amplitude and steepness of the switches (Fig. 2B).
Importantly, we found that these switches regulate biological outcomes and coordinate cell fate decisions in biological systems. Altering the balance between these pathways by expressing a Raf-1 S259A mutant in cultured cell lines stimulates both apoptosis and proliferation by concomitant activation of the MST2 and ERK pathways. Incapacitation of the MST2 pathway by siRNA or hyper-activation of AKT switches signaling from apoptosis to cell transformation and growth confirming the tightly interlinked control of these pathways. We further could validate that these switches exist at an organismal level by experiments in zebrafish embryos, where disruption of the MST2-Raf-2 interaction affected heart development in a switch-like fashion. The rationale for looking at heart development was the observation that Raf-1 mutations altering Ser259 phosphorylation can cause Noonan syndrome, which includes aberrant cardiac development.32-34 Thus, as these switch-like transitions seem to have wide physiological relevance, we tried to elaborate the conditions for these switches and assess whether they are met in different cell lines and tissues.
The conditions for switches
Switches often arise from bistability, a phenomenon where a system can switch between 2 distinct physiological states but cannot rest in between.35 Bistability typically results from mutually activating or repressing regulations, such as positive and double-negative feedback loops.35 The emergence of switches in the MST2-Raf-1 network, however, does not follow these usual means, but is enabled by a core motif comprising 2 reversible phosphorylation cycles linked to protein association/dissociation reactions (Fig. 2C). Since phosphorylation cycles are widespread in signal transduction networks, we expect that this novel switches-generating circuitry could be a common regulatory principle in cellular processes.
Importantly, model analysis allowed us to determine the conditions governing the occurrence of sharp switches. Switches are most abrupt when the (de)phosphorylation reactions of Raf-1 and MST2 operate in the saturated regime, i.e. when the concentrations of Raf-1 and MST2 exceed the Michealis-Menten constants of the respective reactions. Quantifying the concentrations of Raf-1, MST2 and MEK in MCF7 and Hela cells revealed that switching conditions are met in these cells. Under these conditions, switches are sharper for the strong binding between MST2-Raf-1, and a less strong LATS1 mediated feedback loop. Interestingly, the LATS phosphorylation site is conserved in other Raf isoforms, and they feature differential affinities to MST2. A-Raf binds the strongest, followed by Raf-1, whereas B-Raf has barely detectable binding activity.25 Thus, in cells where MST2 is regulated mainly by A-Raf steep switches are predicted. Interestingly, mutant B-RafV600E was recently found to strongly bind MST proteins.36 Therefore, mutant B-Raf expressing cells seem poised to steep switches. The different Raf isoforms are expected to compete with each other for MST binding, and how this competition and their differential affinities shape the switches will be an interesting future research topic.
Our systems analysis also revealed and explained the important roles of Akt in coordinating the balance between mitogenic and apoptotic signaling by the ERK and MST2 pathways. Our results showed that the Akt activation status is crucial in controlling the direction of the MST2-mediated apoptotic switch. High Akt activity switches MST2 signaling OFF, while low Akt activity switches it ON. In cancer Akt is often hyper-activated by mutations of its upstream activator PI3K or loss of PTEN, which dephosphorylates PI3.37,38 A better elucidation of how Akt activity is regulated in these contexts will go a long way in understanding how the apoptotic and proliferative outcomes are integrated in tumor development and progression. It has been suggested that MST can inhibit Akt,39 which would create a feedback loop where Akt and MST suppress each other. This extra layer of regulation could provide additional control to the regulatory machinery of the network, which will require and extended model to be addressed.
Taken together, our mathematical model allowed us to identify and experimentally test the critical elements that govern linear versus switching behavior in the MST2-Raf-1 network, revealing 3 interconnected layers of regulation (Fig. 2A). The first layer is related to the relative concentrations and affinities of the proteins involved in the competing interactions, and the regulation of Raf-1 and MST2 by phosphorylation that changes the binding affinities. The second layer encompasses Akt, which can either promote or suppress MST2 activation and apoptosis depending on its activation profile. The third layer of regulation is the LATS1 mediated feedback phosphorylation of Raf-1 on Ser259, which negatively regulates the activities of both pathways (Fig. 2A). Dephosphorylation of Ser259 is an essential part of Raf-1 activation process,29 which results in a concomitant activation of the mitogenic ERK and pro-apoptotic MST2 pathways. Linking cell proliferation with the risk of apoptosis seems counterintuitive, but is sensible for multicellular organisms where the loss of a cell causes much less damage than its unlicensed proliferation.
Occurrence of switches in common cell lines, tissues and pathological contexts
To examine whether switching is a common feature in other cellular systems and at the tissue level, we gathered protein expression data from quantitative proteomic studies publicly available for a panel of 10 common cell lines and various mouse tissues.40,41 Interestingly, model simulations predicted that switches are to be expected in a multitude of cell lines24 and tissues (Fig. 3A). Although the levels of steepness, characterized by the Hill coefficient of the dose-response curve, for each cell line and tissue varied, overall they characterize the appearance of clear switches in these systems (Fig. 3B and C). This suggests switching may be a robust emergent property across multiple expression backgrounds, which we think is due primarily to the design of the network topology, i.e., how the nodes are wired within the network.
Figure 3.
