

Abstract
Chemically-modified derivatives of cytidine, bearing a 5-(N-substituted-carboxamide) functional group, are new reagents for use in aptamer discovery via the SELEX process (Systematic Evolution of Ligands by EXponential enrichment). Herein, we disclose a practical synthesis of 5-(N-benzylcarboxamide)-2′-deoxycytidine, and the corresponding 5-(N-1-naphthylmethylcarboxamide)- and 5-(N-3-phenylpropylcarboxamide)-2′-deoxycytidine analogs, as both the suitably-protected 3′-O-cyanoethylphosphoramidite reagents (CEP; gram scale) and the 5′-O-triphosphate reagents (TPP; milligram-scale). The key step in the syntheses is a mild, palladium(0)-catalyzed carboxyamidation of an unprotected 5-iodo-cytidine. Use of the CEP reagents for solid-phase oligonucleotide synthesis was demonstrated and incorporation of the TPP reagents by KOD polymerase in a primer extension assay confirmed the utility of these reagents for SELEX. Finally, the carboxyamidation reaction was also used to prepare the nuclease-resistant sugar-variants: 5-(N-benzylcarboxamide)-2′-O-methyl-cytidine and 5-(N-3-phenylpropylcarboxamide)-2′-deoxy-2′-fluoro-cytidine.
Keyword: Aptamer, SELEX, modified nucleotide, cytidine-5-carboxamide
INTRODUCTION
Modified deoxyuridine nucleotides, bearing an N-substituted-carboxamide group at the 5-position (Figure 1), have proven to be valuable tools for improving in vitro selection of protein-binding aptamers (SELEX process)[ 1 – 3 ] and for post-SELEX optimization of binding and pharmacokinetic properties of the selected aptamers.[ 4 – 7 ]
Chemical structures of representative 2′-deoxyuridine-5-carboxamides.
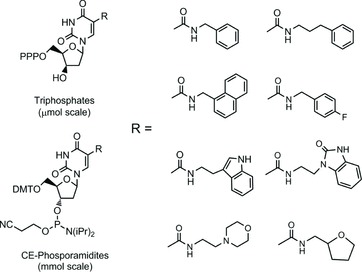
We recently disclosed an efficient, general process for synthesis of uridine-5-carboxamides that could provide multigram quantities of 3′-O-(N,N-diisopropyl-2-cyanoethylphosphoramidite) (CEP) reagents for solid-phase chemical synthesis of aptamers[ 4 ] and which was also applicable to preparation of the corresponding 5′-O-triphosphates (TPP) for enzymatic synthesis.[ 8 , 9 ] It is important to note that the utility of any new mod for SELEX requires synthesis and ready-availability of both the CEP and the TPP reagents. In order to further expand the repertoire of available reagents for in vitro selection and enable the incorporation of a second modified base into randomized DNA libraries, it was desirable to develop a complementary general synthesis of cytidine-5-carboxamide modified nucleotides.
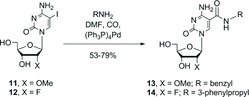
This communication discloses an alternative synthetic route to the cytidine-5-carboxamides which utilizes direct palladium(0)-catalyzed carboxyamidation of unprotected 5-iodo-cytidine as the key constructive step, an approach which was originally investigated by Eaton and coworkers.[ 10 ]
RESULTS AND DISCUSSION
Synthesis of Cytidine-5-Carboxamide Reagents
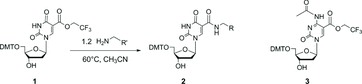
The general synthesis of uridine-5-carboxamides (Figure 2) relied on a common activated ester intermediate, 5-(2,2,2-trifluoroethoxycarbonyl)-2′-deoxyuridine 1, which was originally reported by Matsuda and coworkers.[ 11 ] Treatment of this activated ester with various primary amines (1.2 eq., 60°C, 4 hours) affords the corresponding 5-(N-substituted-carboxamides) 2 in excellent yield. Matsuda had also disclosed the analogous activated ester in the cytidine series, N-acetyl-5-(2,2,2-trifluoroethoxycarbonyl)-2′-deoxycytidine 3.[ 12 ] In our hands, this intermediate was less practically useful for synthesis of cytidine-5-carboxamides due to the lability of the N-acetyl protecting group and the instability of the N-acetyl-5-iodo-cytidine synthetic precursors. Due to these problematic side reactions, many attempts to scale-up the intermediate 3 beyond the reported milligram-scale[ 12 ] were unsuccessful.
Figure 2 . General synthetic process for uridine-5-carboxamides via Matsuda's “activated ester” intermediate 1.
We next examined a direct carboxyamidation approach. Eaton's process for carboxyamidation of 5-iodo-cytidine monophosphate[ 10 ] was adapted for 5-iodocytidine nucleosides, which are more suitable precursors for the desired CEP and TPP reagents (via 5′-O-DMT-protected intermediates). All parameters in the Eaton process were examined. We found DMF to be the best solvent for homogeneous nucleoside carboxyamidation and observed that added base (DBU or DABCO) could be omitted without effect on rate or product distribution. We identified reduction of carbon monoxide pressure from 50 psi to ≤1 atm as a critical parameter to limit formation of 2-ketocarboxamide byproducts.[ 13 , 14 ] At higher carbon monoxide pressure, 2-ketocarboxamides were observed as major byproducts (ca. 20–50%). Thus, commercially-available 5-iodo-2′-deoxycytidine 4 was readily converted into the corresponding 5-N-substituted-carboxamide 5a–c by treatment with the requisite aromatic primary amine RCH2NH2 (6–12 eq), carbon monoxide (≤1 atm), and (Ph3P)4Pd (2 mol%) in N,N-dimethylformamide (DMF) at room temperature for 24–48 hours (Figure 3). A catalyst formed in situ from dba2Pd (2 mol%) and Ph3P (8.8 mol%) also gave good results. The modified nucleoside products 5a–c were readily purified by recrystallization from alcohol, greatly facilitating multi-gram production of these key intermediates.
Figure 3 . Synthesis scheme for 5-(N-substituted-carboxamide)-2′-deoxycytidine CEP reagents by carboxyamidation of 5-iodo-2′-deoxycytidine.
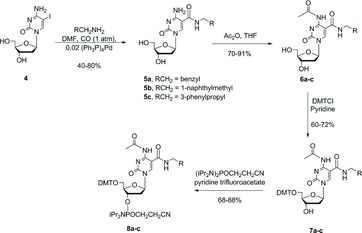
For preparation of CEP reagents, the 5-N-substituted-carboxamides 5a–c were selectively N-protected by stirring with acetic anhydride (no base) in tetrahydrofuran (THF), and then 5′-O-protected as the (4,4′-dimethoxytrityl)-derivatives 7a–c by reaction with 4,4′-dimethoxytrityl chloride in pyridine.[ 15 ] Synthesis of the high purity (>98.0%) CEP reagents 8a–c was completed by pyridinium trifluoroacetate-catalyzed condensation of the 3′-alcohol with 2-cyanoethyl-N,N,N′,N′-tetraisopropyl phosphoramidite[ 16 , 17 ] and final purification by silica gel flash chromatography with degassed solvents.[ 18 ]
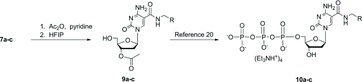
For preparation of TPP reagents 10a–c (Figure 4), the 5′-O-DMT-protected nucleosides 7a–c were peracetylated with acetic anhydride in pyridine, followed by cleavage of the DMT and 4-N-acetyl protecting groups with 1,1,1,3,3,3-hexafluoro-2-propanol.[ 19 ] The resulting crystalline 3′-O-acetate nucleosides 9a–c were readily converted into the crude 5′-O-triphosphates by the Ludwig–Eckstein process.[ 20 ] These chemically-modified nucleotides were purified by a two-stage process: anion exchange chromatography (AEX), followed by reversed-phase preparative HPLC, in order to obtain high purity (>90%).
Figure 4 . Synthesis scheme for 5-(N-substituted-carboxamide)-2′-deoxycytidine-5′-O-triphosphate reagents.
The carboxyamidation reaction was also suitable for preparation of nuclease-resistant ribo-sugar analogues[ 21 ] (Figure 5), for example, 5-(N-benzylcarboxamide)-2′-O-methyl-cytidine 13 and 5-(N-3-phenylpropylcar-boxamide)-2′-deoxy-2′-fluoro-cytidine 14.
Figure 5 . Synthesis scheme for 2′-substituted-cytidine nucleosides by carboxyamidation of a 5-iodocytidine.
Solid-Phase Oligonucleotide Synthesis with CEP Reagents 8a–c
The incorporation efficiency of the 5-carboxamide modified 2′-deoxycytidine phosphoramidites 8a–c was evaluated by solid-phase synthesis of a series of model sequences that incorporated zero (control) through five consecutive modified cytidines as an insertion in a standard model DNA 34-mer.
