

Abstract
The BRCA1 tumor suppressor plays an important role in homologous recombination (HR)-mediated DNA double-strand-break (DSB) repair. BRCA1 is phosphorylated by Chk2 kinase upon γ-irradiation, but the role of Chk2 phosphorylation is not understood. Here, we report that abrogation of Chk2 phosphorylation on BRCA1 delays end resection and the dispersion of BRCA1 from DSBs but does not affect the assembly of Mre11/Rad50/NBS1 (MRN) and CtIP at DSBs. Moreover, we show that BRCA1 is ubiquitinated by SCFSkp2 and that abrogation of Chk2 phosphorylation impairs its ubiquitination. Our study suggests that BRCA1 is more than a scaffold protein to assemble HR repair proteins at DSBs, but that Chk2 phosphorylation of BRCA1 also serves as a built-in clock for HR repair of DSBs. BRCA1 is known to inhibit Mre11 nuclease activity. SCFSkp2 activity appears at late G1 and peaks at S/G2, and is known to ubiquitinate phosphodegron motifs. The removal of BRCA1 from DSBs by SCFSkp2-mediated degradation terminates BRCA1-mediated inhibition of Mre11 nuclease activity, allowing for end resection and restricting the initiation of HR to the S/G2 phases of the cell cycle.
Keywords: BRCA1, cell cycle, DNA double-strand break repair, end-resection, SCF
Abbreviations
- BRCA1
Breast Cancer Susceptibility Gene 1
- HR
homologous Recombination
- NHEJ
Non-Homologous End Joining
- DSB
Double-Strand Break
- SCF
Skp1-Cul1-F box protein complex
- MRN
Mre11-Rad50-Nbs1 complex
Introduction
Germline mutations of the BRCA1 tumor suppressor gene account for a significant number of hereditary breast cancer cases and 80% of families whose members develop both breast and ovarian cancers. A wealth of evidence has established central roles for BRCA1 in DNA double-strand-break (DSB) repair and cell cycle checkpoint control that are likely to underlie its tumor suppression function.1,2
Upon the induction of DSBs, BRCA1 is phosphorylated by multiple kinases, including Chk2, and forms nuclear foci at sites of damaged DNA.3-6 Current understanding of BRCA1 in DSB repair is that BRCA1 serves as a scaffold protein that recruits multiple repair proteins to the break sites.2 While this “scaffold protein” model conceptualizes a general role of BRCA1 in facilitating formation of the multiple protein complexes involved in DSB repair, the current paradigm regarding BRCA1 function in DNA repair lacks a more substantive and insightful understanding of the protein at the mechanistic level. What is the exact role of BRCA1, especially the role of its phosphorylation, in DSB repair?
DSB can be repaired by either homologous recombination (HR) or nonhomologous end-joining (NHEJ).7 HR is a high-fidelity repair pathway and therefore it is the preferred repair mechanism in order to maintain genome integrity. However, HR repair is believed to occur during late S and G2 phases of the cell cycle, when sister chromatids are available to serve as homologous templates.8-10 While NHEJ is generally more error-prone, it is available throughout the cell cycle and is especially important when HR is not available, due to cell cycle stages or a deficiency in the HR repair machinery. How do cells coordinate HR repair with S/G2 phases to maximize the chance of faithful DNA repair at a time when sister chromatids are available?
BRCA1 forms multiple protein complexes in response to DSBs.2 These complexes are involved in the repair process, per se, and cell cycle checkpoint activation (S and G2-M checkpoints),11 or both. One of these complexes, MRN (Mre11-Rad50-NBS1)-BRCA1-CtIP, has been shown to promote HR-mediated DNA repair by initiating DNA end resection, an early step in HR that represents a point of commitment to HR repair. Mre11 possesses intrinsic exo- and endo-nuclease activities, which likely facilitate initial processing of DNA ends for subsequent extensive end resection. The ortholog of CtIP in budding yeast, Sae2, also possesses a nuclease activity that is involved in end resection,12-14 and an intrinsic nuclease activity of its mammalian counterpart has recently been demonstrated.15,16 In addition, it has been shown that CtIP stimulates the nuclease activity of the MRN complex in mammalian cells.17,18 BRCA1 recruits CtIP to the complex after CtIP is phosphorylated.19-21 Loss of BRCA1 impairs the assembly of MRN-BRCA1-CtIP at DSB sites and, therefore, subsequent end resection.22,23 Paradoxically, BRCA1 has also been shown to inhibit Mre11 nuclease activity in vitro.24 This seemingly paradoxical role of BRCA1 in end resection has never been addressed.
In response to DSBs, BRCA1 is hyperphosphorylated by multiple DNA repair/checkpoint kinases, including ATM, ATR, and Chk2. In particular, Chk2 phosphorylates human BRCA1 at Ser988 (or Ser971 in mouse Brca1).5 Knock-in (KI) mice containing a homozygous Brca1 mutation at aa971 from serine to alanine (S971A) have increased tumor incidence when exposed to γ-irradiation (γIR).25 Furthermore, cells derived from the S971A KI mice display partially defective G2-M checkpoint in response to γIR.25,26 Human cells harboring the S988A mutation in BRCA1 have a normal S-phase checkpoint, but impaired HR repair of DSBs.26 These data suggest that Chk2 phosphorylation of BRCA1 plays an important role in DSB repair and the G2-M checkpoint. Abrogation of BRCA1 phosphorylation by Chk2 results in increased genome instability and tumorigenesis. However, the precise mechanism by which S988 phosphorylation promotes HR repair is not understood. Here, we report that abrogation of Chk2 phosphorylation at S971 in mouse Brca1 (S971A) delays end resection. The Brca1(S971A) mutation does not appear to affect the assembly of MRN and CtIP at DSBs, but specifically delays dispersion of Brca1 from DSBs. We show that BRCA1 is ubiquitinated by SCFSkp2 and abrogation of Chk2 phosphorylation at S988 of human BRCA1 impairs its ubiquitination. SCFSkp2 activity, which appears at late G1 and peaks at S/G2, is known to ubiquitinate phosphodegron motifs.27,28 Our study suggests that BRCA1 plays a dual role in HR. After the assembly of repair proteins at DSBs, BRCA1 is likely degraded by SCFSkp2 at S/G2 phases. The consequent removal of BRCA1 from DSBs terminates BRCA1-mediated inhibition of Mre11, allowing for end resection and ensuring the initiation of HR to S/G2 phases of the cell cycle.
Results
Abrogation of Chk2 Phosphorylation on Mouse Brca1 Confers PARP Inhibitor Hypersensitivity
To determine whether the increased genomic instability associated with Brca1S971A/S971A mice results from a deficiency in the HR pathway, primary mouse embryonic fibroblasts (MEFs) carrying either Brca1+/+ or Brca1S971A/S971A were treated with a poly(ADP-ribose) polymerase (PARP) inhibitor (KU-0058948) and their survival was examined in a clonogenic assay.29-32 Two pairs of MEFs from independent breedings of mice were used: the A2/A7 pair (p53−/−background, breeding between Brca1S971A/+p53−/− mice) and the B8/B9 pair (p53+/− background, breeding between Brca1S971A/+p53+/+ and Brca1S971A/+p53−/− mice). In a p53−/− background, Brca1S971A/S971A MEFs displayed moderate, but statistically significant, sensitivity to the PARP inhibitor as compared to Brca1+/+ MEFs (Fig. 1). In a p53+/− background, Brca1S971A/S971A MEFs were slightly more sensitive to the PARP inhibitor than wtMEFs, but the difference was statistically insignificant (data not shown). This could be due to the robust growth observed in primary MEFs with null p53, while primary MEFs with p53+/− background tend to grow more slowly. The moderate sensitivity of Brca1S971A/S971A MEFs to the PARP inhibitor is consistent with the notion that HR is partially impaired in Brca1S971A/S971A cells.
Figure 1.