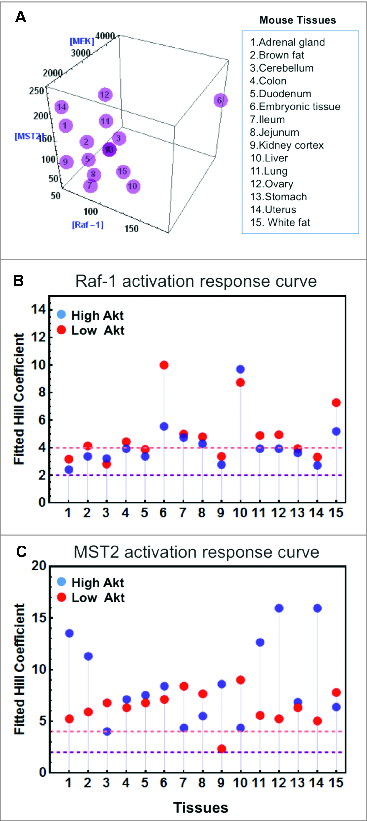
Existence of switches in mouse tissues. (A) Three-dimensional plot showing the concentrations of Raf-1, MST2 and MEK1/2 in 15 mouse tissues.41 (B and C) Hill coefficients of Raf-1 and MST2 activation in response to increasing Ras activation (RasGTP) were derived, as in.24 As Akt activity can influence the switching behavior, Hill coefficients were calculated under low (red dots) and high (blue dots) Akt activities. Hill coefficients of >2 indicate switch-like behavior, and Hill coefficients >4 strong switches. Model description and parameter values are same as in.24
The MST pathway is often deregulated in human cancer. Intriguingly, deregulation comes about mainly by epigenetic changes rather than somatic mutations of the pathway components. This is particularly notable in breast cancer where RASSF1A is epigenetically silenced in about 90% of the cases due to promoter hyper-methylation42. Similarly, although MST1/2 and LATS1/2 are rarely affected by somatic mutations,43 their promoters are often hyper-methylated in many cancers44,45 This prompted us to ask how the switches may be perturbed under the pathological context where RASSF1A is silenced. This scenario can be simulated by assuming a low level of RASSF1A expression in the model. Interestingly, the model predicts that under high Akt activity, down-regulated RASSF1A led to a more abrupt switching OFF of MST2 activity compared to a graded response at high RASSF1A level (Fig. 4A). As a result, to achieve the same level of MST2 inhibition cells would require much lower active Ras signal when RASSF1A is downregulated. This result is in keeping with the observed alterations in cancer cells, which include the activation of Akt dependent survival signals and silencing of RASSF1A expression.
Figure 4.
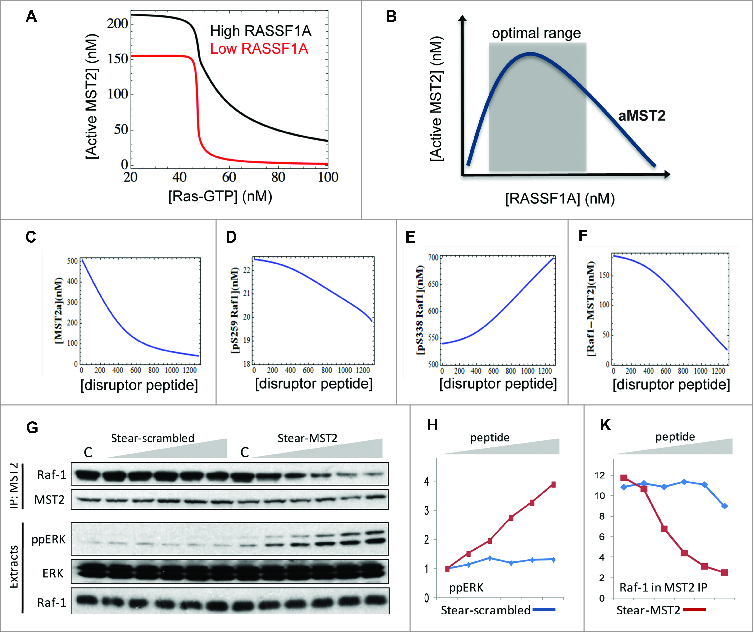
Model predictions of 1A perturbation and effect of the disruptor peptide. (A) Simulations of MST2 activation in response to increasing Ras-GTP under high and down-regulated RASSF1A expression. (B) Illustration of the biphasic property of MST2 activation dependence on RASS1F level. (C–F) Simulations of various model species in response to increasing level of the MST2-Raf-1 binding disruptor peptide. Parameter values for the kinetic rates of binding between the peptide and inactive MST2 are kf = 0.1 nM−1s−1, kr = 0.001 s−1, kact = 0.02 nM−1 using the same published model in.24 The other parameter values are given in Table M3 of ref.24 (G) HeLa cells were incubated with increasing concentrations (0–10 μM) of stearylated scrambled (Stear-scrambled) or MST2 (Stear-MST2) disruptor peptides for 1 hour. Raf-1 and Mst2 immunoprecipitates and 10 μg of cellular extracts were analyzed by Western blotting using the indicated antibodies. (H and K) Blots were quantitated by laser densitometry and analyzed using the Image J software.
Interestingly, our combined analysis uncovered an unexpected dual role for RASSF1A in regulating MST2 activation.24 At low expression, RASSF1A stimulates MST2 activity, whereas it inhibits MST2 activity at high expression (Fig. 4B). This is consistent with a proposed view that RASSF1A functions as a scaffold protein, which are hallmarked by such biphasic activation characteristics. This observation signals that caution is required when one is to assess the role of RASSF1A without having quantitative knowledge of its concentration in cells, further highlighting the importance of a quantitative analysis.