GAGTGACCGTCTGCCTGX0-5CAGACGACGAGCGGGA
where X = modified cytidine
The results indicate that full-length synthetic yields for 1 to 3 sequential couplings of the modified cytidine phosphoramidites 8a–c were comparable to unmodified DNA phosphoramidites (Table 5). Some loss of yield was observed for 4 or 5 sequential couplings of modified cytidines; however, significant amounts of full length product were obtained and confirmed in all cases.
Incorporation of Cytidine-5-Carboxamide TPP Reagents by KOD Polymerase
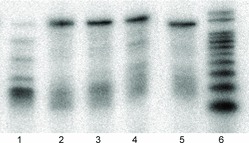
The three new cytidine-5-carboxamide TPP 10a–c were found to be readily utilized as substrates by KOD exonuclease-minus DNA polymerase in the primer extension assay. All three modified cytidine triphosphates were incorporated at least as efficiently as natural, unmodified 2′-deoxycytidine triphosphate in this assay (Figure 6).
Figure 6 . Polyacrylamide gel image of Primer Extension Assay with dNTP's, as described in Material and Methods. Lane 1: dAdGdT (negative control); Lane 2: dAdGdTdC (control); Lane 3: dAdGdT + 10a; Lane 4: dAdGdT + 10b; Lane 5: dAdGdT + 10c; Lane 6: 20/200 DNA Ladder.
CONCLUSIONS
A practical process for synthesizing cytidine-5-carboxamide modified nucleotides, as both 5′-O-triphosphates and 3′-O-CE phosphoramidites, has been developed which provides valuable new reagents for in vitro selection and post-SELEX optimization of aptamers. SELEX experiments are currently in progress using cytidine-5-carboxamide modified nucleotides alone, or combined with uridine-5-carboxamide modified nucleotides (“dual-mod SELEX”) and results will be reported in due course.
EXPERIMENTAL
The starting materials: 5-iodo-2′-deoxycytidine; 5-iodo-2′-O-methyl-cytidine; 5-iodo-2′-deoxy-2′-fluorocytidine were purchased from ChemGenes Corporation (Wilmington, MA 01887, USA) or Thermo Fisher Scientific Inc. (Waltham, MA 02454, USA). Carbon monoxide (safety: poison gas) at 99.9% purity was purchased from Specialty Gases of America (Toledo, OH 43611, USA). All other reagents were purchased from Sigma-Aldrich (Milwaukee, WI 53201, USA) and were used as received. Thin layer chromatography (TLC) was performed on glass-backed silica gel60 plates with a fluorescent indicator. NMR spectra were recorded on Bruker instruments at the University of Colorado at Boulder NMR facility (MHz and solvents listed below) and processed with SpinWorks software, ver 3.1.8.1 (University of Manitoba, Winnipeg, Cananda). Mass spectra (ESI-) were determined by electrospray (negative ion mode) using flow injection (95% 100 mM ammonium acetate: 5% acetonitrile, pH 6.9) with an Agilent 6130 Quadrupole LC/MS system. UV spectra and quantitations were measured with a Hewlett Packard 8452A Diode Array Spectrophotometer. Melting points were determined in open glass tubes using a Thomas Hoover 6427-H10 Capillary Melting-Point apparatus.
5-(N-Benzylcarboxamide)-2′-deoxycytidine (5a). An argon-filled 1L round-bottom flask was charged with: 5-iodo-2′-deoxycytidine (30 g, 85 mmol); benzylamine (109.3 g, 1020 mmol, 12 eq); and anhydrous N,N-dimethylformamide (DMF, 205 mL). The mixture was rapidly magnetically-stirred until all the solids had dissolved. The resulting solution was degassed by two cycles of evacuation to 50 mm and refilling with argon. A mixture of bis(dibenzylidineacetone)palladium(0) (978 mg, 1.7 mmol, 0.02 eq) and triphenylphosphine (1.92 g, 7.3 mmol, 0.086 eq) was added and the resulting fine black suspension was rapidly stirred, evacuated to 50 mm and filled with carbon monoxide (1 atm) from a rubber balloon. The mixture was stirred at room temperature (∼20°C) and periodically refilled with carbon monoxide. After 26 hours, the reaction was found to be complete by TLC analysis (silica gel, eluent: 15% methanol/85% dichloromethane (v/v), Rf(SM) = 0.3, Rf(5a) = 0.4). The reaction mixture was diluted with ethyl acetate (205 mL), filtered, and rinsed forward with 65% ethyl acetate/35% DMF (100 mL). The clear green filtrate was concentrated on a rotary evaporator (50–80°C, 1–2 mm) until all the solvents and most of the benzylamine had distilled. The dark orange residue (∼75 g) was dissolved in hot abs. ethanol (650 mL) and rapidly hot-filtered to remove a small amount of insoluble flakes (∼2 g). The clear filtrate was allowed to cool with slow stirring and the product crystallized as needles. After stirring overnight, the slurry was filtered and the cake washed with ice-cold ethanol (100 mL). After drying in vacuo, the product 5a was obtained as a white, crystalline solid: 22.0 g, 72% yield, mp 193–4°C. 1H NMR (500 MHz, d6-DMSO): δ = 8.73 (t, J = 5.8 Hz, 1H), 8.42 (s, 1H), 8.06 (bs, 1H), 7.75 (bs, 1H), 7.32 (m, 4H), 7.25 (m, 1H), 6.14 (t, J = 6.5 Hz, 1H), 5.24 (d, J = 4.4 Hz, 1H), 5.03 (t, J = 5.5 Hz, 1H), 4.42 (dd, J = 15.4, 7.2 Hz, 1H), 4.41 (dd, J = 15.4, 7.3 Hz, 1H), 4.26 (m, J = 4.3 Hz, 1H), 3.83 (dd, J = 7.9, 4.3 Hz, 1H), 3.64 (m, 1H), 3.58 (m, 1H), 2.19 (m, 2H). 13C NMR (500 MHz, d6-DMSO): δ = 165.88 (1C), 163.97 (1C), 153.99 (1C), 144.26 (1C), 139.64 (1C), 128.80 (2C), 127.60 (2C), 127.30 (1C), 99.20 (1C), 88.08 (1C), 86.29 (1C), 70.44 (1C), 61.50 (1C), 42.72 (1C), 40.62 (1C). MS m/z: calcd for C17H19N4O5, 359.36; found, 359.1 [M–H]−.
4-N-Acetyl-5-(N-benzylcarboxamide)-2′-deoxycytidine (6a). A 1L round bottom flask was charged with 5a (20.0 g, 55.4 mmol), and anh. tetrahydrofuran (THF, 500 mL). The well-stirred mixture was treated with acetic anhydride (26.4 mL, 277 mmol, 5 eq) and the mixture was heated at 50°C for 20 hours. TLC analysis of an aliquot (homogenized by dissolving in 50% methanol/50% dichloromethane) showed that the reaction was complete (eluent: 15% methanol/85% dichloromethane (v/v), Rf(5a) = 0.4, Rf(6a) = 0.6). The slurry was cooled to 5–10°C for 1 hour, filtered, and washed with cold THF (40 mL). Drying in vacuo afforded the product 6a as white, crystalline needles, 20.4 g, 91% yield, mp 173–4°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.35 (s, 1H), 8.98 (t, J = 5.7 Hz, 1H), 8.73 (s, 1H), 7.34 (d, J = 4.4 Hz, 4H), 7.26 (m, J = 4.3 Hz, 1H), 6.10 (t, J = 6.1 Hz, 1H), 5.28 (d, J = 4.4 Hz, 1H), 5.09 (t, J = 5.4 Hz, 1H), 4.44 (dd, J = 15.3, 8.1 Hz, 1H), 4.43 (dd, J = 15.2, 8.1 Hz, 1H), 4.28 (dt, J = 9.8, 4.0 Hz, 1H), 3.91 (dd, J = 7.9, 4.0 Hz, 1H), 3.68 (m, 1H), 3.60 (m, 1H), 2.41 (s, 3H), 2.34 (m, 1H), 2.22 (m, 1H). 13C NMR (500 MHz, d6-DMSO): δ = 171.27 (1C), 165.49 (1C), 159.77 (1C), 153.19 (1C), 146.24 (1C), 139.16 (1C), 128.82 (2C), 127.76 (2C), 127.41 (1C), 100.32 (1C), 88.67 (1C), 87.50 (1C), 70.11 (1C), 61.17 (1C), 43.00 (1C), 40.78 (1C), 26.70 (1C). MS m/z: calcd for C19H21N4O6, 401.40; found, 401.1 [M–H]−.