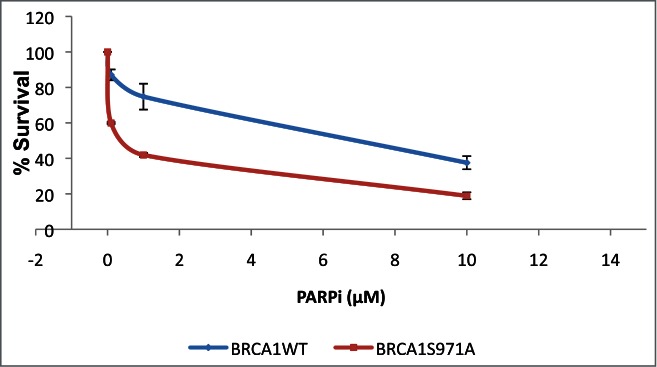
Primary Brca1S971A/S971A MEFs are more sensitive than Brca1+/+ MEFs to PARP inhibitor KU-0058948 in p53−/− background. Normal student t-test was used for statistical analysis. p = 0.02, 0.08 and 0.05 for PARPi at 0.1, 1 and 10μM, respectively. p = 0.0008 for overall curves(n = 3).
Assembly of the MRN-BRCA1-CtIP complex is not impaired by abrogation of Chk2 phosphorylation on BRCA1
To investigate how the Brca1(S971A) mutation impairs HR, 2 pairs of MEFs (Brca1+/+p53+/− vs. Brca1S971A/S971Ap53+/−) from independent breedings were immortalized and their impact on repair protein assembly was examined. Both Brca1+/+ and Brca1S971A/S971A MEFs were subjected to laser microirradiation and the localization of several repair proteins at DSBs was visualized by indirect immunofluorescence.33,34 Assembly of Mre11, NBS1, CtIP and 53BP1 at DSBs was statistically indistinguishable between Brca1+/+ and Brca1S971A/S971A cells at 10min, 30min and 60min time points after microirradiation (Fig. 2A). Although the percentages of Mre11+/γH2AX+ cells were similar in wt and mutant MEFs, the intensity of Mre11 stripes in Brca1S971A/S971A MEFs tended to be stronger. This could explain why more Mre11 foci were observed in HCC1937/BRCA1-S988A than in HCC1937/BRCA1-wt cells in an earlier report.26 Consistent with the immunofluorescence results, a similar amount of either CtIP or NBS1 was coimmunoprecipitated with BRCA1 when FLAG-tagged human BRCA1(wt) or BRCA1(S988A) was transiently transfected into HEK293T cells and immunoprecipitated with an anti-FLAG antibody (Fig. 2B). These data suggest that abrogation of Chk2 phosphorylation on BRCA1 did not affect recruitment of the MRN complex, 53BP1 or CtIP to DSBs.
Figure 2.
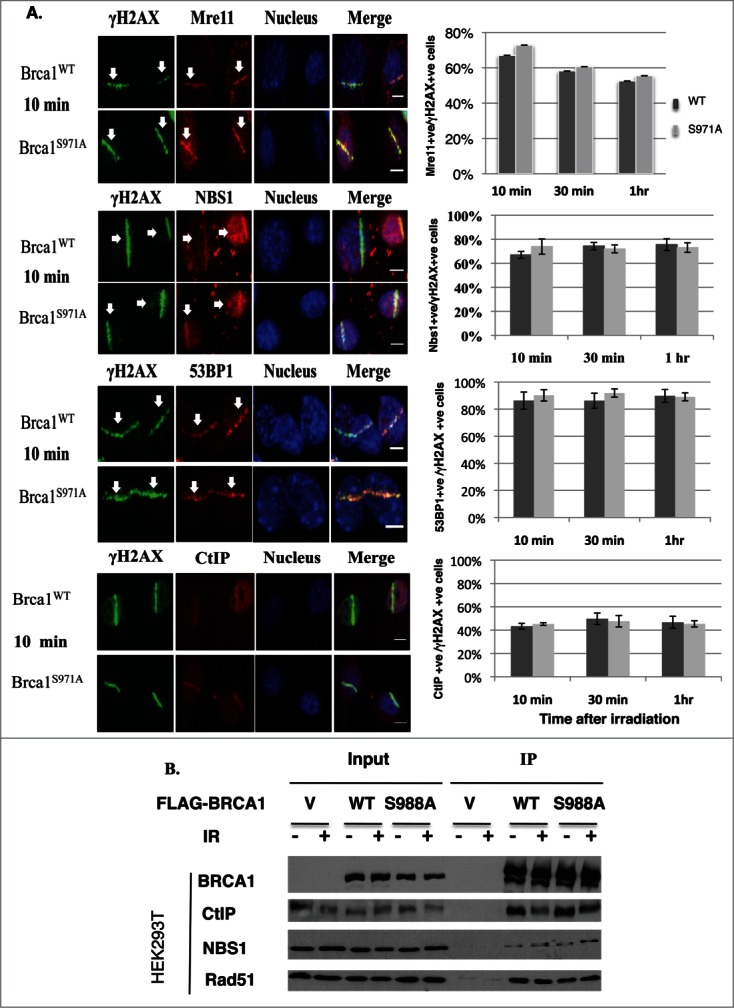
Abrogation of Chk2 phosphorylation on BRCA1 has no impact on the assembly of repair complexes upon DSBs. (A) Comparison of Mre11, CtIP, 53BP1 and Nbs1 recruitment at DSB laser stripes in Brca1+/+ and Brca1S971A/S971A MEFs. Left panels show laser stripes of Mre11, CtIP, 53BP1, Nbs1 (red) and γH2AX (green) at 10min point. Right panels show summary of 3 independent experiments for each individual protein. “+ve” stands for “positive.” For Mre11: 10 min (n = 352 total cells for wt, n = 340 total cells for S971A, p = 0.23); 30 min(n = 313 and 323, p = 0.50); 60 min (n = 315 and 308, p = 0.47). For Nbs1: 10 min (n = 297 and 265, p = 0.37), 30 min (n = 287 and 272, p = 0.62), 60 min (n = 251 and 258, p = 0.72). For 53BP1: 10 min (n = 452 and 535, p = 0.67); 30 min (n = 464 and 451, p = 0.16); 60 min (n = 490 and 462,p = 0.91). For CtIP: 10 min (n = 467 and 462, p = 0.53); 30 min (n = 429 and 534,p = 0.78); 60 min (n = 452 and 422, p = 0.84). Experiments conducted with overnight BrdU incorporation followed by microirradiation at 47% laser output. Similar results obtained from 55% laser output with no BrdU incorporation. Scale bar, 10μm. (B) Interaction between BRCA1 and CtIP, NBS1 or Rad51 is comparable for BRCA1(wt) and BRCA1(S988A) 30–60 min after IR in FLAG-BRCA1 coimmunoprecipitation.