A detailed mechanistic picture of how RASSF1A facilitates MST2 activation still remains elusive. Although MST2 homo-dimerization has been suggested as a key event leading to full activation of MST2, it is not clear exactly how RASSF1A aids the formation of this dimer, and whether it is possible for RASSF1A to form stable trimers with 2 MST2 molecules through the SARAH domain. Computational methods like molecular modeling and atomistic molecular dynamics may shed new light on these questions. Such insights will help to construct more accurate and predictive mechanistic models of RASSF1A mediated MST2 activation, the understanding of which is essential for future therapeutic strategies targeting RASSF1A.
Disrupting the MST2-Raf-1 interaction complex as a promising therapeutic strategy
Shutting down apoptotic signals by means of genetic or epigenetic changes is a strategy commonly employed by tumor cells to initiate and maintain unlicensed proliferation. This is notable in many types of cancer where the silencing of the tumor suppressor RASSF1A27,42 impedes MST2-mediated apoptosis. Approaches to reactivate suppressed apoptotic pathways are conceptually attractive, but challenging therapeutic avenue to halt tumor progression and possibly re-sensitize tumor cells to existing drugs. The concept that dynamically changing PPIs can coordinate signaling and “compute” specific biological outcomes suggests that targeting PPIs could be an appealing way to manipulate network behavior for therapeutic purposes. MST2 is naturally locked in an inhibitory complex by Raf-1 from where it is released and activated by RASSF1A. Using pharmacological agents to break this lock to release MST2 could potentially replace the function of RASSF1A and lead to enhanced MST2 activation and apoptosis in tumor cells. We combined model predictions and experimentation to examine this intriguing idea.
Using peptide arrays we mapped the Raf-1 binding site to a small interface in the SARAH domain of MST2, and designed a cell permeable disruptor peptide based on this sequence.24 Using modeling we asked whether the peptide could induce a dose-dependent disruption of the MST2-Raf-1 complex, and if so what the dose-response curve would look like? Since the synthetic peptide sequence overlaps with the dimerization domain of MST2, we assume in the model that the peptide disrupts the MST2-Raf-1 interaction by binding directly to the inactive MST2 monomer. The model predicted that treatment of cells with increasing disruptor peptide concentrations efficiently dissociates the MST2-Raf-1 binding in a dose-dependent fashion (Fig. 4C). The peptide also strongly impedes MST2 activation (Fig. 4D), as the occupation of the MST2 dimerization domain by the disruptor peptide prevents MST2 dimerization and activation. Moreover, model simulations suggested a linear increase of active pS338 Raf-1 and ERK, but a decrease of inactive pS259 Raf-1, in response to increasing peptide level (Fig. 4E and F). Because MST2 binding protects pS259 Raf-1 from dephosphorylation, disruption of the MST2-Raf-1 complex ceases to protect this site against phosphatases resulting in reduced pS259 Raf-1 and increased Raf-1 activation, as predicted by the model. Importantly, follow-up experiments showed excellent congruency between data and model predictions (Fig. 4G–K), confirming the ability of the interfering peptide to efficiently activate the ERK pathway by disrupting the MST2-Raf-1 complex. Targeted disruption of protein-protein interaction is thus a viable way to manipulate network behavior.
Although efficient in disrupting the MST2-Raf-1 binding, the designed peptide did not activate MST2 due to also preventing MST2 dimerization. However, one can design an alternative peptide that binds to the MST2 binding site in Raf-1 instead. We also mapped the Raf-1 domains involved in MST2 binding, which showed MST2 binds Raf-1 through 2 distinct sites that overlap the RBD and partially the MEK-binding domain.24 This suggests that a peptide containing either the RBD or MEK-binding sequence in Raf-1 (or both with a linker) may be able to interfere with both MST2 binding and Ras (or MEK) binding to Raf-1. Such a peptide would kill 2 birds with one stone by releasing MST2 to trigger apoptosis while blocking Raf-1 activation to inhibit proliferation, a dual property much desired in any anti-cancer agent.