5′-O-(4,4′-Dimethoxytrityl)-4-N-acetyl-5-(N-benzylcarboxamide)-2′-deoxycytidine (7a). A 250 mL round bottom flask was charged with 6a (4.82 g, 12 mmol) and anhydrous pyridine (40 mL). The resulting colorless solution was magnetically-stirred as 4,4′-dimethoxytrityl chloride (4.47 g, 13.2 mmol, 1.1 eq) was added in five portions over one hour. The orange-yellow solution was stirred for 30 minutes more, quenched with ethanol (4.2 mL, 72 mmol), and concentrated on the rotovap (1–2 mm, ≤35°C). The resulting sticky orange residue (∼13 g) was partitioned with ethyl acetate (100 mL) and cold, saturated aq. sodium bicarbonate (50 mL). The organic layer was dried with sodium sulfate, filtered and concentrated to leave a yellow foam. Purification by flash chromatography (silica gel, eluent: 1% triethylamine/99% ethyl acetate) afforded 7a as a white solid foam, 6.1 g, 72% yield, mp 96–8°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.44 (s, 1H), 9.12 (t, J = 5.5 Hz, 1H), 8.46 (s, 1H), 7.38 (d, J = 7.4 Hz, 2H), 7.25 (m, 12H), 6.84 (m, 4H), 6.10 (t, J = 6.1 Hz, 1H), 5.34 (d, J = 4.7 Hz, 1H), 4.20 (m, 3H), 4.05 (m, 1H), 3.72 (d, J = 1.7 Hz, 6H), 3.41 (dd, J =10.5, 6.0 Hz, 1H), 3.20 (dd, J =10.4, 3.5 Hz,1H), 2.44 (s, 3H), 2.39 (m, 1H), 2.25 (m, 1H). 13C NMR (500 MHz, d6-DMSO): δ = 171.28 (1C), 165.38 (1C), 159.88 (1C), 158.53 (2C), 153.07 (1C), 146.13 (1C), 145.26 (1C), 138.91 (1C), 136.00 (1C), 135.98 (1C), 130.16 (2C), 130.11 (2C), 128.78 (2C), 128.28 (2C), 128.13 (2C), 127.84 (2C), 127.46 (1C), 127.14 (1C), 113.60 (4C), 100.32 (1C), 88.04 (1C), 86.86 (1C), 86.19 (1C), 70.69 (1C), 60.22 (1C), 55.43 (1C), 55.42 (1C), 43.03 (1C), 40.70 (1C), 26.76 (1C). MS m/z: calcd for C40H39N4O8, 703.77; found, 703.2 [M–H]−.
5′-O-(4,4′-Dimethoxytrityl)-4-N-acetyl-5-(N-benzylcarboxamide)-2′-deoxycytidine-3′-O-(N,N-diisopropyl-O-2-cyanoethylphosphoramidite) (8a). A 250 mL round bottom flask was charged with: 7a (11.0 g, 15.6 mmol); anhydrous dichloromethane (40 mL); 2-cyanoethyl-N,N,N′,N′-tetraisopropylphosphordiamidite (5.9 mL, 18.7 mmol, 1.2 eq); and finally, pyridine trifluoroacetate (3.61 g, 18.7 mmol, 1.2 eq). After 30 minutes, TLC analysis showed that the reaction was complete (eluent: 75% ethyl acetate/25% hexanes (v/v), Rf(7a) = 0.2, Rf(8a) = 0.7/0.8 [two isomers]). The entire reaction mixture was applied to a silica gel flash column preconditioned with 1% triethylamine/64% ethyl acetate/35% hexanes (until the eluent is basic), then eluted with 65% ethyl acetate/35% hexanes (argon-sparged). The product-containing fractions were protected from air in sealed jars under argon and concentrated to afford 8a as a colorless solid foam, 10.8 g, 76% yield, mp 70–81°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.47 (s, 1H), 9.11 (bs, 1H), 8.57/8.54 (s, 1H), 7.37 (m, 2H), 7.24 (m, 12H), 6.84 (m, 4H), 6.10 (m, 1H), 4.40 (m, 1H), 4.21 (m, 3H), 3.70 (m, 8H), 3.55 (m, 2H), 3.28 (m, 2H), 2.75 (t, J = 5.9 Hz, 1H), 2.64 (t, J = 5.9 Hz, 1H), 2.55 (m, 1H), 2.42 (m, 4H), 1.11 (m, 9H), 0.98 (d, J = 6.8 Hz, 3H). 31P NMR (500 MHz, d6-DMSO): δ = 147.55/147.37 (s, 1P). MS m/z: calcd for C45H56N6O9P, 903.99; found, 903.3 [M–H]−.
5-(N-1-Napthylmethyl)-2′-deoxycytidine-5-carboxamide (5b). Prepared as described for 5a, using 1-naphthylmethylamine (6 eq) in place of benzylamine, and with a reaction time of 48 hours at room temperature. After concentrating the reaction mixture, the residue was extracted with diisopropyl ether (∼40 mL/g) to remove most of the excess 1-naphthylmethylamine. The residue was crystallized from hot methanol (50 mL/g), with hot filtration, to afford the product 5b as a white solid, 40% yield, mp >220°C (chars w/o melting). 1H NMR (500 MHz, d6-DMSO): δ = 8.81 (t, J = 5.5 Hz, 1H), 8.43 (s, 1H), 8.14 (d, J = 4.4, 1H), 8.09 (bs, 1H), 7.96 (m, 1H), 7.79 (m, 1H), 7.75 (bs, 1H), 7.53 (m, 4H), 6.14 (t, J = 6.6 Hz, 1H), 5.24 (d, J = 4.3 Hz, 1H), 5.01 (t, J = 5.6 Hz, 1H), 4.90 (dd, J = 15.6, 13.4 Hz, 1H), 4.89 (dd, J = 15.5, 13.2 Hz, 1H), 4.26 (m, J = 4.1 Hz, 1H), 3.84 (dd, J = 8.4, 4.4 Hz, 1H), 3.58 (m, 2H), 2.20 (m, 2H). 13C NMR (500 MHz, d6-DMSO): δ = 165.45 (1C), 163.58 (1C), 153.57 (1C), 143.93 (1C), 136.07 (1C), 134.20 (1C), 133.32 (1C), 128.61 (1C), 127.59 (1C), 126.34 (1C), 125.89 (1C), 125.53 (1C), 125.07 (1C), 123.36 (1C), 98.82 (1C), 87.71 (1C), 85.99 (1C), 70.13 (1C), 61.16 (1C), 42.42 (1C), 40.14 (1C). MS m/z: calcd for C21H21N4O5, 409.42; found, 409.1 [M–H]−.
4-N-Acetyl-5-(N-1-naphthylmethylcarboxamide)-2′-deoxycytidine(6b). A 100 mL round bottom flask was charged with 5b (1.17 g, 2.85 mmol) and anh. tetrahydrofuran (26 mL) and stirred to form a gray-white slurry. Acetic anhydride (1.4 mL, 14.3 mmol, 5 eq) was added to the mixture dropwise while stirring at room temperature. The reaction mixture was stirred and heated to 50°C for 21 hours. An aliquot was sampled for TLC analysis (eluent: 10% methanol/90% dichloromethane (v/v), Rf(5b) = 0.61, Rf(6b) = 0.12) which indicated that the reaction was complete. The reaction flask was transferred to an ice bath and stirred for >1 hour. The mixture was then filtered and the filter cake was rinsed with chilled isopropyl ether. The resulting solids were collected and further evaporated in vacuo to yield fine gray-white crystals (1.01 g), 78.2% yield, mp 167-8°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.35 (s, 1H), 9.07 (t, J = 4.6 Hz, 1H), 8.74 (s, 1H), 8.15 (d, J = 8.7 Hz, 1H), 7.96 (m, 1H), 7.87 (m, 1H), 7.53 (m, 4H), 6.11 (t, J = 6.2 Hz, 1H), 5.29 (d, J = 4.2 Hz, 1H), 5.08 (t, J = 5.4 Hz, 1H), 4.92 (dd, J = 15.5, 10.1 Hz, 1H), 4.91 (dd, J = 15.7, 9.7 Hz, 1H), 4.28 (dt, J = 9.4, 3.8 Hz, 1H), 3.92 (dd, J = 7.6, 3.9 Hz, 1H), 3.64 (m, 1H), 3.58 (m, 1H), 2.42 (s, 3H), 2.35 (m, 1H), 2.22 (m, 1H). 13C NMR (500 MHz, d6-DMSO): δ = 171.27 (1C), 165.53 (1C), 159.77 (1C), 153.20 (1C), 146.30 (1C), 136.48 (1C), 134.17 (1C), 133.74 (1C), 131.26 (1C), 129.02 (1C), 128.12 (1C), 126.83 (1C), 126.33 (1C), 125.95 (1C), 123.80 (1C), 100.38 (1C), 88.74 (1C), 87.63 (1C), 70.25 (1C), 61.29 (1C), 41.13 (1C), 40.92 (1C), 26.71 (1C). MS m/z: calcd for C23H23N4O6, 451.46; found, 451.1 [M–H]−.