Abrogation of Chk2 phosphorylation on BRCA1 delays end resection
Although the assembly of the MRN/BRCA1/CtIP complex at DSBs was not affected, recruitment of Rad51 was delayed in Brca1S971A/S971A cells (Fig. 3A and B). Both Brca1+/+ and Brca1S971A/S971A cells were cultured in the presence of BrdU for 2 hours before microirradiation. When microirradiated at 47% laser output, DSBs were generated in a BrdU-dependent manner (Fig. S1). The transient incorporation of BrdU, followed by immediate microirradition at 47% output, ensured that DSBs were generated only in cells at S/G2 phases, thus allowing us to monitor HR repair specifically within the S/G2 phase cells of an asynchronous cell populations without resorting to synchronizing manipulations. This was verified by co-staining BrdU with cyclin-A, an established S/G2 phase marker. Indeed, 85–90% of BrdU-stripe+ cells (from 3 independent experiments) were cyclin-A+ (data not shown). ∼10% of the BrdU-stripe+ cells showed a level of cyclin-A staining indistinguishable from the background, possibly due to lower cyclin-A levels corresponding to the early S phase. Interestingly, at th 10min time point, nearly 80% of γH2AX laser lines co-stained with Rad51 in Brca1+/+ cells, while less than half of γH2AX laser lines co-stained with Rad51 in Brca1S971A/S971A cells. However, there was no difference in Rad51 recruitment between Brca1+/+ and Brca1S971A/S971A cells at the 30min and 60min time points. Rad51 is believed to be recruited to DSBs, partly through BRCA1, after end resection.35 Because the in vivo association between Rad51 and wtBRCA1 or BRCA1S988A was indistinguishable (Fig. 2B), we speculated that the delayed Rad51 recruitment might be due to delayed end resection in mutant cells. We therefore examined ssDNA generated by end resection in wt and mutant MEFs. When BrdU is present in DNA, ssDNA generated by end resection exposes BrdU, which can be specifically detected with a BrdU antibody without denaturing DNA.17,18,22,36 Both wt and mutant cells were transiently labeled with BrdU for 2 hours before microirradiation at 55% laser output, under which DSBs could be generated in a BrdU-independent manner (Fig. S1). It is important to generate DSBs in a BrdU-independent manner in this context to ensure that the intensity of the BrdU lines reflects the extent of end resection only, not the amount of DSBs generated. Similar to Rad51 recruitment, significantly fewer BrdU-laser lines (meaning ssDNA) that co-stained with γH2AX were present in Brca1S971A/S971A cells relative to Brca1+/+ cells at the 10min and 30min time points (Fig. 3C and D). Although the difference persisted even at the 60min time point, the difference was not statistically significant. Furthermore, the average intensity of ssDNA (BrdU) laser lines was weaker in Brca1S971A/S971A cells than in Brca1+/+ cells (Fig. 3E), suggesting that end resection was delayed/slower in the absence of S971 phosphorylation. Similar results were obtained when cells were incubated with BrdU for more than 24 hours (data not shown). The cell cycle distributions of Brca1+/+ and Brca1S971A/S971A MEFs after 2hrs of BrdU incorporation were similar (Fig. 4B, left panel). It has been shown that BRCA1-S988 phosphorylation also affects mitotic progression and mitotic checkpoint arrest, in addition to its involvement in DSB repair;37,38 therefore, abrogation of Chk2 phosphorylation could lead to the accumulation of mitotic cells in Brca1S971A/S971A MEFs and reduction of G2 cells in the G2/M population, which could in turn contribute to lower end resection activity. We therefore examined the mitotic populations of Brca1+/+ and Brca1S971A/S971A MEFs using a mitosis-specific marker, phospho-histone H3Ser10, in flow cytometry analysis. Although there was an increase in M phase population in Brca1S971A/S971A MEFs, the overall M-phase cells in the total cell population were marginal (Fig. 4B right panel). Taken together, these data suggest that the defect in end resection was not due to a significantly lower percentage of S/G2 cells in mutant MEFs. Consistent with the notion that end resection is delayed in Brca1S971A/S971A cells, phosphorylation of Chk1 and RPA2 at 10 min post γ-irradiation was much weaker in mutant than in wt cells, while upstream events such as phosphorylation of ATM, Chk2 and H2AX were similar (Fig. 4A). The weaker phosphorylation of Chk1 and RPA2 in mutant cells persists throughout the 60min time course (Fig. S2), suggesting that the initial response to DSBs might play a bigger role in signaling checkpoint activation. The weaker activation of Chk1 phosphorylation in Brca1S971A/S971A MEFs could be the reason that the G2/M checkpoint is partially impaired in S971A knock-in mice.25
Figure 3.
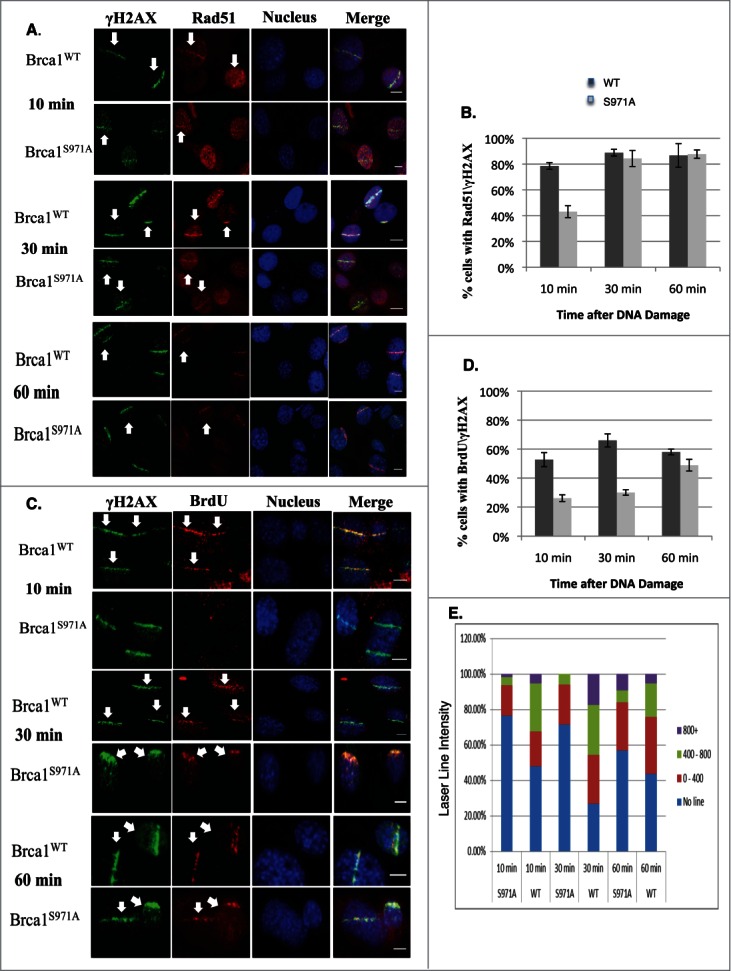
Brca1S971A/S971A MEF has delayed end-resection in response to DSBs. (A) Rad51 laser stripes (red) and γH2AX (green) from S/G2 cells at 10 min, 30 min and 60 min time points. Scale bar, 10μm. (B) Average from 6 independent experiments (3 experiments from each pair of MEFs). Ten min (n = 338and303, p = 2.87E-05); 30 min (n = 377and383, p = 0.26); 60 min (n = 426and330, p = 0.45). (C) Representative BrdU (ssDNA) stripes (red) and γH2AX (green) from total cell population at 10, 30 and 60 min time points. 55% laser output and 2 hrs of BrdU incorporation. (D) Average from 6 independent experiments (3 experiments/pair of MEFs). Ten min (n = 740and640, p = 0.000584); 30 min (n = 710and617, p = 2.65E-05); 60 min (n = 682and605, p = 0.069). (E) Quantification of BrdU stripe intensity from one representative experiment. Intensities are displayed with arbitrary units defined by Image J software.
Figure 4.

(A) Activation of phospho-Chk1 and phospho-RPA, not phosphorylation of ATM, Chk2 and H2AX, is impaired in Brca1S971A/S971A MEFs 10 min after γIR (10 Gy). (B) The cell cycle distribution of Brca1+/+ and Brca1S971A/S971A MEFs using FACS analysis. Left panel: MEFs are cultured in the presence of BrdU for 2 hours and BrdU positive/S phase cells are identified by a BrdU antibody. Right panel: M-phase population in normal growing MEFs is detected by a phospho-histone H3Ser10 antibody. One representative experiment from 3 independent experiments.