The role of phosphatases in the MST2-Raf-1 network
The regulation of the MST2-Raf-1 network is further complicated by the involvement of protein phosphatases. Early studies reported activation of MST1 and MST2 in response to cellular stress or the use of phosphatase inhibitors such as okadaic acids,4 which led to okadaic acids often used as an experimental reagent to activate the MST/Hippo pathway.46 A major phosphatase that regulates many nodes of the MST2-Raf-1 signaling network is the protein phosphatase 2A (PP2A).47,48
We previously showed that in addition to interfering with MST2 dimerization, Raf-1 suppresses the activation of MST2 by recruiting a phosphatase, most likely PP2A, to dephosphorylate MST2 on its activation sites.3 Previous experimental work suggested that PP2A also dephosphorylates Raf-1 on its inhibitory Ser259,49,50 and that Ser259 phosphorylation is maintained by MST2 by stabilizing the expression of the PP2A catalytic subunit.51 The negative function of PP2A toward MST2 is additionally mediated via the prevention of RASSF1A-induced MST2 auto-phosphorylation and activation.52 Besides the MST kinases, PP2A also targets other members of the MST2 pathway as substrates, e.g., the co-transcriptional factor TAZ and YAP, which are direct targets of the LATS kinases downstream of MST2. Upon activation of the MST2 pathway, TAZ and YAP are phosphorylated by LATS1 which triggers their relocation from the nucleus to the cytoplasm. PP2A and the protein phosphatase 1 (PP1) have been reported to associate with and dephosphorylate TAZ and YAP.36,53,54 Moreover, PP2A may directly affect LATS kinases and Mob1, a scaffold protein that binds to and stimulates LATSs, as their phosphorylation status is enhanced following okadaic acid treatment.52,55 Finally, PP2A has been long known to dephosphorylate Akt and inhibits its activation.56
The extensive and complex involvement of PP2A raises an important, yet non-trivial question as to how it integrates the regulation of multiple substrates to coordinate dynamics at the network level? Answering this question may shed new light on whether targeting PP2A could be a viable therapeutic option.57 Figure 5A overlays the existing network regulations with the various positive and negative regulations induced by PP2A (highlighted in blue). Interestingly, this integrated wiring contains numerous intertwined regulatory motifs including double negative feedbacks (DNF), coherent and incoherent feed-forward loops (CFF, IFF) that involve different network nodes (Fig. 5B–E). The co-existence of these motifs may confer extremely rich dynamics to network behavior and important coordinating roles to PP2A. Indeed in a context-dependent manner PP2A could regulate opposing, life and death decision, through Akt.58 Due to the high nonlinearity generated by not one but multiple control motifs, model-based analysis will be instrumental in guiding experiments to dissect the functional role of PP2A in the MST2-Raf-1 signaling machinery.
Figure 5.
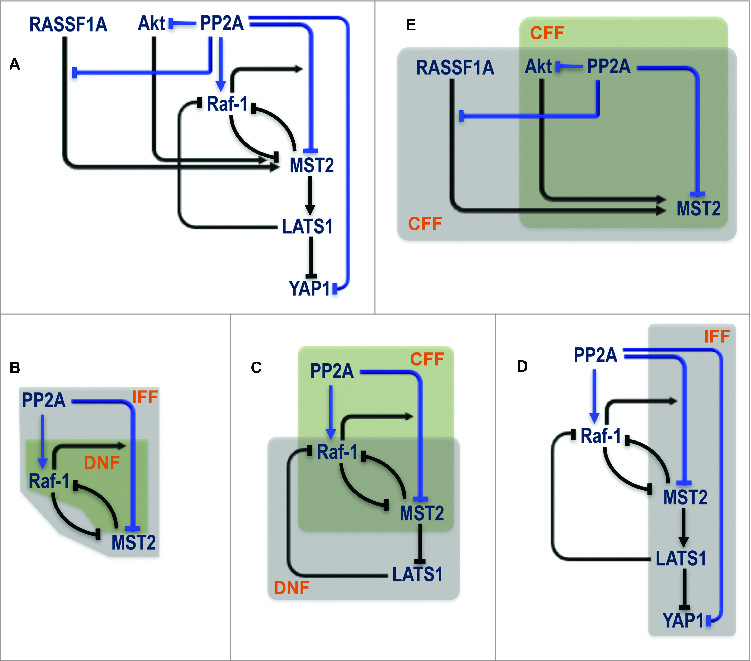
PP2A centered regulation in the MST2-Raf-1 network. (A) Existing network regulation overlaid with novel regulation induced by PP2A. (B–E) Various overlapping control motifs identified for different part of the PP2A related circuitry. IFF = Incoherent Feed Forward; DNF = Double Negative Feedback; CFF = Coherent Feed Forward.
In addition, a novel family of Ser/Thr phosphatases PHLPP is receiving increased attention. PHLPP has been shown to exert a dual function, terminating cell survival through dephosphorylation of pro-survival kinases such as Akt, and promoting apoptosis via MST1 dephosphorylation and enhanced activation.59-61 Moreover, known targets of PHLPP include ERK60 and PHLPP1β interacts directly with Ras to suppress Ras-Raf-MEK-ERK signaling.62 These connections suggest that PHLPP, Akt, MST kinases and the MAPK nodes could constitute an additional layer of control to impose swift balance of proliferation and apoptosis which may be cell type and context dependent. The question how such control plays out within the MST2-Raf-1 crosstalk is still an open issue and is an attractive avenue for future research.
Regulation by E3 ligases and Ubiquitin Proteasome System (UPS) in the MST2-Raf-1 network
Along with phosphatases, which have received much attention as prominent negative regulators of the MST2 pathway,47,48 more recent studies have uncovered the UPS as a prominent mechanism for negative regulation. The UPS down-regulates the pathway signal through various E3 ligases that target specific pathway nodes. The HECT type E3 ubiquitin ligase ITCH is a notable example. ITCH ubiquitinates LATS1 and the downstream tumor suppressor protein p73, which is a pro-apoptotic effector of the MST2 pathway.22 ITCH poly-ubiquitinates these proteins and targets them for degradation,63,64 thereby promoting tumorigenesis.65 Intriguingly, because ITCH contains a consensus LATS phosphorylation motif, it would be interesting to test if ITCH is a genuine LATS1 substrate,63 and whether this results in a mutual cross-regulation between LATS1 and ITCH. Another ubiquitin ligase of the NEDD4-like family, WWP1 E3 was also shown to mediate LATS1 degradation, promoting cell proliferation in breast cancer cells.66 In addition, the RING ubiquitin ligase praja2 can down-regulate MOB1 and thus attenuate MST2 activity.67
Another interesting mutual regulation exists between Akt and the Skp2-SCF E3 ubiquitin ligase complex. On one hand, Akt phosphorylates Skp2 which stimulates the activity of the Skp2-SCF complex.68 On the other hand, the non-proteolytic K63-linked ubiquitination of Akt, which is required for its membrane recruitment and activation (rather than degradation) upon growth factor stimulation, is partly due to Skp2-SCF.69 This creates a 2-way positive regulation between Akt and Skp2-SCF, which could potentially generate a threshold-gated control for Akt-dependent suppression of MST2 pathway. In the past years, we have generated a number of mathematical models to analyze the dynamic properties of specific ubiquitination related systems.70-73 The application of these generic models for the E3 ligases and their substrates specifically involved in the MST2-Raf-1 network with the existing model of MST2-Raf-1 dynamics24 will certainly help illuminate the roles of the E3 ligases and the UPS in controlling the decision making process in the MST2-Raf-1 network.