5′-O-(4,4′-Dimethoxytrityl)-4-N-Acetyl-5-(N-1-naphthylmethylcarboxamide)-2′-deoxycytidine (7b). Prepared as described for 7a, as a colorless solid in 59% yield, mp 148–150°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.40 (s, 1H), 9.35 (bt, 1H), 8.48 (s, 1H), 8.02 (m, 1H), 7.96 (m, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.54 (m, 2H), 7.40 (m, 2H), 7.35 (m, 1H), 7.25 (m, 8H), 6.85 (d, J = 8.9 Hz, 4H), 6.09 (t, J = 6.1 Hz, 1H), 5.32 (d, J = 3.7 Hz, 1H), 4.72 (dd, J = 14.9, 4.8 Hz, 1H), 4.60 (dd, J = 15.1, 3.4 Hz, 1H), 4.16 (dt, J = 10.9, 4.7 Hz, 1H), 4.04 (m, 1H), 3.70 (s, 6H), 3.29 (dd, J = 10.5, 6.4 Hz, 1H), 3.18 (dd, J = 10.4, 7.0 Hz, 1H), 2.43 (s, 3H), 2.38 (m, 1H), 2.26 (m, 1H). 13C NMR (500 MHz, d6-DMSO): δ = 171.25 (1C), 165.37 (1C), 159.88 (1C), 158.52 (1C), 158.54 (1C), 153.03 (1C), 146.32 (1C), 145.31 (1C), 136.05 (1C), 135.97 (1C), 133.92 (1C), 133.74 (1C), 131.22 (1C), 130.20 (2C), 130.11 (2C), 129.05 (1C), 128.30 (2C), 128.15 (2C), 127.16 (1C), 126.81 (1C), 126.33 (1C), 125.85 (1C), 125.80 (1C), 123.65 (1C), 113.61 (4C), 100.49 (1C), 88.12 (1C), 86.79 (1C), 86.17 (1C), 70.59 (1C), 64.40 (1C), 55.41 (2C), 41.01 (1C), 40.70 (1C), 40.82 (1C), 26.76 (1C). MS m/z: calcd for C44H41N4O8, 753.83; found, 753.21 [M–H]−.
5′-O-(4,4′-Dimethoxytrityl)-4-N-Acetyl-5-(N-1-naphthylmethylcarboxamide)-2′-deoxycytidine -3′-O-(N,N-diisopropyl-O-2-cyanoethylphosphoramidite) (8b). Prepared as described for 8a, as a white solid foam, 88% yield, mp 88–96°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.41 (s, 1H), 9.13 (bs, 1H), 8.56/8.54 (s, 1H), 8.01 (m, 1H), 7.95 (m, 1H), 7.85 (m, 1H), 7.53 (m, 2H), 7.37 (m, 2H), 7.24 (m, 9H), 6.83 (m, 4H), 6.06 (m, 1H), 4.66 (m, 2H), 4.39 (m, 1H), 4.16 (m, 1H), 3.68 (m, 8H), 3.52 (m, 2H), 3.28 (m, 1H), 3.20 (m, 1H), 2.74 (t, J = 5.8 Hz, 1H), 2.63 (t, J = 5.9 Hz, 1H), 2.45 (m, 5H), 1.09 (m, 9H), 0.96 (d, J = 6.8 Hz, 3H). 31P NMR (500 MHz, d6-DMSO): δ = 146.93/146.69 (s, 1P). MS m/z: calcd for C53H58N6O9P, 954.05; found, 953.3 [M–H]−.
5-(N-3-Phenylpropylcarboxamide)-2′-deoxycytidine (5c). Prepared as described for 5a (40 mmol-scale), using 3-phenylpropylamine (6 eq) in place of benzylamine and a reaction time of 48 hours at room temperature. After removal of the solvents on the rotovap, the residue was triturated with diethyl ether (∼30 mL/g) to extract the excess 3-phenylpropylamine and the residue was dissolved in hot ethanol, stirred at room temperature for 18 hours, followed by stirring at 0°C for 1 hour. The resulting mixture was filtered and the mother liquor was evaporated resulting in a brown resin. This was partitioned between dichloromethane and water. After standing and stirring at room temperature, white feathery crystals formed in the organic layer, and in the aqueous layer as well. The layers were filtered and the filter cake was washed with diethyl ether to afford 5c as a fluffy white solid (10.78 g), 69.5% yield, mp 120-2°C. 1H NMR (500 MHz, d6-DMSO): δ = 8.39 (s, 1H), 8.13 (t, J = 5.3 Hz, 1H), 8.05 (bs, 1H), 7.71 (bs, 1H), 7.28 (t, J = 7.4, 2H), 7.22 (d, J = 7.0, 2H), 7.17 (t, J = 7.4, 1H), 6.13 (t, J = 6.4 Hz, 1H), 5.22 (d, J = 4.3 Hz, 1H), 5.07 (t, J = 5.5 Hz, 1H), 4.26 (dt, J = 9.4, 4.1 Hz, 1H), 3.83 (dd, J = 7.8, 3.9 Hz, 1H), 3.66 (m, 1H), 3.58 (m, 1H), 3.19 (dd, J = 12.9, 6.7 Hz, 2H), 2.61 (t, J = 7.5 Hz, 2H), 2.19 (m, 2H), 1.78 (m, J = 7.4 Hz, 2H). 13C NMR (500 MHz, d6-DMSO): δ = 165.34 (1C), 163.56 (1C), 153.60 (1C), 143.53 (1C), 141.70 (1C), 128.38 (2C), 128.33 (2C), 125.78 (1C), 98.99 (1C), 87.63 (1C), 85.86 (1C), 69.82 (1C), 60.96 (1C), 40.36 (1C), 38.58 (1C), 32.63 (1C), 32.63 (1C). MS m/z: calcd for C19H23N4O5, 387.42; found, 387.1 [M–H]−.
4-N-Acetyl-5-(N-3-phenylpropyl)carboxamide-2′-deoxycytidine (6c). A solution of 5c (10.8 g, 28 mmol) in anh.THF (100 mL) was stirred and treated dropwise with acetic anhydride (3 eq). The solution was stirred for 18 hours at room temperature affording a thin suspension. The mixture was slowly diluted by dropwise addition of diisopropyl ether (35 mL). The solids were isolated by filtration and dried in vacuo to afford 6c as a white solid (8.44 g), 70.5% yield, mp 128–9°C. 1H NMR (400 MHz, d6-DMSO): δ = 11.34 (s, 1H), 8.69 (s, 1H), 8.41 (t, J = 5.2 Hz, 1H), 7.23 (m, 5H), 6.09 (t, J = 6.0 Hz, 1H), 5.15 (bs, 2H), 4.27 (m, 1H), 3.90 (dd, J = 9.6, 3.8 Hz,1H), 3.68 (m, 1H), 3.59 (m, 1H), 3.21 (dd, J = 12.3, 7.0 Hz 2H), 2.62 (m, 2H), 2.40 (s, 3H), 2.33 (m, 1H), 2.21 (m, 1H), 1.79 (m, 2H). 13C NMR (400 MHz, d6-DMSO): δ = 171.21 (1C), 165.34(1C), 159.73 (1C), 153.21 (1C), 146.01 (1C), 142.08 (1C), 128.80 (2C), 128.75 (2C), 126.22 (1C), 99.14 (1C), 88.61 (1C), 87.41 (1C), 69.88 (1C), 61.05 (1C), 41.04 (1C), 39.22 (1C), 33.01 (1C), 30.97 (1C), 26.67 (1C). MS m/z: calcd for C21H25N4O6, 429.45; found, 429.1 [M–H]−.
5′-O-(4,4′-Dimethoxytrityl)-4-N-acetyl-5-(N-3-phenylpropyl)carboxamide-2′-deox-ycytidine (7c). Prepared from 6c, as described for 7a, as a white solid foam, 64.6% yield, mp 92–5°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.38 (s, 1H), 8.56 (t, J = 5.2 Hz, 1H), 8.37 (s, 1H), 7.35 (d, J = 7.4 Hz, 2H), 7.21 (m, 12H), 6.80 (m, 4H), 6.11 (t, J = 6.0 Hz, 1H), 5.32 (d, J = 4.8 Hz, 1H), 4.16 (dt, J = 10.8, 4.7 Hz, 1H), 4.04 (m, 1H), 3.70 (d, J = 2.2 Hz, 6H), 3.26 (dd, J = 10.6, 6.1 Hz, 1H), 3.21 (dd, J = 10.5, 3.3 Hz, 1H), 3.03 (m, 1H), 2.95 (m, 1H), 2.49 (s, 2H), 2.43 (s, 3H), 2.39 (m, 1H), 2.23 (m, 1H), 1.57 (m, 2H). 13C NMR (500 MHz, d6-DMSO): δ = 170.77 (1C), 164.81 (1C), 159.41 (1C), 158.07 (1C), 158.06 (1C), 152.69 (1C), 145.18 (1C), 144.81 (1C), 141.53 (1C), 135.51 (1C), 135.50 (1C), 129.70 (2C), 129.61 (2C), 128.32 (2C), 128.29 (2C), 127.81 (2C), 127.66 (2C), 126.66 (1C), 125.79 (1C), 113.13 (4C), 100.37 (1C), 87.47 (1C), 86.40 (1C), 85.72 (1C), 70.03 (1C), 59.80 (1C), 54.99 (1C), 54.97 (1C), 40.48 (1C), 38.92 (1C), 32.64 (1C) 32.29 (1C), 26.27 (1C). MS m/z: calcd for C42H43N4O8, 731.83; found, 731.2 [M–H]−.