Abrogation of Chk2 phosphorylation on Brca1 delays Brca1 dispersion from DSBs
Earlier work reported that, although BRCA1(wt) and BRCA1(S988A) both form damage-induced foci, mutant BRCA1 is preferentially retained at foci after γ-irradiation at a time when wtBRCA1 forms a diffused pattern.5 We therefore examined the recruitment and retention of Brca1 at DSBs in response to microirradiation. Because Brca1 could be recruited to DSBs during the G1 as well as the S/G2 phases, as judged by Brca1/Cyclin A co-staining or by microirradiation of synchronized MEFs (data not shown), we focused on Brca1 recruitment/retention during the S/G2 phases. WT and mutant MEFs were irradiated at 47% laser output after 2hrs of BrdU incorporation to generate DSBs in S/G2 cells only and Brca1 laser stripes were examined at 10min, 30min and 60min after microirradiation. Both wt and mutant Brca1 were recruited to DSBs similarly at 10min; however, at 60min, wt Brca1 laser stripes were significantly weaker while mutant Brca1 laser stripes remained strong (Fig. 5A), which is consistent with the foci “retention” reported earlier in human BRCA1(S988A) cells.5 Significantly, both wt and mutant Brca1 laser stripes gradually decreased their intensity over a 60min time course, but the intensity of mutant Brca1 stripes decreased more slowly than wtBrca1 stripes (Fig. 5B).
Figure 5.
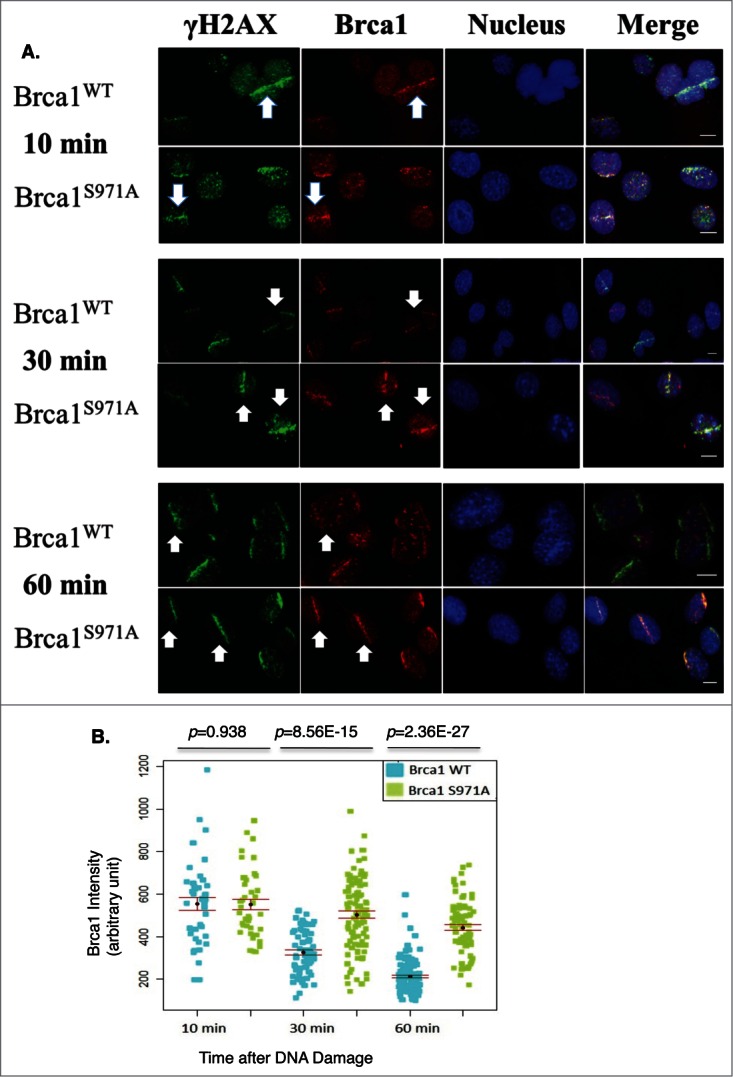
Retention/Slower dispersion of Brca1(S971A) at DSBs. (A) Brca1 (red) stripes at 10, 30, and 60 min time points after microirradiation. 47% laser output and 2 hrs of BrdU incorporation. Scale bar, 10 μm. (B) The average intensity of Brca1 stripes over a 60 min time course. Quantification of each Brca1 stripe with arbitrary units defined by Image J software and shown in a dot plot.
BRCA1 has been shown to preferentially bind branched DNA and to inhibit Mre11 nuclease activity.24 The slow “dispersion” of Brca1(S971A) from DSBs prompted us to test whether BRCA1 phosphorylation by Chk2 leads to dissociation of BRCA1 from DSBs. To test this hypothesis, the DNA binding activity of a human BRCA1 fragment encompassing the Chk2 phosphorylation site was examined in a reconstituted in vitro assay.24 Upon in vitro phosphorylated with purified Chk2, this BRCA1 fragment displayed the same DNA binding affinity as the corresponding unphosphorylated fragment (Fig. S3), suggesting that Chk2 phosphorylation did not affect BRCA1 binding to DNA.
Abrogation of Chk2 phosphorylation impairs BRCA1 ubiquitination
If the “dispersion” of wt BRCA1 from DSBs is due to degradation of the protein, then the “retention” of mutant BRCA1 at DSBs may reflect a requirement for Chk2 phosphorylation in the degredation of BRCA1. Interestingly, extracts of MEFs harbor 2 bands that could be recognized by Brca1 antibodies from 2 independent sources (Fig. 6A and data not shown). The intensity of the top band increased upon γIR and it shifted to a lower position after λ phosphatase treatment (Fig. S4), suggesting that the top band is phosphorylated Brca1. Noticeably, the steady-state levels of phospho-Brca1 were markedly increased in Brca1S971A/S971A MEFs relative to Brca1+/+ MEFs over a 60-minutes time course after γ-irradiation (Fig. 6A and B). We therefore examined whether Chk2 phosphorylation affects BRCA1 ubiquitination. HEK293T cells were transiently co-transfected with HA-ubiquitin and either FLAG-tagged BRCA1(wt) or BRCA1(S988A), and subjected to γIR in the presence of MG-132. Cells were harvested one hour later and BRCA1 was immunoprecipitated by a FLAG antibody. Upon immunoblotting with an HA antibody, in vivo ubiquitination of BRCA1 was revealed as a high molecular weight smear migrating above the parental BRCA1 band. Fig. 6C shows that FLAG-BRCA1(S988A) was consistently less ubiquitinated than FLAG-BRCA1(wt).
Figure 6.
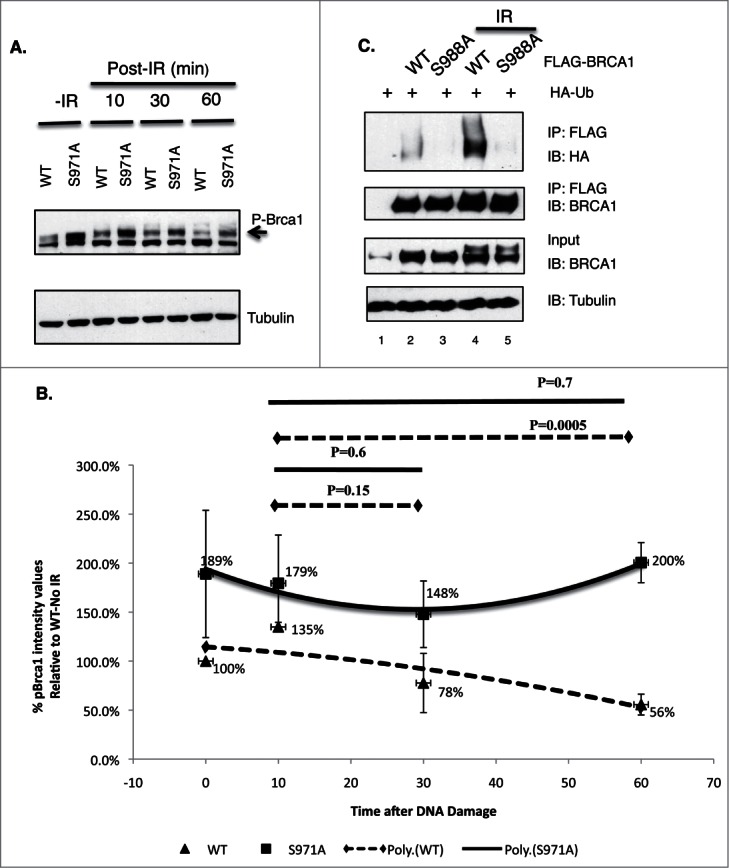
The impact of Chk2 phosphorylation on BRCA1 stability and ubiquitination. (A) Comparison of phosphorylated Brca1 level in Brca1+/+ and Brca1S971A/S971A MEFs upon γIR (10 Gy). One representative experiment from 4 independent experiments. Brca1 is detected by an antibody recognizing mouse Brca1 (a gift of Richard Baer). (B) Quantification of phospho-Brca1 levels normalized by tubulin levels. Average of 4 independent experiments. Trend line represents polynomial fit of data. P = 0.001 using pairwise t-test. (C) Comparison of in vivo ubiquitination of FLAG-tagged BRCA1(wt) and BRCA1(S988A).