Conclusions and Outlook
The accumulated work on the MST2-Raf-1 signaling crosstalk paradigm has unveiled a conceptually novel and thought-provoking notion, i.e. that signaling networks use dynamically changing PPIs as devices that compute biochemical and biological decisions. A key dynamic component enabling switch-like decision making is the combination of competing PPIs and phosphorylations that change their affinities. Through the evolution of diverse interaction domains proteins are delicately directed to form higher order structures that operate as molecular machines. While production machines, such as the ribosome, are stable assemblies, it now transpires that PPIs are also used in the dynamic setting of signal transduction networks for computing cell fate decisions. The dynamic element is performing the computing tasks and usually results from posttranslational modifications that regulate the binding affinities. It is this beautiful arrangement that underlines the various competing protein formations, dynamically modulated by phosphorylation, which enable the Boolean computing ability of the MST2-Raf-1 network. Since protein bindings and phosphorylation are widespread in cellular processes, it is expected that many similar decision making apparatuses are waiting to be discovered.
A systematic analysis combining mathematical modeling and experimentation has been instrumental in gaining a holistic understanding of the MST2-Raf-1 signaling machinery. Given the complex wiring architecture, model-based analyses were required to tease out the emergent network properties and identifying the governing conditions. As novel regulators, such as the phosphatases and E3 ligases, come into play and further complicates the network regulatory landscape, quantitative systems analysis will again be needed for deciphering the properties and principles of the mechanisms that integrate these various regulatory signals. Mathematical modeling will also be valuable in assessing the viability of potential therapeutic strategies by predicting the responses to drugs targeting the network. We, therefore, expect to see modeling and model-based analysis to continue being at the forefront of not just MST2-Raf-1 research, but biomedical research in general.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The research leading to these results has received funding from the Science Foundation Ireland under grant No. 06/CE/B1129 (LKN, DG, BNK,WK); the European Union Seventh Framework Program (FP7/2007- 2013) ASSET project under grant agreement number FP7- HEALTH-2010–259348 (BNK, WK); PRIMES project under grant agreement No. FP7-HEALTH-2011–278568 (LNK, BNK, WK) and University College Dublin's Seed Funding program (LKN).
References
- 1. Guo M, Hay BA. Cell proliferation and apoptosis. Curr Opin Cell Biol 1999; 11:745-52; PMID:10600713; http://dx.doi.org/ 10.1016/S0955-0674(99)00046-0 [DOI] [PubMed] [Google Scholar]
- 2. Hipfner DR, Cohen SM. Connecting proliferation and apoptosis in development and disease. Nat Rev Mol Cell Biol 2004; 5:805-15; PMID:15459661; http://dx.doi.org/ 10.1038/nrm1491 [DOI] [PubMed] [Google Scholar]
- 3. O'Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 2004; 306:2267-70; PMID:15618521; http://dx.doi.org/ 10.1126/science.1103233 [DOI] [PubMed] [Google Scholar]
- 4. Avruch J, Zhou D, Fitamant J, Bardeesy N, Mou F, Barrufet LR. Protein kinases of the Hippo pathway: regulation and substrates. Semin Cell Dev Biol 2012; 23:770-84; PMID:22898666; http://dx.doi.org/ 10.1016/j.semcdb.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pan D. The hippo signaling pathway in development and cancer. Dev Cell 2010; 19:491-505; PMID:20951342; http://dx.doi.org/ 10.1016/j.devcel.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Neill E, Kolch W. Taming the Hippo: Raf-1 controls apoptosis by suppressing MST2Hippo. Cell Cycle 2005; 4:365-7; PMID:15701972; http://dx.doi.org/ 10.4161/cc.4.3.1531 [DOI] [PubMed] [Google Scholar]
- 7. Bossuyt W, Chen CL, Chen Q, Sudol M, McNeill H, Pan D, Kopp A, Halder G. An evolutionary shift in the regulation of the Hippo pathway between mice and flies. Oncogene 2014; 33:1218-28; PMID:23563179; http://dx.doi.org/ 10.1038/onc.2013.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAPTAZ in mechanotransduction. Nat 2011; 474:179-83; PMID:21654799; http://dx.doi.org/ 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- 9. Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009; 16:425-38; PMID:19878874; http://dx.doi.org/ 10.1016/j.ccr.2009.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014; 25:831-45; PMID:24882515; http://dx.doi.org/ 10.1016/j.ccr.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X, Yu K, Hao Y, Li DM, Stewart R, Insogna KL, Xu T. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol 2004; 6:609-17; PMID:15220930; http://dx.doi.org/ 10.1038/ncb1140 [DOI] [PubMed] [Google Scholar]