5′-O-(4,4′-Dimethoxytrityl)-4-N-acetyl-5-(N-3-phenylpropylcarboxamide)-2′deoxy-cytidine-3′-O-(N,N-diisopropyl-O-2-cyanoethylphosphoramidite) (8c). Prepared from 7c, as described for 8a, A 500 mL round bottom flask containing 7c (7.11g, 9.70 mmol) under an argon atmosphere was charged with anh. dichloromethane (97 mL). Anhydrous N,N-diisopropylethylamine (3.4 mL, 19.4 mmol) was added to the flask, and mixture was chilled to 0°C while stirring. Over the course of a half hour, 2-cyanoethyldiisopropyl chlorophosphoramidite (2.6 mL, 11.6 mmol) was added dropwise to the rapidly stirring mixture. The mixture was allowed to slowly warm to room temperature while it stirred. After 17 hours, the reaction was sampled for TLC, which showed that the reaction was complete (eluent: 75% ethyl acetate/25% hexanes (v/v), Rf(7c) = 0.10, Rf(8c) = 0.46/0.56 [two isomers]). The reaction mixture was transferred to a 250 mL separatory funnel using toluene and quenched by washing with cold, argon sparged 2% sodium bicarbonate solution (2×, 400 mL/ wash). The organic layer was collected and evaporated until the majority of the dichloromethane had been removed. The organic layer was returned to the separatory funnel with chilled argon-sparged toluene and washed with chilled argon-sparged deionized water (2×, 400 mL/wash). The organic layer was then diluted with chilled argon-sparged ethyl acetate and washed with brine (1×, 400 mL). The organic layer was collected, dried over sodium sulfate, filtered and evaporated. The worked-up reaction mixture was dissolved with dichloromethane and loaded onto a pre-conditioned column and eluted with chilled, argon-sparged mobile phase (80% ethyl acetate/20% hexanes) and product containing fractions were collected in sealed argon-purged bottles to limit product contact with air. Product containing fractions were evaporated at <40°C to yield a white foam (6.16 g), 68.0% yield, mp 69–79°C. 1H NMR (500 MHz, d6-DMSO): δ = 11.40 (s, 1H), 8.56 (m, 1H), 8.47/8.43 (s, 1H), 7.35 (m, 2H), 7.20 (m, 12H), 6.80 (m, 4H), 6.08 (m, 1H), 4.40 (m, 1H), 4.18 (m, 1H), 3.70 (m, 8H), 3.53 (m, 2H), 3.30 (m, 2H), 3.00 (m, 2H), 2.75 (t, J = 5.9 Hz, 1H), 2.63 (t, J = 5.9 Hz, 1H), 2.56 (m, 1H), 2.50 (m, 2H), 2.40 (m, 4H), 1.59 (m, 2H), 1.11 (m, 9H), 0.97 (d, J = 6.8 Hz, 3H). 31P NMR (500 MHz, d6-DMSO): δ = 147.60/147.43 (s, 1P) MS m/z: calcd for C51H60N6O9P, 932.05; found, 931.4 [M–H]−.
5-(N-Benzylcarboxamide)-2′-O-methyl-cytidine (13). Prepared from 5-iodo-2′-O-methylcytidine as described for 5a, except that the product was crystallized from hot 2-propanol (12 mL/g) affording 13 as a felty white solid, 79% yield. 1H NMR (500 MHz, d6-DMSO): δ = 8.57 (t, J = 5.9 Hz, 1H), 8.57 (s, 1H), 8.05 (bs, 1H), 7.85 (bs, 1H), 7.33 (m, 4H), 7.25 (m, 1H), 5.85 (d, J = 3.1 Hz, 1H), 5.27 (t, J = 5.4 Hz, 1H), 5.11 (d, J = 6.8 Hz, 1H), 4.43 (dd, J = 15.4, 10.4 Hz, 1H), 4.40 (dd, J =15.3, 10.4 Hz, 1H), 4.17 (dd, J = 6.7, 5.2 Hz, 1H), 3.87 (dt, J = 6.8, 3.2 Hz,1H), 3.80 (dd, J = 5.0, 3.2 Hz,1H), 3.77 (m, 1H), 3.61 (ddd, J = 12.2, 5.3, 3.4 Hz, 1H), 3.44 (s, 3H). 13C NMR (500 MHz, d6-DMSO): δ = 165.43 (1C), 163.57 (1C), 153.49 (1C), 143.99 (1C), 139.16 (1C), 128.40 (2C), 127.22 (2C), 126.90 (1C), 98.97 (1C), 87.95 (1C), 84.28 (1C), 83.05 (1C), 67.67 (1C), 59.92 (1C), 57.70 (1C), 42.40 (1C). MS m/z: calcd for C18H21N4O6, 389.39; found, 389.1 [M–H]−.
5-(N-3-Phenylpropyl)-2′-deoxy-2′-fluoro-cytidine (14). Prepared from 5-iodo-2′-deoxy-2′-fluoro-cytidine as described for 5c, as felty white solid (53% yield). 1H NMR (500 MHz, d6-DMSO): δ = 8.52 (s, 1H), 8.07 (bs, 1H), 7.95 (t, J = 5.4 Hz,1H), 7.85 (bs, 1H), 7.22 (t, J = 7.4, 5H), 5.91 (d, J = 17.6 Hz, 1H), 5.58 (d, J = 6.6 Hz, 1H), 5.32 (t, J = 5.3 Hz, 1H), 4.99 (dd, J = 53.2, 3.9 Hz, 1H), 4.27 (m, 1H), 3.92 (d, J = 8.3 Hz,1H), 3.86 (m, 1H), 3.58 (ddd, J =12.5, 5.4, 2.9 Hz, 1H), 3.19 (dd, J = 12.7, 5.3 Hz, 2H), 2.61 (t, J = 7.5 Hz, 2H), 1.78 (m, J = 7.3 Hz, 2H). 13C NMR (500 MHz, d6-DMSO): δ = 165.22 (s, 1C), 163.74 (s, 1C), 153.34 (s, 1C), 143.82 (s, 1C), 141.73 (s, 1C), 128.42 (s, 2C), 128.35 (s, 2C), 125.81 (s, 1C), 99.26 (1C), 94.02 (d, J = 736.6 Hz, 1C), 88.65 (d, J = 134.7 Hz, 1C), 83.07 (s, 1C), 67.00 (d, J = 65 Hz, 1C), 59.24 (s, 1C), 38.65 (s, 1C), 32.64 (s, 1C), 30.73 (s, 1C). 19F NMR (400 MHz, d6-DMSO): δ = −200.82 (ddd, J = 19.0, 6.2, 56.6 Hz, 1F). MS m/z: calcd for C19H22N4O5, 405.41; found, 405.1 [M–H]−.
Solid-Phase Oligonucleotide Synthesis
An ABI 3900 automated DNA synthesizer (Applied Biosystems, Foster City, CA) was used with conventional phosphoramidite methods[ 22 ] with minor changes to the coupling conditions for modified phosphoramidites 8a–c (Table 4). Reagent 8a was used as a 0.1 M solution in dichloromethane/acetonitrile (1/1) and reagents 8b and 8c were used as 0.1 M solutions in acetonitrile. Solid support was an ABI style fritted column packed with controlled pore glass (CPG, Prime Synthesis, Aston, PA) loaded with 3′-DMT-dT succinate with 1000 Å pore size. Deprotection was accomplished by treatment with 20% diethylamine in acetonitrile[ 23 ] followed by gaseous methylamine cleavage and deprotection for 2 hours at 35°C.[ 24 , 25 ] Identity and percent full length (%FL) product were determined on an Agilent 1290 Infinity with an Agilent 6130 Quadrupole mass spectrometry detector using an Acquity OST C18 column 1.7 μm, 2.1 × 100 mm (Waters Corp., Milford, MA), using a gradient of 0 to 25 percent B in 11 minutes (Buffer A: 100 mM 1,1,1,3,3,3-hexafluoro-2-propanol, 8.6 mM triethylamine, pH 8.25; Buffer B: 10% Buffer A in 90 % acetonitrile).[ 26 ]
Synthesis of 5′-O-Triphosphate Reagents 10a–c
The Ludwig-Eckstein process[ 20 ] was used to convert the 3′-O-acetyl-protected intermediates 9a–c into crude 5′-O-triphosphates 10a–c. A two-stage preparative HPLC purification was used for these chemically-modified nucleotides.