Our earlier work revealed that BRCA1 protein levels are regulated by ubiquitin E3 ligase SCF complexes.39 In particular, we identified 3 F-box proteins that interact with BRCA1 (FBXO44, Skp2 and FBXO5) and showed that siRNA-mediated depletion of any one of these 3 factors stabilized BRCA1.39 To determine whether BRCA1 is a direct substrate of these SCF complexes, in vitro ubiquitination assays were conducted using immunopurified full-length FLAG-BRCA1. Fig. 7A shows that all 3 SCF complexes were capable of ubiquitinating full-length BRCA1 in an E2-dependent manner. We then investigated whether the SCF complexes could localize at DSBs. Interestingly, both Skp1 and Skp2 were recruited to DSBs (Fig. 7B), while FBXO44 and FBXO5 were not.39 About 6–15% of γH2AX stripes contained Skp1/Skp2 stripes, suggesting that the recruitment of Skp1/2 might be transient and occur during specific cell cycle stages. Furthermore, knockdown of Skp2 by siRNA also impaired BRCA1 ubiquitination in vivo (Fig. 7C). Similar to Brca1S971A/S971A MEFs, treatment of Brca1+/+ MEFs with a Skp2 inhibitor, Skp2-C25,40 led to less efficient end resection (Fig. 7D), although the effect with Skp2-C25 was more severe.
Figure 7.
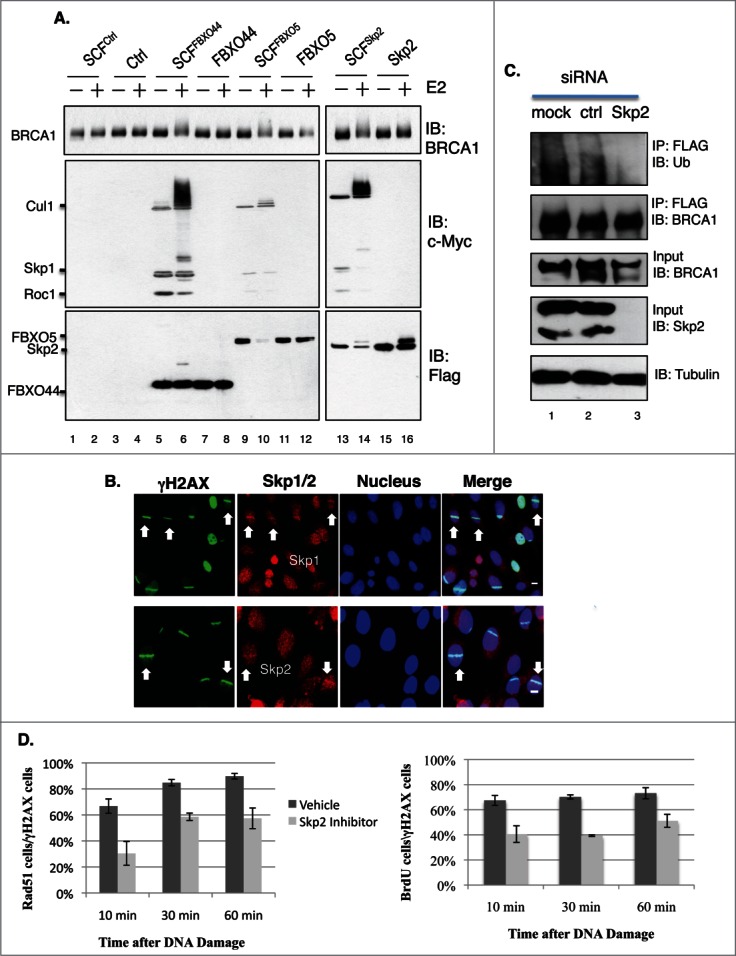
(A) BRCA1 is ubiquitinated in vitro by SCFSkp2. FLAG-BRCA1 immunoprecipitated from HEK293T is used as the substrate. Myc-tagged Skp1, Cul1 and Roc1 are coexpressed in HEK293T in the presence of FLAG-tagged F-box proteins and coimmunoprecipitated with FLAG-FBX proteins as the source of SCF E3 complex. (B) Endogenous Skp1 and Skp2 (red) are localized at DSBs in asynchronously growing U2OS cells. 55% laser output and no BrdU incorporation. Scale bar, 10μm. (C) Knockdown of Skp2 impairs BRCA1 ubiquitination in a HEK293T cell line that stably expresses FLAG-BRCA1, as described in.51 FLAG-BRCA1 is immunoprecipitated by a FLAG antibody. (D) A Skp2 inhibitor (Skp2-C25) impairs end resection in Brca1+/+ MEFs upon microirradiation. Skp2-C25 is added at a final concentration of 10 μM and 15 min before microirradiation. Average from 3 independent experiments. For Rad51: 10 min (n = 133 for no inhibitor, n = 179 for with inhibitor, p = 0.0267); 30 min (n = 159and219, p = 0.0086); 60 min (n = 157 and 209, p = 0.0169). For BrdU: 10 min (n = 241 for no inhibitor, n = 206 for with inhibitor, p = 0.049); 30 min (n = 276 and 302, p = 6.8E-05); 60 min (n = 269and246, p = 0.034).
Discussion
In this study, we show that abrogation of Chk2 phosphorylation on BRCA1 delays end resection, an early step in HR-mediated DSB repair. We demonstrate that, although Chk2 phosphorylation of BRCA1 has no effect on repair complex assembly, it controls the timing of end resection. Our study reveals that DSB-induced Chk2 phosphorylation promotes ubiquitination of BRCA1 by the E3 ligase, SCFSkp2. SCFSkp2 activity is known to appear in late G1 and peak in S/G2, and to ubiquitinate the phosphodegron motif.27,28 It is also known that BRCA1 inhibits Mre11 nuclease activity.24 Therefore, SCFSkp2 may preferentially ubiquitinate BRCA1 after it has been phosphorylated by Chk2 at DSBs. The ubiquitination of phospho-BRCA1 leads to degradation of BRCA1 at S/G2, unleashing Mre11 nuclease activity, and thus ensuring that end resection/HR occurs during S/G2 (Fig. S5). Of note, BRCA1(S988A) is still ubiquitinated to some extent, but much more weakly compared with BRCA1(wt) ubiquitination. This difference could lead to slower degradation/removal of the mutant BRCA1 at DSBs, explaining why end resection in Brca1S971A/S971A MEFs is delayed instead of completely abolished. Interestingly, the Brca1(S971A) mutation displays ∼50% of Brca1(wt) efficiency on end resection at 10min post microirradiation. Likewise, a similar reduction in end resection was observed in MEFs containing an Exo1 gene deletion or a knock-in mutation that inactivates Mre11 nuclease activity.36,41 Thus, the S988A point mutation of BRCA1 reveals a dual function of BRCA1 that cannot be uncovered upon analysis of BRCA1 null mutations. In a BRCA1-null background, assembly of the HR complex is severely impaired and end resection is compromised. As a result, the BRCA1-null cells mainly rely on NHEJ to repair DSBs, which would contribute to genome instability and tumorigenesis. This study demonstrates that BRCA1 is not just a scaffold protein for assembling the desired repair complexes at DSB sites; instead it serves as a built-in clock that restricts HR to the S/G2 phases of the cell cycle. Indeed, the proper timing of HR activity with respect to cell cycle progression may entail several complementary mechanisms that include Chk2 phosphorylation of BRCA1, 53BP1/RIF1-mediated suppression of end resection in G142-44 and cell cycle-dependent phosphorylation of CtIP.18,20 Also, Chk2 phosphorylation of BRCA1 in response to DSBs is a key “automation” design feature in this intricate “clock." While other interpretations can be envisioned, the model presented here (Fig.S5) describes a novel function for Chk2 phosphorylation of BRCA1 that is consistent with the existing data from both mouse and human studies.