- 12. Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006; 125:987-1001; PMID:16751106; http://dx.doi.org/ 10.1016/j.cell.2006.03.046 [DOI] [PubMed] [Google Scholar]
- 13. You B, Yan G, Zhang Z, Yan L, Li J, Ge Q, Jin JP, Sun J. Phosphorylation of cardiac troponin I by mammalian sterile 20-like kinase 1. The Biochem J 2009; 418:93-101; PMID:18986304; http://dx.doi.org/ 10.1042/BJ20081340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wen W, Zhu F, Zhang J, Keum YS, Zykova T, Yao K, Peng C, Zheng D, Cho YY, Ma WY, et al. MST1 promotes apoptosis through phosphorylation of histone H2AX. J Biol Chem 2010; 285:39108-16; PMID:20921231; http://dx.doi.org/ 10.1074/jbc.M110.151753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hergovich A. Regulation and functions of mammalian LATSNDR kinases: looking beyond canonical Hippo signalling. Cell Biosci 2013; 3:32; PMID:23985307; http://dx.doi.org/ 10.1186/2045-3701-3-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 2003; 113:507-17; PMID:12757711; http://dx.doi.org/ 10.1016/S0092-8674(03)00355-6 [DOI] [PubMed] [Google Scholar]
- 17. Barry ER, Camargo FD. The Hippo superhighway: signaling crossroads converging on the HippoYap pathway in stem cells and development. Curr Opin Cell Biol 2013; 25:247-53; PMID:23312716; http://dx.doi.org/ 10.1016/j.ceb.2012.12.006 [DOI] [PubMed] [Google Scholar]
- 18. Varelas X, Wrana JL. Coordinating developmental signaling: novel roles for the Hippo pathway. Trends Cell Biol 2012; 22:88-96; PMID:22153608; http://dx.doi.org/ 10.1016/j.tcb.2011.10.002 [DOI] [PubMed] [Google Scholar]
- 19. Romano D, Matallanas D, Frederick DT, Flaherty KT, Kolch W. One Hippo and many masters: differential regulation of the Hippo pathway in cancer. Biochem Soc Trans 2014; 42:816-21; PMID:25109963; http://dx.doi.org/ 10.1042/BST20140030 [DOI] [PubMed] [Google Scholar]
- 20. Matallanas D, Romano D, Al-Mulla F, O'Neill E, Al-Ali W, Crespo P, Doyle B, Nixon C, Sansom O, Drosten M, et al. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol Cell 2011; 44:893-906; PMID:22195963; http://dx.doi.org/ 10.1016/j.molcel.2011.10.016 [DOI] [PubMed] [Google Scholar]
- 21. Matallanas D, Romano D, Hamilton G, Kolch W, O'Neill E. A Hippo in the ointment: MST signalling beyond the fly. Cell Cycle 2008; 7:879-84; PMID:18414046; http://dx.doi.org/ 10.4161/cc.7.7.5630 [DOI] [PubMed] [Google Scholar]
- 22. Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, Baccarini M, Vass JK, Kolch W, O'Neill E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell 2007; 27:962-75; PMID:17889669; http://dx.doi.org/ 10.1016/j.molcel.2007.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Romano D, Matallanas D, Weitsman G, Preisinger C, Ng T, Kolch W. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res 2010; 70:1195-203; PMID:20086174; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, Kolch W. Protein interaction switches coordinate Raf-1 and MST2Hippo signalling. Nat Cell Biol 2014; 16:673-84; PMID:24929361; http://dx.doi.org/ 10.1038/ncb2986 [DOI] [PubMed] [Google Scholar]
- 25. Rauch J, O'Neill E, Mack B, Matthias C, Munz M, Kolch W, Gires O. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res 2010; 70:1679-88; PMID:20145135; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hesson LB, Cooper WN, Latif F. The role of RASSF1A methylation in cancer. Dis Markers 2007; 23:73-87; PMID:17325427; http://dx.doi.org/ 10.1155/2007/291538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Avruch J, Xavier R, Bardeesy N, Zhang XF, Praskova M, Zhou D, Xia F. Rassf family of tumor suppressor polypeptides. J Biol Chem 2009; 284:11001-5; PMID:19091744; http://dx.doi.org/ 10.1074/jbc.R800073200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, Kolch W. Raf family kinases: old dogs have learned new tricks. Genes Cancer 2011; 2:232-60; PMID:21779496; http://dx.doi.org/ 10.1177/1947601911407323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. Regulation of Raf-1 activation and signalling by dephosphorylation. EMBO J 2002; 21:64-71; PMID:11782426; http://dx.doi.org/ 10.1093/emboj/21.1.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dumaz N, Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem 2003; 278:29819-23; PMID:12801936; http://dx.doi.org/ 10.1074/jbc.C300182200 [DOI] [PubMed] [Google Scholar]
- 31. Dhillon AS, Pollock C, Steen H, Shaw PE, Mischak H, Kolch W. Cyclic AMP-dependent kinase regulates Raf-1 kinase mainly by phosphorylation of serine 259. Mol Cell Biol 2002; 22:3237-46; PMID:11971957; http://dx.doi.org/ 10.1128/MCB.22.10.3237-3246.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kobayashi T, Aoki Y, Niihori T, Cave H, Verloes A, Okamoto N, Kawame H, Fujiwara I, Takada F, Ohata T, et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat 2010; 31:284-94; PMID:20052757; http://dx.doi.org/ 10.1002/humu.21187 [DOI] [PubMed] [Google Scholar]