3′-O-Acetyl-5-(N-1-benzylcarboxamide)-2′-deoxycytidine (9a). An argon purged 50 mL round bottom flask was charged with 7a (900 mg, 1.35 mmol), anh. pyridine (9 mL) and acetic anhydride (0.63 mL, 6.75 mmol). After 18 hours at room temperature, the solvent was evaporated in vacuo (1 mm, 30°C) to yield a tan foam which was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (9 mL) and heated at 50°C under argon. After 6 hours, the reaction mixture was poured into a rapidly stirring mixture of methanol (15 mL) and toluene (10 mL). The resulting orange solution was concentrated (1 mm, 30°C) to yield a red oily residue which, upon mixing with ethyl acetate (6 mL), gave a crystalline slurry. Crystallization was further enhanced with the addition of hexanes (1 mL). The mixture was stirred overnight and filtered, washing the filter cake with 50:50 ethyl acetate: hexanes. The product 9a was isolated, after drying, as a pale gray solid (405 mg), 75% yield, mp 193–4°C. 1H NMR (500 MHz, d6-DMSO): δ = 8.82 (t, J = 5.8 Hz, 1H), 8.41 (s, 1H), 8.09 (bs, 1H), 7.81 (bs, 1H), 7.33 (m, 4H), 7.25 (m, 1H), 6.17 (dd, J = 8.0, 6.0 Hz, 1H), 5.24 (dt, J = 6.2, 1.8 Hz, 1H), 5.13 (t, J = 5.6 Hz, 1H), 4.43 (dd, J = 15.4, 13.3 Hz, 1H), 4.19 (dd, J =15.3, 13.2 Hz, 1H), 4.06 (dd, J = 5.9, 3.7 Hz, 1H), 3.65 (m, 2H), 2.45 (m, 1H), 2.34 (ddd, J = 14.2, 5.9, 1.5 Hz, 1H), 2.07 (s, 3H). 13C NMR (500 MHz, d6-DMSO): δ = 170.05 (1C), 165.34 (1C), 163.54 (1C), 153.44 (1C), 143.94 (1C), 139.20 (1C), 128.34 (2C), 127.16 (2C), 126.85 (1C), 99.06 (1C), 85.96 (1C), 85.06 (1C), 74.63 (1C), 61.18 (1C), 42.28 (1C), 37.31 (1C), 20.85 (1C). MS m/z: calcd for C19H21N4O6, 401.40; found, 401.1 [M–H]−.
3′-O-Acetyl-5-(N-1-naphthylmethylcarboxamide)-2′-deoxycytidine (9b). Prepared from 7b, as described for 9a, as a pale gray crystalline solid, 54% yield, mp 222–4°C. 1H NMR (500 MHz, d6-DMSO): δ = 8.89 (t, J = 5.8 Hz,1H), 8.44 (s, 1H), 8.14 (d, J = 8.4 Hz, 1H), 8.11 (bs, 1H), 7.96 (dd, J = 8.6, 1.3 Hz, 1H), 7.86 (dd, J = 6.6, 2.8 Hz, 1H), 7.84 (bs, 1H), 7.57 (m, 2H), 7.49 (m, 2H), 6.15 (dd, J = 8.2, 6.0 Hz, 1H), 5.23 (dt, J = 6.2, 1.9 Hz, 1H), 5.13 (t, J = 5.8 Hz, 1H), 4.94 (dd, J = 15.5, 5.8 Hz, 1H), 4.86 (dd, J = 15.7, 5.4 Hz, 1H), 4.05 (dt, J = 8.1, 1.8 Hz, 1H), 3.62 (m, 2H), 2.45 (m, 1H), 2.33 (ddd, J = 12.6, 6.1, 1.8 Hz, 1H), 2.06 (s, 3H). 13C NMR (500 MHz, d6-DMSO): δ = 170.51 (1C), 165.78 (1C), 164.02 (1C), 153.90 (1C), 144.52 (1C), 134.57 (1C), 133.74 (1C), 131.27 (1C), 129.02 (1C), 128.03 (1C), 126.80 (1C), 126.31 (1C), 125.94 (1C), 125.54 (1C), 123.78 (1C), 99.52 (1C), 86.60 (1C), 85.55 (1C), 70.12 (1C), 61.67 (1C), 40.80 (1C), 37.75 (1C), 21.32 (1C). MS m/z: calcd for C23H23N4O6, 451.46; found, 451.1 [M–H]−.
3′-O-Acetyl-5-(N-3-phenylpropylcarboxamide)-2′-deoxycytidine (9c). Prepared from 7c, as described for 9a, as a white crystalline solid, 74% yield, mp 186–9°C. 1H NMR (500 MHz, d6-DMSO): δ = 8.37 (s, 1H), 8.26 (t, J = 5.3 Hz, 1H), 8.06 (bs, 1H), 7.79 (bs, 1H), 7.23 (m, 5H), 6.16 (dd, J = 7.9, 6.1 Hz, 1H), 5.24 (dt, J =4.2, 2.1 Hz, 1H), 5.18 (t, J = 5.6 Hz, 1H), 4.06 (dd, J = 5.7, 3.6 Hz, 1H), 3.65 (m, 2H), 3.19 (dd, J = 12.8, 6.2 Hz, 2H), 2.62 (t, J = 7.5 Hz, 2H), 2.43 (m, 1H), 2.34 (m,1H), 2.06 (s, 3H), 1.78 (m, J = 7.4 Hz, 2H). 13C NMR (500 MHz, d6-DMSO): δ = 170.52 (1C), 165.68 (1C), 164.00 (1C), 153.94 (1C), 144.09 (1C), 142.13 (1C), 128.80 (2C), 127.75 (2C), 126.21 (1C), 99.80 (1C), 86.42 (1C), 85.06 (1C), 75.07 (1C), 61.63 (1C), 39.12(1C), 37.91 (1C), 33.04 (1C), 31.18 (1C), 21.31 (1C). MS m/z: calcd for C21H25N4O6, 429.45; found, 429.1 [M–H]−.
Nucleoside Triphosphate Purification by Two-Stage Preparative HPLC
The crude triphosphates 10a–c were purified via two orthogonal preparative HPLC techniques: anion exchange chromatography to separate the nucleoside triphosphate from other nucleoside by-products (such as diphosphate and monophosphate) and reversed-phase chromatography to remove residual by-products of reaction reagents.
Anion exchange chromatography was performed in two injections for each 0.5 mmol reaction using an HPLC column packed with Source 15Q resin, eluting with a linear elution gradient of two triethylammonium bicarbonate buffers (Table 1). The desired triphosphate was usually the final material to elute from the column, as a broad peak with 10–12 minutes’ width on the HPLC chromatogram. Fractions were analyzed and product-containing fractions were combined and evaporated in a Genevac HT4 series II evaporator to produce a colorless to light tan resin. This was reconstituted in deionized water and applied in a single injection for reversed phase purification on a Novapak HRC18 prep column eluting with a linear gradient of acetonitrile in triethylammonium acetate buffer (Table 2). Fractions containing pure triphosphate were combined and evaporated to produce a colorless to light tan resin.
Table 1 . Anion-exchange (AEX) purification conditions for modified nucleotide triphosphates.
| HPLC system | Waters 625HPLC/486 detector @ 240/278 nm |
| Column | Source 15Q 196 mL (GE Healthcare PN: 17—1947-05) |
| Mobile Phase | A: 10 mM Triethylammonium bicarbonate/10% Acetonitrile, B: 1 M Triethylammonium bicarbonate/10% Acetonitrile |
| Gradient (% Buffer B) | 5%–70% |
| Run Time; flow rate; fraction size | 50 minutes; 35 mL/minute; 50 mL |
| Analytical Column | Dionex DNA-Pac PA100 column (Thermo Scientific, PN: 043010) |
Table 2 . Reversed-phase (RP) HPLC purification conditions for modified nucleotide triphosphates.
| HPLC system | Waters 625HPLC/486 detector @ 240 nm |
| Column | Waters Novapak HRC18, 19 mm × 300 mm (PN WAT025822) |
| Mobile Phase | A: 100 mM Triethylammonium acetate B: 100% Acetonitrile |
| Gradient (% Buffer B) | 0%–50% |
| Run Time; flow rate; fraction size | 30 minutes; 12 mL/minute; 25 mL |
| Analytical Column | Waters Symmetry column (PN: WAT054215) |
Final pure triphosphates 10a–c were reconstituted in deionized water for analysis and quantitated using a Hewlett Packard 8452A Diode Array Spectrophotometer at 240 nm (Table 3).