SCFSkp2-dependent degradation of BRCA1 may only affect a subset of the cellular BRCA1 pool, namely those BRCA1 polypeptides that are in a complex with MRN/CtIP in the close vicinity of DSBs. BRCA1 may not be affected in other repair complexes localized at other sub-chromosomal compartments. The BRCA1 stripes/foci detected by immunofluorescence after DSB formation reflect the combination of multiple BRCA1-containing complexes, including the mutually exclusive complexes A (BRCA1-Abraxas-Rap80), B (BRCA1-Brip1), and C (BRCA1-CtIP-MRN), as well as the BRCA1-PALB2-BRCA2-Rad51 complex.2 We propose that Chk2-induced degradation is restricted to BRCA1 polypeptides of complex C (BRCA1-CtIP-MRN) only. We further speculate that degradation is likely achieved due to recognition of complex C by the Skp1/2 ligase. Phosphorylation of BRCA1 by Chk2 alone does not appear to affect the interaction between BRCA1 and Skp2 as both BRCA1(wt) and BRCA1(S988A) bind to Skp2 equally well. This is reasonable, given that the interactions between BRCA1 and Skp2 occur at the N- and the C-termini of BRCA1, both of which are distal to the Chk2 phosphorylation site (Fig. S6). Recently, Skp2 has been shown to localize at foci after DNA damage and ubiquitinate NBS1 with lysine-63 polyubiquitination linkage, which did not lead to NBS1 degradation.45 It is conceivable that other factors also play a role in SCFSkp2 ubiquitination specificity, in terms of both substrate recruitment and lysine linkage. It is also possible that SCFSkp2 may not be the only E3 involved in ubiquitination and clearance of BRCA1 from damage sites, as several other ubiquitin E3 ligases are present at DSBs.
Several recent studies suggest that BRCA1, in combination with POH1, is required to remove 53BP1 from DSB ends in order to promote HR.46-48 Since 53BP1 directly inhibits HR and promotes NHEJ, the presence of BRCA1 would be desirable throughout S/G2 phases. Our model of BRCA1 dispersion from the DSBs before the initiation of end resection would appear to be inconsistent with its function in displacing 53BP1 from DSBs during S/G2 phases. It is conceivable that, before the commitment of HR at end resection, the presence of BRCA1 is critical to prevent 53BP1 binding to DSB ends in order to promote HR. When end resection is initiated, the HR pathway is presumably committed. This irreversible choice is likely accompanied and achieved by multiple changes including protein compositions and chromatin conformations at DSBs that would no longer “attract” 53BP1 to DSBs ends. A key question remains how these seemingly conflicting processes in response to DSBs are controlled temporally and spatially to achieve maximum genome stability.
In addition to its role in displacing 53BP1 from DSBs ends, the function of BRCA1 proposed in this study also provides an alternative/additional explanation how inactivation of 53BP1 rescues the DSB repair and tumor suppression defects associated with BRCA1 loss.49 In the absence of p53BP1, NHEJ is presumably inefficient. So why is BRCA1 no longer needed for HR when 53BP1 is absent? We speculate that the HR pathway may still function during S/G2 in BRCA1/53BP1 double knockout cells, although at a lower efficiency, because CtIP can still be recruited to DSBs in the absence of BRCA1, by virtue of its independent interaction with NBS1.18,23,50 When 53BP1 is absent, there is no strong competition between HR and NHEJ pathways. As a result, there is no direct impact for slower assembly of MRN-CtIP complex at DSBs. In other words, the role of BRCA1 in controlling the timing of the HR pathway described in this study could be nonessential in the absence of NHEJ-promoting proteins such as 53BP1.49 It will be important to investigate in the future how BRCA1 coordinates with 53BP1/Rif1 and CtIP to ensure the timing of HR upon DNA damage and what other factors are involved in ubiquitination/degradation of phosphorylated BRCA1 by SCFSkp2.
Materials and Methods
PARP inhibitor sensitivity assay: Primary MEF cells from exponentially-growing cultures were seeded at various densities in 6-well plates and, 18–20 hours later, various doses (0.1 μM, 1 μM, 10 μM) of KU-0058948, a PARP inhibitor (KuDOS Pharmaceuticals, now belongs to Astra Zeneca. KU-058948 is similar, but not identical to, Olaparib) were added. The drug was left on continuously for the duration of the experiment. Seven–10 d later the cells were stained with 0.5% crystal violet in 20% ethanol, the colonies were counted and results were normalized to the plating efficiency.
Laser Microirradiation: MEFs were plated on 8 well Nunc LabTek II chamber slides at multiple densities per well (between 15,000 cells/well to 40,000 cells/well) and pre-sensitized with 50 μM BrdU for a duration of either 2 hours or overnight (as indicated). The well with ∼70–80% cell confluency was selected for microirradiation. About 150–200 cells were microirradiated within 10 minutes using a randomly drawn pattern. The microirradiation was performed using an MMI Cell Cut laser micro-dissection system consisting of a 390 nm ND-YAG laser that is coupled to the optical path of the microscope.
Immunofluorescence: Chamber slides were fixed at the indicated times after microirradiation with either a 3% Paraformaldehyde/2%Sucrose solution in Phosphate Buffered Saline (PBS/pH 7.4) for 15 minutes at room temperature or 70%Methanol/30%Acetone for 15 minutes at −20°C. They were washed thrice in PBS for 10 minutes each. The samples were then permeablized in 0.5% Triton X-100 buffer (10 mM PIPES, pH 6.8, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM sucrose, and 0.5% Triton X-100) with 10% FBS for 5 minutes on ice. They were then washed thrice in 10% FBS (in PBS) for 10 minutes each. The samples were blocked in 10% FBS for an hour. The samples were incubated with primary antibodies for durations of either 2 hours or overnight. For co-staining of 2 antibodies, serial incubation of the 2 different primary antibodies was performed. The primary antibodies were then detected by incubation with secondary antibodies tagged with Alexa-488 (1:1000), Alexa-546 (1:1000) or Alexa-647 (1:1000) (Molecular Probes, Life Technologies) for 2 hours. The cells were then washed twice with PBS and the DNA was counter-stained with ToPro-3 Iodide (1:500) or DAPI (1:10,000). The slides were then mounted with a 24×50, #1.5 coverslip using either Slowfade gold with DAPI (Life Technologies) or Vectashield with DAPI (Vector laboratories). All immunostained slides were visualized using a Nikon TE-2000, Zeiss LSM510 or Nikon Sweptfield confocal microscope.