- 33. Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, Momma K, Katayama H, Nakagawa M, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet 2007; 39:1013-7; PMID:17603482; http://dx.doi.org/ 10.1038/ng2078 [DOI] [PubMed] [Google Scholar]
- 34. Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet 2007; 39:1007-12; PMID:17603483; http://dx.doi.org/ 10.1038/ng2073 [DOI] [PubMed] [Google Scholar]
- 35. Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol 2006; 7:165-76; PMID:16482094; http://dx.doi.org/ 10.1038/nrm1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee SJ, Lee MH, Kim DW, Lee S, Huang S, Ryu MJ, Kim YK, Kim SJ, Hwang JH, Oh S, et al. Cross-regulation between oncogenic BRAF(V600E) kinase and the MST1 pathway in papillary thyroid carcinoma. PloS One 2011; 6:e16180; PMID:21249150; http://dx.doi.org/ 10.1371/journal.pone.0016180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang BH, Liu LZ. PI3KPTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res 2009; 102:19-65; PMID:19595306; http://dx.doi.org/ 10.1016/S0065-230X(09)02002-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27:5497-510; PMID:18794884; http://dx.doi.org/ 10.1038/onc.2008.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cinar B, Fang PK, Lutchman M, Di Vizio D, Adam RM, Pavlova N, Rubin MA, Yelick PC, Freeman MR. The pro-apoptotic kinase Mst1 and its caspase cleavage products are direct inhibitors of Akt1. EMBO J 2007; 26:4523-34; PMID:17932490; http://dx.doi.org/ 10.1038/sj.emboj.7601872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics 2012; 11:M111 014050; PMID:22278370; http://dx.doi.org/ 10.1074/mcp.M111.014050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geiger T, Velic A, Macek B, Lundberg E, Kampf C, Nagaraj N, Uhlen M, Cox J, Mann M. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol Cell Proteomics 2013; 12:1709-22; http://dx.doi.org/ 10.1074/mcp.M112.024919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci 2007; 120:3163-72; PMID:17878233; http://dx.doi.org/ 10.1242/jcs.010389 [DOI] [PubMed] [Google Scholar]
- 43. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer 2013; 13:246-57; PMID:23467301; http://dx.doi.org/ 10.1038/nrc3458 [DOI] [PubMed] [Google Scholar]
- 44. Seidel C, Schagdarsurengin U, Blumke K, Wurl P, Pfeifer GP, Hauptmann S, Taubert H, Dammann R. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol Carcinog 2007; 46:865-71; PMID:17538946; http://dx.doi.org/ 10.1002/mc.20317 [DOI] [PubMed] [Google Scholar]
- 45. Takahashi Y, Miyoshi Y, Takahata C, Irahara N, Taguchi T, Tamaki Y, Noguchi S. Down-regulation of LATS1 and LATS2 mRNA expression by promoter hypermethylation and its association with biologically aggressive phenotype in human breast cancers. Clin Cancer Res 2005; 11:1380-5; PMID:15746036; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-1773 [DOI] [PubMed] [Google Scholar]
- 46. Hata Y, Timalsina S, Maimaiti S. Okadaic Acid: a tool to study the hippo pathway. Marine Drugs 2013; 11:896-902; PMID:23493077; http://dx.doi.org/ 10.3390/md11030896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ribeiro PS, Josue F, Wepf A, Wehr MC, Rinner O, Kelly G, Tapon N, Gstaiger M. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol Cell 2010; 39:521-34; PMID:20797625; http://dx.doi.org/ 10.1016/j.molcel.2010.08.002 [DOI] [PubMed] [Google Scholar]
- 48. Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal 2013; 6:rs15; PMID:24255178; http://dx.doi.org/ 10.1126/scisignal.2004712 [DOI] [PubMed] [Google Scholar]
- 49. Abraham D, Podar K, Pacher M, Kubicek M, Welzel N, Hemmings BA, Dilworth SM, Mischak H, Kolch W, Baccarini M. Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J Biol Chem 2000; 275:22300-4; PMID:10801873; http://dx.doi.org/ 10.1074/jbc.M003259200 [DOI] [PubMed] [Google Scholar]
- 50. Jaumot M, Hancock JF. Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14-3-3 interactions. Oncogene 2001; 20:3949-58; PMID:11494123; http://dx.doi.org/ 10.1038/sj.onc.1204526 [DOI] [PubMed] [Google Scholar]
- 51. Kilili GK, Kyriakis JM. Mammalian Ste20-like kinase (Mst2) indirectly supports Raf-1ERK pathway activity via maintenance of protein phosphatase-2A catalytic subunit levels and consequent suppression of inhibitory Raf-1 phosphorylation. J Biol Bhem 2010; 285:15076-87; PMID:20212043; http://dx.doi.org/ 10.1074/jbc.M109.078915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guo C, Zhang X, Pfeifer GP. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J Biol Chem 2011; 286:6253-61; PMID:21199877; http://dx.doi.org/ 10.1074/jbc.M110.178210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, et al. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 2011; 144:782-95; PMID:21376238; http://dx.doi.org/ 10.1016/j.cell.2011.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu CY, Lv X, Li T, Xu Y, Zhou X, Zhao S, Xiong Y, Lei QY, Guan KL. PP1 cooperates with ASPP2 to dephosphorylate and activate TAZ. J Biol Chem 2011; 286:5558-66; PMID:21189257; http://dx.doi.org/ 10.1074/jbc.M110.194019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moreno CS, Lane WS, Pallas DC. A mammalian homolog of yeast MOB1 is both a member and a putative substrate of striatin family-protein phosphatase 2A complexes. J Biol Chem 2001; 276:24253-60; PMID:11319234; http://dx.doi.org/ 10.1074/jbc.M102398200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci 1999; 24:186-91; PMID:10322434; http://dx.doi.org/ 10.1016/S0968-0004(99)01375-4 [DOI] [PubMed] [Google Scholar]