Table 5 . Synthetic DNA sequences incorporating 2′-deoxycytidines-5-carboxamides.
| Sequence |
LC/MS Data |
||||
|---|---|---|---|---|---|
| Cytidine Mod/ | HPLC Data | FL Expected | FL Observed | ||
| (Phosphoramidite) | Xn n = | %FL | Mass (amu) | Mass (amu) | Δ (amu) |
| Benzyl/8a | 0 | 65 | 10533.8 | 10531.2 | 2.6 |
| 1 | 60 | 10955.1 | 10953.6 | 1.5 | |
| 2 | 64 | 11376.4 | 11375.4 | 1.0 | |
| 3 | 65 | 11797.7 | 11797.9 | 0.2 | |
| 4 | 52 | 12219.0 | 12220.2 | 1.2 | |
| 5 | 52 | 12640.4 | 12642.5 | 2.1 | |
| 1-Naphthylmethyl/8b | 0 | 60 | 10533.8 | 10531.3 | 2.5 |
| 1 | 67 | 11005.1 | 11003.3 | 1.7 | |
| 2 | 64 | 11476.4 | 11475.8 | 0.6 | |
| 3 | 54 | 11947.7 | 11948.2 | 0.5 | |
| 4 | 47 | 12419.0 | 12420.2 | 1.2 | |
| 5 | 49 | 12890.3 | 12892.9 | 2.6 | |
| 3-Phenylpropyl/8c | 0 | 69 | 10533.8 | 10531.3 | 2.4 |
| 1 | 59 | 10983.1 | 10981.3 | 1.8 | |
| 2 | 68 | 11432.5 | 11431.6 | 0.9 | |
| 3 | 41 | 11881.9 | 11882.0 | 0.1 | |
| 4 | 42 | 12331.2 | 12332.2 | 1.0 | |
| 5 | 49 | 12780.6 | 12782.8 | 2.2 | |
Table 3 . 2′-Deoxycytidine-5′-O-triphosphate yields and purities.
| Extinction | Yield | Yield | Purity: | Purity: | |
|---|---|---|---|---|---|
| Triphosphate | Coefficient (ref 9) | μmoles | Percent | Analytical AEX | Analytical RP-HPLC |
| 10a | 13,700 cm−1 M−1 | 43 | 9% | No data | 92.6% |
| 10b | 20,000 cm−1 M−1 | 121 | 24% | 95.5% | 98.2% |
| 10c | 13,700 cm−1 M−1 | 92 | 18% | 98.3% | 98.2% |
Table 4 . ABI 3900 coupling cycle parameters for oligonucleotide synthesis (50 nmol scale).
| Step | Operation | Purpose | Reps | Reagent | Volume, μL | Wait, sec |
|---|---|---|---|---|---|---|
| Pre | Prep Support | wash | 3 | ACN | 200 | 0 |
| Prep Support | detritylation | 2 | Deblock | 50 | 0 | |
| 1 | Coupling cycle | detritylation | 3 | Deblock | 50 | 3 |
| 2 | Coupling cycle | wash | 1 | ACN | 195 | 0 |
| 3 (ATG) | Coupling cycle | coupling | 2 | Activator, amidite | 36+19 | 30+175 |
| 3 (8a–c) | Coupling cycle | coupling | 3 | Activator, amidite | 36+19 | 60+250 |
| 4 | Coupling cycle | capping | 1 | Cap A, B | 15+15 | 5 |
| 5 | Coupling cycle | oxidation | 1 | oxidizer | 35 | 3 |
| 8 | Coupling cycle | wash | 1 | ACN | 190 | 0 |
| post | Finalize Oligo | detritylation | 2 | Deblock | 140 | 0 |
| Finalize Oligo | wash | 4 | ACN | 199 | 0 | |
| Finalize Oligo | dry support | 1 | ACN; Ar | 199 | 0 |
Key:ACN AcetonitrileDeblock 10% Dichloroacetic acid in tolueneActivator 0.3 M 5-Benzylmercaptotetrazole and 0.5% N-methylimidazole in ACNOxidizer 0.025 M Iodine in 44.9% ACN/45% pyridine/10.1% waterCap A 10% Acetic Anhydride in pyridine and tetrahydrofuranCap B 16% 1-Methylimidazole in tetrahydrofuranAr Dry argon flush for 20 s
5-(N-1-Benzylcarboxamide)-2′-deoxycytidine-5′-O-triphosphate (10a). 1H NMR (300 MHz, D2O): δ = 8.45 (s, 1H), 7.25 (m, 5H), 6.14 (t, J = 6.9 Hz, 1H), 4.57 (m, J = 2.9 Hz, 1H), 4.43 (dd, J = 20.2, 15.4 Hz, 2H), 4.17 (m, 3H), 2.39 (m, 1H), 2.27 (m, 1H). 31P NMR (300 MHz, D2O): δ = −9.96 (d, J = 50.0 Hz, 1P), −11.43 (d, J = 50.8 Hz, 1P), −23.24(t, J = 50.5Hz, 1P MS m/z: calcd for C17H21N4O14P3, 599.04; found, 599.1 [M–H]−.
5-(N-1-Naphthylmethylcarboxamide)-2′-deoxycytidine-5′-O-triphosphate (10b): 1H NMR (500 MHz, D2O): δ = 8.12 (s, 1H), 7.98 (d, J = 8.5 Hz, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.58 (m, 1H), 7.40 (m, 1H), 7.33 (m, 1H), 7.24 (m, 1H), 5.87 (t, J = 6.7 Hz, 1H), 4.66 (d, J = 8.1 Hz, 2H), 4.40 (m, J = 3.0 Hz, 1H), 4.04 (m, 3H), 2.21 (ddd, J = 14.1, 6.0, 3.4 Hz, 1H), 2.06 (m, 1H). 13C NMR (500 MHz, D2O): δ = 165.95 (s, 1C), 163.15 (s, 1C), 155.22 (s, 1C), 143.33 (s, 1C), 133.35 (s, 1C), 133.17 (s, 2C), 130.55 (s, 2C), 128.66 (s, 1C), 127.84 (s, 1C), 126.65 (s, 1C), 126.13 (s, 1C), 125.64 (s, 1C), 125.12 (s, 1C), 123.11 (s, 1C), 100.55 (s, 1C), 86.88 (s, 1C), 85.87 (d, J = 55.95 Hz, 1C), 70.76 (s, 1C), 65.38 (d, J = 36 Hz, 1C), 41.19 (s, 1C), 39.61 (m, 1C). 31P NMR (500 MHz, D2O): δ = −10.99 (d, J = 82.4 Hz, 1P), −11.61 (d, J = 84.9 Hz, 1P), −23.47 (t, J = 83.5 Hz, 1P). MS m/z: calcd for C21H24N4O14P3, 649.36; found, 649.0 [M–H]−.
5-(N-3-Phenylpropylcarboxamide)-2′-deoxycytidine-5′-O-triphosphate (10c): 1H NMR (500 MHz, D2O): δ = 8.07 (s, 1H), 7.11 (m, 4H), 6.98 (m, 1H), 6.00 (t, J = 6.5 Hz, 1H), 4.44 (m, J = 3.0 Hz, 1H), 4.06 (m, 3H), 3.21 (m, 1H), 3.13 (m, 1H), 2.50 (t, J = 7.5 Hz, 2H), 3.13 (ddd, J = 14.1, 10.9, 3.1 Hz, 1H), 2.13 (m, 1H), 1.76 (m, 2H). 13C NMR (500 MHz, D2O): δ = 165.85 (s, 1C), 163.50 (s, 1C), 155.73 (s, 1C), 142.94 (s, 1C), 142.40 (s, 1C), 128.55 (s, 2C), 128.40 (s, 2C), 125.72 (s, 1C), 101.15 (s, 1C), 86.93 (s, 1C), 85.96 (d, J = 55.2 Hz, 1C), 70.90 (s, 1C), 65.38 (d, J = 37.6 Hz, 1C), 39.88 (s, 1C), 39.55 (s, 1C), 32.74 (s, 1C), 26.68 (s, 1C). 31P NMR (500 MHz, D2O): δ = −11.00 (d, J = 82.7 Hz, 1P), −11.09 (d, J = 85.7 Hz, 1P), −23.53 (t, J = 84.3 Hz, 1P). MS m/z: calcd for C19H25N4O14P3, 627.35; found, 627.0 [M–H]−.