Cell cycle Distribution by flow cytometry: For BrdU/PI labeling, wt and mutant MEFs were pulse-labeled with 50 μM BrdU (Sigma) for 2hrs. After trypsinization, cells were washed with cold PBS and fixed by 90% prechilled ethanol. Cells were then treated with 2N HCl at RT for 20 min, followed by PBS/0.5% BSA/0.5% Tween-20 wash and neutralization with 0.1M Boric Acid/pH8.5 at RT for 5 min. Cells were washed again and stained with Alexa Fluor 488 conjugated anti-BrdU (1:100, Invitrogen) for 1 hr at 37°C in the dark. Cells were washed and incubated in PBS containing 25 μg/ml propidium iodide (PtdIns) (Sigma) and 25 μg/ml RNase A (Roche) for 30 min followed by flow cytometry analysis. For detection of phosphorylated histone H3Ser10, ethanol-fixed cells were permeablized with 0.25% Triton X-100 in PBS for 15min on ice, washed and stained with anti-phospho-Ser10 histone H3 antibody (1:50, Cell Signaling) for 1 hr at 37°C, followed by incubation of an Alexa Fluor 488-conjugated anti-rabbit secondary antibody (1:500, Invitrogen) for another 1 hr at 37°C in dark. Cells were washed and incubated with PI for 30 min prior to flow cytometry analysis. All data were collected using FACScalibar (Becton Dickinson) and analyzed by Flowjo software.
Antibodies: The following antibodies were used for immunofluorescence – Mouse anti-γH2AX 1:1000 (Millipore, #05–636), Rabbit anti- γH2AX 1:1000 (Cell Signaling, #9718), Rabbit anti-53BP1 1:500 (Bethyl, A300–272A), Rabbit anti-Mre11 1:200 (Novus, NB100 – 142), Rabbit anti-Rad51 1:100 (sc-8349, Santa Cruz), Mouse anti-BrdU 1:100 (RPN202, GE Life Sciences), Rabbit anti-pRPA2-T21 1:25 (ab61065, Abcam), Rabbit anti-Brca1 1:500 (Richard Baer), Rabbit anti-CtIP 1:50 (a kind gift from Richard Baer50), Skp2 1:50 (LS-B2487, Lifespan Biosciences), Skp1 1:100 (sc-7163, Santa Cruz). The following antibodies were used for immunoblotting – Rabbit anti-pRPA2-S4/S8 1:1000 (A300–245A, Bethyl), Rabbit anti-Chk1-S345 1:100 (#2348, Cell Signaling), Total RPA 1:100 (sc-56770, Santa Cruz), Total Chk1 1:100 (sc-56291, Santacruz).
Imaging and Statistical Analysis: For the purpose of intensity quantification, multiple Z-sections totaling ∼10–12 μm were obtained and collapsed into a single image representing average pixel intensity using Image J. The straight line tool (line width = 20) was used to draw a line over all γH2AX stripes and the intensities of the corresponding BRCA1 or BrdU stripes were measured. All statistical analyses were performed either using Microsoft Excel or R (www.r-project.org). Unpaired students t-test was performed to generate ‘p’ values indicative of statistical significance. Statistical significance for mouse tumor incidence was determined using a Chi-square test.
Immunoprecipitation: For immunoprecipitation, 18 h after plasmid DNA transfection, proteasome inhibitor MG132 was added into the medium to a final concentration of 5μM. Approximately 24 h after transfection, cells were harvested by scraping and lysed by rotating at 4°C for 30 min with 1ml/60-mm dish high salt lysis buffer (HSL: 50 mM Tris-HCl, pH 8.0, 1% Nonidet P-40, 500 mM NaCl) or RIPA Buffer (150 mM NaCl, 50 mM Tris pH8.0, 1% NP-40, 0.5% DOC, 0.1% SDS), with a freshly added phosphatase inhibitors and protease inhibitors cocktail (20 mM NaF, 1 mM Na4P2O7, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin, 1 ug/ml leupeptin, 1 ug/ml aprotinin). Lysed cells were passed through 21 G needle 6 times, and then clarified by centrifugation at 16,000 × g at 4°C for 15 min. The supernatants (1 ml) were mixed with 30 μl of anti-FLAG M2 beads (Sigma, Cat. # A2220, 50% slurry) at 4°C overnight. The proteins bound to the beads were washed 3 times with either high salt lysis buffer or RIPA buffer and boiled in 2x Laemmli SDS loading buffer for 5 min. The samples were resolved by SDS-PAGE followed by immunoblotting.
In vivo and in vitro ubiquitination assays: For in vivo ubiquitination assays, HEK293T cells were transfected with HA-Ub and either pcDNA3-FLAG-BRCA1(wt) or pcDNA3-FLAG-BRCA1(S988A). Eighteen hours after DNA transfection, MG132 was added to the transfected cells at a final concentration of 5 μM. Cells were harvest 24 hours after transfection. The full-length BRCA1 was immunoprecipitated with a FLAG antibody and was analyzed using 3–8% SDS-PAGE, followed by immunoblotting with α-HA and α-BRCA1 Ab1. For in vitro ubiquitination assays, recombinant SCFControl and SCFFBX complexes were isolated from transfected 293T cells, as previously described51 with some modifications. Components of SCF complex (Myc-SKP1, Myc-Cul1, Myc-Roc1, and FLAG-tagged FBX proteins) were expressed in 293T cells and immunopurified with 30 μl of anti-FLAG M2 beads (50% slurry). Briefly, cells were harvested 24 hrs after transfection and lysed by rotating at 4°C for 30 min with 1 ml/60-mm dish NP-40 buffer I (50 mm Tris-HCl, pH 7.5, 0.5% Nonidet P-40, 150 mm NaCl, 1 mM EDTA, 2 mM DTT) supplemented with 20 mM NaF, 1 mM Na4P2O7, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 2 μg/ml pepstatin, 2 ug/ml leupeptin, and 2 ug/ml aprotinin. Lysed cells were passed through 21 G needle 6 times, and then clarified by centrifugation at 16,000 × g at 4°C for 15 min. The supernatants (900 μl) were mixed with 30 μl of anti-FLAG M2 beads (50% slurry) at 4°C overnight. The proteins bound to the beads were washed 3 times with NP-40 buffer I, NP-40 buffer II (50 mm Tris-HCl, pH 7.5, 0.5% Nonidet P-40, 300 mm NaCl, 1 mM EDTA, 2 mM DTT), NP-40 buffer III (25 mm Tris-HCl, pH 7.5, 0.01% Nonidet P-40, 50 mm NaCl, 1 mM EDTA, 10% Glycerol) and used in an in vitro Ub ligation assay containing 50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 2 mM NaF, 10 nM okadaic acid, 2 mM ATP, 0.6 mM DTT, 2 μg of ubiquitin (Boston Biochem, U-100), 100 ng E1 (Boston Biochem, E-304), and 400 ng E2-UbcH5b (Boston Biochem, E2–622) as described previously.51
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Steve P Jackson, Anyndja Dutta, Sang Eun Lee and Paul Hasty for valuable input, Brian Allen and Sabrina Smith for technical assistance.