- 57. Nguyen LK, Matallanas D, Croucher DR, von Kriegsheim A, Kholodenko BN. Signalling by protein phosphatases and drug development: a systems-centred view. FEBS J 2013; 280:751-65; PMID:22340367; http://dx.doi.org/ 10.1111/j.1742-4658.2012.08522.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Andrabi S, Gjoerup OV, Kean JA, Roberts TM, Schaffhausen B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc Natl Acad Sci U S A 2007; 104:19011-6; PMID:18006659; http://dx.doi.org/ 10.1073/pnas.0706696104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. O'Neill AK, Niederst MJ, Newton AC. Suppression of survival signalling pathways by the phosphatase PHLPP. FEBS J 2013; 280:572-83; PMID:22340730; http://dx.doi.org/ 10.1111/j.1742-4658.2012.08537.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W, Stein J, Stein GS, Iglehart JD, Shi Q, et al. Mst1 is an interacting protein that mediates PHLPPs' induced apoptosis. Mol Cell 2010; 38:512-23; PMID:20513427; http://dx.doi.org/ 10.1016/j.molcel.2010.03.017 [DOI] [PubMed] [Google Scholar]
- 61. Warfel NA, Newton AC. Pleckstrin homology domain leucine-rich repeat protein phosphatase (PHLPP): a new player in cell signaling. J Biol Chem 2012; 287:3610-6; PMID:22144674; http://dx.doi.org/ 10.1074/jbc.R111.318675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shimizu K, Okada M, Nagai K, Fukada Y. Suprachiasmatic nucleus circadian oscillatory protein, a novel binding partner of K-Ras in the membrane rafts, negatively regulates MAPK pathway. J Biol Chem 2003; 278:14920-5; PMID:12594205; http://dx.doi.org/ 10.1074/jbc.M213214200 [DOI] [PubMed] [Google Scholar]
- 63. Ho KC, Zhou Z, She YM, Chun A, Cyr TD, Yang X. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability [corrected]. Proc Natl Acad Sci U S A 2011; 108:4870-5; http://dx.doi.org/ 10.1073/pnas.1101273108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ 2007; 14:743-51; PMID:17110958; http://dx.doi.org/ 10.1038/sj.cdd.4402063 [DOI] [PubMed] [Google Scholar]
- 65. Salah Z, Melino G, Aqeilan RI. Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res 2011; 71:2010-20; PMID:21212414; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3516 [DOI] [PubMed] [Google Scholar]
- 66. Yeung B, Ho KC, Yang X. WWP1 E3 ligase targets LATS1 for ubiquitin-mediated degradation in breast cancer cells. PloS One 2013; 8:e61027; PMID:23573293; http://dx.doi.org/ 10.1371/journal.pone.0061027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lignitto L, Arcella A, Sepe M, Rinaldi L, Delle Donne R, Gallo A, Stefan E, Bachmann VA, Oliva MA, Tiziana Storlazzi C, et al. Proteolysis of MOB1 by the ubiquitin ligase praja2 attenuates Hippo signalling and supports glioblastoma growth. Nat Commun 2013; 4:1822; PMID:23652010; http://dx.doi.org/ 10.1038/ncomms2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ecker K, Hengst L. Skp2: caught in the Akt. Nat Cell Biol 2009; 11:377-9; PMID:19337320; http://dx.doi.org/ 10.1038/ncb0409-377 [DOI] [PubMed] [Google Scholar]
- 69. Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng Z, Huang HY, Tsai KK, Flores LG, Shao Y, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell 2012; 149:1098-111; PMID:22632973; http://dx.doi.org/ 10.1016/j.cell.2012.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nguyen LK, Kolch W, Kholodenko BN. When ubiquitination meets phosphorylation: a systems biology perspective of EGFRMAPK signalling. Cell Commun Signal 2013; 11:52; PMID:23902637; http://dx.doi.org/ 10.1186/1478-811X-11-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nguyen LK, Munoz-Garcia J, Maccario H, Ciechanover A, Kolch W, Kholodenko BN. Switches, excitable responses and oscillations in the Ring1BBmi1 ubiquitination system. PLoS Comput Biol 2011; 7:e1002317; PMID:22194680; http://dx.doi.org/ 10.1371/journal.pcbi.1002317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nguyen LK, Dobrzynski M, Fey D, Kholodenko BN. Polyubiquitin chain assembly and organization determine the dynamics of protein activation and degradation. Frontiers Physiol 2014; 5:4; PMID:24478717; http://dx.doi.org/ 10.3389/fphys.2014.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nguyen LK, Zhao Q, Varusai T, Kholodenko BN. Ubiquitin chain specific auto-ubiquitination triggers sustained oscillation, bistable switches and excitable firing. IET Systems Biol 2014; http://dx.doi.org/10.1049/iet-syb.2014.0024 [DOI] [PMC free article] [PubMed] [Google Scholar]