Primer Extension Assay
The modified nucleoside triphosphates were evaluated as substrates for KOD exonuclease-minus DNA polymerase in a primer extension assay using a standard template that contained all possible triple nucleotide combinations. The template sequence (TriLink Biotechnologies, San Diego, CA 92121, USA) was:
5′-TTTTTTTTCTTCTTCTCCTTTCTCTTCCCAAAATCACACGGACCCAGGGCATTCTAGATATGGTTTACGCTCAAGCGAACTTGCCGTCCTGAGTGTAAAGAGGGAAAgagggcagggtgtggcatatatat-3′
The primer sequence (IDT Technologies, Coralville, IA 52241, USA) was:
5′-atatatatgccacaccctgccctc-3′
In brief, 10 pmoles of primer were labeled with 10 pmoles of 32P-ATP at 37°C for 30 minutes with 3′ phosphatase minus T4 polynucleotide Kinase (New England Biolabs, Ipswich, MA 01938, USA) in 7 mM Tris-HCl, pH 7.6 @ 25°C, 10 mM MgCl2, 5 mM dithiothreitol, and purified by passage through two Sephadex G-50 cartridges (GE Healthcare). The 30 μL primer extension reactions contained 120 mM Tris-HCl, pH 7.8, 10 mM KCl, 7 mM MgSO4, 6 mM (NH4)2SO4, 0.001 % BSA, 0.1% Triton X-100, 3 pmoles of template, 6 pmoles of primer, and 7.5 Units of KOD exonuclease-minus DNA polymerase (EMD Novagen). The reactions were incubated at 96°C for 30 seconds and 65°C for 1 hour in a 96-well plate in an MJ thermocycler (Bio-Rad, Pleasanton, CA 94566, USA).
5 μL samples were analyzed on 8% acrylamide, 7 M urea, 1× TBE gels (Life Technologies, Grand Island, NY 14012, USA) and exposed for 1 hour on an imaging plate before scanning in a FujiFilm FLA3000 phosphorimager (GE Healthcare).
REFERENCES
- Gold L. Ayers D. Bertino J. Bock C. Bock A. Brody E.N. Carter J. Dalby A. Eaton B. Fitzwater T. Aptamer-based proteomic technology for biomarker discovery. PLoS ONE. 2010;5(12):e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein M. Synthesis of deoxynucleoside triphosphates that include proline, Urea, or sulfonamide groups and their polymerase incorporation into DNA. Chem., A Eur. J. 2012;18:13320–13330. doi: 10.1002/chem.201201662. [DOI] [PubMed] [Google Scholar]
- Imaizumi Y. Kasahara Y. Fujita H. Kitadume S. Ozaki H. Endoh T. Kuwahara M. Sugimoto N. Efficacy of base-modification on target binding of small molecule DNA aptamers. J. Am. Chem. Soc. 2013;135(25):9412–9419. doi: 10.1021/ja4012222. [DOI] [PubMed] [Google Scholar]
- Davies D.R. Gelinas A.D. Zhang C. Rohloff J.C. Carter J.D. O’Connell D. Waugh S.M. Wolk S.M. Mayfield W.S. Burgin A.B. Edwards T.E. Stewart L.J. Gold L. Janjic N. Jarvis T.C. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. PNAS. 2012;190(49):19971–19976. doi: 10.1073/pnas.1213933109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.Y. Kang H. Ryu S.H. Lee D.S. Lee J.H. Kim S. Bioimaging of nucleolin aptamer-containing 5-(N-benzylcarboxamide)-2′-deoxyuridine more capable of specific binding to targets in cancer cells. J. Biomed. Biotechnol. 2010 doi: 10.1155/2010/168306. article ID 168306, 9 pages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr C.E. Eaton B.E. Netzel T.L. Synthesis of N,N-dialkylaniline-2′-deoxyuridine conjugates for DNA-mediated electron transfer studies. Nucleos., Nucleot. Nucl. Acids. 2000;19(5&6):851–866. doi: 10.1080/15257770008033027. [DOI] [PubMed] [Google Scholar]
- Gaballah S.T. Kerr C.E. Eaton B.E. Netzel T.L. Synthesis of 5-(2,2′-Bipyridyl- and 2,2′-Bipyridinediiumyl)-2′-deoxyuridine Nucleosides: Precursors to Metallo-DNA Conjugates. Nucleos., Nucleot. Nucl. Acids. 2002;21(8&9):547–560. doi: 10.1081/NCN-120015068. [DOI] [PubMed] [Google Scholar]
- Vaught J.D. Dewey T. Eaton B.E. T7 RNA Polymerase Transcription with 5-Position Modified UTP Derivatives. J. Am. Chem. Soc. 2004;126:11231–11237. doi: 10.1021/ja049009h. [DOI] [PubMed] [Google Scholar]
- Vaught J.D. Bock C. Carter J. Fitzwater T. Otis M. Schneider D. Rolando J. Waugh S. Wilcox S.K. Eaton B.E. Expanding the chemistry of DNA for in vitro selection. J. Am. Chem. Soc. 2010;132(12):4141–4151. doi: 10.1021/ja908035g. [DOI] [PubMed] [Google Scholar]
- Tu C. Dewey T. Eaton B. Palladium Catalyzed Nucleoside Modification Methods Using Nucleophiles and Carbon Monoxide. US Patent. 1999;5:945. 527 (Aug. 31. [Google Scholar]
- Nomura Y. Ueno Y. Matsuda A. Site-specific introduction of functional groups into phosphodiester oligodeoxynucleotides and their thermal stability and nuclease-resistance properties. Nucleic Acids Res. 1997;25(14):2784–2791. doi: 10.1093/nar/25.14.2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura Y. Haginoya N. Ueno Y. Matsuda A. Nucleosides and Nucleotides. 161. Incorporation of 5-(N-aminoalkyl)carbamoyl-2′-deoxycytidines into oligodeoxynucleotides by a convenient post-synthetic modification method. Bioor. Med. Chem. Lett. 1996;6(23):2811–2816. [Google Scholar]
- Uozumi Y. Arii T. Watanabe T. Double Carbonylation of Aryl Iodides with Primary Amines under Atmospheric Pressure Conditions Using the Pd/Ph3P/DABCO/THF System. J. Org. Chem. 2001;66:52772–5274. doi: 10.1021/jo0156924. [DOI] [PubMed] [Google Scholar]
- Takacs A. Petz A. Kollar L. Palladium-catalyzed Aminocarbonylation of Iodoarenes and Iodoalkenes with Aminophosphonate as N-Nucleophile. Tetrahedron. 2008;64:8726–8730. [Google Scholar]
- Ross B.S. Han M. Ravikumar V.T. Efficient Large-Scale Synthesis of 5′-O-Dimethoxytrityl-N4-Benzoyl-5-methyl-2′-deoxycytidine. Nucleos., Nucleot. Nucl. Acids. 2006;25:765–770. doi: 10.1080/15257770600726059. [DOI] [PubMed] [Google Scholar]
- Sanghvi Y.S. Guo Z. Pfundheller H.M. Converso A. Improved Process for the Preparation of Nucleosidic Phosphoramidites Using a Safer and Cheaper Activator. Org. Process Res. Dev. 2000;4:175–181. [Google Scholar]
- Good results were also obtained with 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite and N,N,-diisopropylethylamine, see example compound 8c [Google Scholar]
- Still W.C. Kahn M. Mitra A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978;43:2923–2925. [Google Scholar]
- Leonard N.J. Neelima, N. 1,1,1,3,3,3-Hexafluoro-2-propanol for the Removal of the 4,4′-Dimethoxytrityl protecting group from the 5′-Hydroxyl of acid-Sensitive nucleosides and nucleotides. Tetrahedron Lett. 1995;36(43):7833–7836. [Google Scholar]
- Ludwig J. Eckstein F. Rapid and efficient synthesis of nucleoside 5′-O-(1-Thiotriphosphates), 5′-triphosphates and 2′,3′-Cyclophosphorothioates using 2-Chloro-4H-1,3,2-benzodioxaphophorin-4-one. J. Org. Chem. 1989;54:631–635. [Google Scholar]
- Ito T. Ueno Y. Komatsu Y. Matsuda A. Synthesis, thermal stability and resistance to enzymatic hydrolysis of the oligonucleotides containing 5-(N-aminohexyl)carbamoyl-2′-O-methyluridines. Nucleic Acids Res. 2003;31(10):2514–2523. doi: 10.1093/nar/gkg374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Review Beaucage S.L. Iyer R.P. Advances in the synthesis of oligonucleotides by the phosphoramidite approach. Tetrahedron. 1992;48(12):2223–2311. [Google Scholar]
- Hsiung H.M. Process for de-cyanoethylating blocked nucleotides. US Patent 4,426,517 (Jan. 17, 1984) and references cited therein. [Google Scholar]
- Kempe T. Recovery of oligonucleotides by gas phase cleavage. US Patent. 1996;5:514. 789 (May 7. [Google Scholar]
- Iyer R.P. Yu D. Xie J. Zhou W. Agrwal S. The use of gaseous ammonia for the deprotection and cleavage steps during the solid-phase synthesis of oligonucleotides and analogs. Bioor. Med. Chem. Lett. 1997;7(11):1443–1448. [Google Scholar]
- Gilar M. Analysis and purification of synthetic oligonucleotides by reversed-phase high-performance liquid chromatography with photodiode array and mass spectrometry detection. Anal. Biochem. 2001;298:196–206. doi: 10.1006/abio.2001.5386. [DOI] [PubMed] [Google Scholar]