Funding
The study was supported by National Institutes of Health Grant (to YH) R01CA118578 and the cancer center support grant P30CA054174.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res 2005; 3:531-539; PMID:16254187; http://dx.doi.org/ 10.1158/1541-7786.MCR-05-0192 [DOI] [PubMed] [Google Scholar]
- 2. Huen MSY, Shirley MHS, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mole Cell Biol 2010; 11:139-148; http://dx.doi.org/ 10.1038/nrm2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997; 90:425-435; PMID:9267023; http://dx.doi.org/ 10.1016/S0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 4. Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of Brca1 in the DNA damage response to double-strand breaks. Science 1999; 286:1162-1166; PMID:10550055; http://dx.doi.org/ 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 5. Lee J-S, Collins KM, Brown AL, Lee C-H, Chung JH. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 2000; 404:201-204; PMID:10724175; http://dx.doi.org/ 10.1038/35004614. [DOI] [PubMed] [Google Scholar]
- 6. Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997; 88:265-275; PMID:9008167; http://dx.doi.org/ 10.1016/S0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 7. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 2010; 11:196-207; PMID:20177395; http://dx.doi.org/ 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37-45; PMID:16327781; http://dx.doi.org/ 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 9. Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO 2004; 23:4868-4875; http://dx.doi.org/ 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004; 431:1011-1017; PMID:15496928; http://dx.doi.org/ 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu B, Kim S-T, Kastan MB. Involvement of Brca1 in S-phase and G2-phase checkpoints after ionizing irradiation. Mol and Cellx Biol 2001; 21:3445-3450; http://dx.doi.org/ 10.1128/MCB.21.10.3445-3450.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell 2007; 28:638-651; PMID:18042458; http://dx.doi.org/ 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu Z, Chung W-H, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008; 134:981-994; PMID:18805091; http://dx.doi.org/ 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008; 455:770-774; PMID:18806779; http://dx.doi.org/ 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hailong Wang, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A. CtIP Maintains Stability at Common Fragile Sites and Inverted Repeats by End Resection-Independent Endonuclease Activity. Mol Cell 2014; 54:1012-1021; PMID:24837675; http://dx.doi.org/ 10.1016/j.molcel.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nodar Makharashvili, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP, et al. Catalytic and Noncatalytic Roles of the CtIP Endonuclease in Double-Strand Break End Resection. Mol Cell 2014; 54:1022-1033; PMID:24837676; http://dx.doi.org/ 10.1016/j.molcel.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009; 459:460-463; PMID:19357644; http://dx.doi.org/ 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature 2007; 450:509-514; PMID:17965729; http://dx.doi.org/ 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu X, Chen J. BRCA1-CtIP interaction requires phosphorylation of CtIP at Ser327 by CDK, which occurs in the late S and G2 phases of the cell cycle. Mol and Cell Biol 2004; 24:9478-9486; http://dx.doi.org/ 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell 2007; 28:134-146; PMID:17936710; http://dx.doi.org/ 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 2009; 284:9558-9565; PMID:19202191; http://dx.doi.org/ 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlegel BP, Jodelka FM, Nunez R. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res 2006; 66:5181-5189; PMID:16707442; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3209. [DOI] [PubMed] [Google Scholar]
- 23. Chen L, Nievera CJ, Lee AY-L, Wu X. Cell cycle-dependent complex formation of BRCA1-CtIP-MRN is important for DNA double-strand break repair. J Biol Chem 2008; 283:7713-7720; PMID:18171670; http://dx.doi.org/ 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 24. Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M. Direct DNA binding by Brca1. Proc Natl Acad Sci U S A 2001; 98:6086-6091; PMID:11353843; http://dx.doi.org/ 10.1073/pnas.111125998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim SM, Cao L, Li C, Xu X, Huber LJ, Chodosh LA, Deng CX. Uterus hyperplasia and increased carcinogen-induced tumorigenesis in mice carrying a targeted mutation of the Chk2 phosphrylation site in Brca1. Mol Cell Biol 2004; 24:9498-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol 2004; 24:708-718; PMID:14701743; http://dx.doi.org/ 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev 1999; 13:1181-1189; PMID:10323868; http://dx.doi.org/ 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and b-TrCP: tipping the scales of cancer. Nat Rev Cancer 2008; 8:438-449; PMID:18500245; http://dx.doi.org/ 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting DNA repair defect in BRCA1 mutant cells as a therapeutic strategy. Nature 2005; 434:917-921; PMID:15829967; http://dx.doi.org/ 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 30. Sun C, Zhang F, Xiang T, Chen Q, Pandita TK, Huang Y, Hu MC, Yang Q. Phosphorylation of ribosomal protein S6 confers PARP inhibitor resistance in BRCA1-deficient cancers. Oncotarget 2014; 5:3375-3385; PMID:24831086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi Y, Battelli C, Watson J, Liu J, Curtis J, Morse AN, Matulonis UA, Chowdhury D, Konstantinopoulos PA. Sublethal concentrations of 17-AAG suppress homologous recombination DNA repair and enhance sensitivity to carboplatin and olaparib in HR proficient ovarian cancer cells. Oncotarget 2014; 5:2678-2687; PMID:24798692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bonanno L, Costa C, Majem M, Sanchez JJ, Gimenez-Capitan A, Rodriguez I, Vergnenegre A, Massuti B, Favaretto A, Rugge M, et al. The predictive value of 53BP1 and BRCA1 mRNA expression in advanced non-small-cell lung cancer patients treated with first-line platinum-based chemotherapy. Oncotarget 2013; 4:1572-1581; PMID:24197907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sophie E, Polo Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409-433; PMID:21363960; http://dx.doi.org/ 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol 2003; 5:255-260. [DOI] [PubMed] [Google Scholar]
- 35. Ciccia A, Elledge SJ. The DNA Damage Response: Making It Safe to Play with Knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schaetzlein S, Kodandaramireddy NR, Ju Z, Lechel A, Stepczynska A, Lilli DR, Clark AB, Rudolph C, Kuhnel F, Wei K, et al. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell 2007; 130:863-877; PMID:17803909; http://dx.doi.org/ 10.1016/j.cell.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, Jacob R, Dittmar G, Weichert W, Petersen I, et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol 2010; 12:492-499; PMID:20364141; http://dx.doi.org/ 10.1038/ncb2051. [DOI] [PubMed] [Google Scholar]
- 38. Chabalier-Taste C, Racca C, Dozier C, Larminat F. BRCA1 is regulated by Chk2 in response to spindle damage. Biochim Biophys Acta 2008; 1783:2223-2233; PMID:18804494; http://dx.doi.org/ 10.1016/j.bbamcr.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 39. Lu Y, Li J, Cheng D, Parameswaran B, Zhang S, Jiang Z, Yew PR, Peng J, Ye Q, Hu Y. The F-box protein FBXO44 mediates BRCA1 ubiquitination and degradation. J Biol Chem 2012; 287:41014-41022; PMID:23086937; http://dx.doi.org/ 10.1074/jbc.M112.407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chan C-H, Morrow JK, Li CF, Gao Y, Jin G, Moten A, Stagg LJ, Ladbury JE, Cai Z, Xu D, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013; 154:556-568; PMID:23911321; http://dx.doi.org/ 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buis J, Wu Y, Deng Y, Leddon J, Westfield G, Eckersdorff M, Sekiguchi JM, Chang S, Ferguson DO. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 2008; 135:85-96; PMID:18854157; http://dx.doi.org/ 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol Cell 2013; 49:858-871; PMID:23333305; http://dx.doi.org/ 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol Cell 2013; 49:872-883; PMID:23333306; http://dx.doi.org/ 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 44. Feng L, Fong K, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem 2013; 288:11135-11143; PMID:23486525; http://dx.doi.org/ 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu J, Zhang X, Zhang L, Wu CY, Rezaeian AH, Chan CH, Li JM, Wang J, Gao Y, Han F, et al. Skp2 E3 Ligase Integrates ATM Activation and Homologous Recombination Repair by Ubiquitinating NBS1. Mole Cell 2012; 46:1-11; PMID:22500733; http://dx.doi.org/ 10.1016/j.molcel.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 2012; 125:3529-3534; PMID:22553214; http://dx.doi.org/ 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Butler LR, Densham RM, Jia J, Garvin AJ, Stone HR, Shah V, Weekes D, Festy F, Beesley J, Morris JR. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO 2012; 31:3918-3934; http://dx.doi.org/ 10.1038/emboj.2012.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kakarougkas A, Ismail A, Katsuki Y, Freire R, Shibata A, Jeggo PA. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res 2013; 41:10298-10311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-254; PMID:20362325; http://dx.doi.org/ 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Reczek CR, Szabolcs M, Stark JM, Ludwig T, Baer R. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J Cell Biol 2013; 201:693-707; PMID:23712259; http://dx.doi.org/ 10.1083/jcb.201302145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001; 413:316-322; PMID:11565034; http://dx.doi.org/ 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.