

Abstract
The Sinorhizobium meliloti periplasmic ExoR protein and the ExoS/ChvI two-component system form a regulatory mechanism that directly controls the transformation of free-living to host-invading cells. In the absence of crystal structures, understanding the molecular mechanism of interaction between ExoR and the ExoS sensor, which is believed to drive the key regulatory step in the invasion process, remains a major challenge. In this study, we present a theoretical structural model of the active form of ExoR protein, ExoRm, generated using computational methods. Our model suggests that ExoR possesses a super-helical fold comprising 12 α-helices forming six Sel1-like repeats, including two that were unidentified in previous studies. This fold is highly conducive to mediating protein–protein interactions and this is corroborated by the identification of putative protein binding sites on the surface of the ExoRm protein. Our studies reveal two novel insights: (a) an extended conformation of the third Sel1-like repeat that might be important for ExoR regulatory function and (b) a buried proteolytic site that implies a unique proteolytic mechanism. This study provides new and interesting insights into the structure of S. meliloti ExoR, lays the groundwork for elaborating the molecular mechanism of ExoRm cleavage, ExoRm–ExoS interactions, and studies of ExoR homologs in other bacterial host interactions.
Keywords: ExoR, Sinorhizobium meliloti Rm1021, sel1-like repeats, superhelical fold, molecular modeling, computational analyses, ExoS
Introduction
Most bacteria, including parasitic and symbiotic species, rely on two-component signal transduction systems for detecting and adapting to changes in their environment.1 A group of Gram-negative bacteria rely on the ExoR-ExoS/ChvI (RSI) pathway, to transit from a free-living to host-invading form.2 This pathway is best understood in Sinorhizobium meliloti, the model organism for bacterium–plant symbiosis. ExoS and ChvI form a typical two-component system that consists of a membrane-integral histidine kinase, ExoS, and an associated cytoplasmic response regulator, ChvI.3 In S. meliloti, the activities of the ExoS/ChvI system are regulated by a periplasmic regulatory protein, ExoR, through a direct interaction with ExoS.4 The current model for the ExoR-ExoS/ChvI pathway suggests that ExoS/ChvI system is turned off when the periplasmic domain of ExoS is in a protein complex with the mature periplasmic form of ExoR, ExoRm.5,6 In the ExoRm–ExoS complex, ExoS acts as a phosphatase and keeps ChvI dephosphorylated and inactive. When the ExoRm–ExoS interaction is disrupted through the proteolytic cleavage of ExoRm, ExoS becomes an active kinase, and phosphorylates ChvI directly,3,6 resulting in upregulation of succinoglycan biosynthesis and repression of flagellum biosynthesis, allowing the cells to switch from a free-living to host-invading form.2,4–6
Even though ExoR has been established as a key regulator of the RSI pathway,4–7 its tertiary structure, including structural details and associated functions, remains unknown. Delineating the structure–function correlations of the ExoR protein is critical to understanding the cleavage mechanism of ExoRm, ExoRm–ExoS interactions, and other aspects of its regulatory role in the RSI pathway.5,6 Comparative modeling methods have been successfully applied in revealing key structural details, structure–function relationships, and interactions with putative binding partners, including those of helical repeat proteins (e.g., repeats of proteasome-binding protein PA2008 and repeats of transcriptional activator-like [TAL] effectors),9 confirmed by subsequent crystal structures of TAL effector–DNA complexes.10 In this study, we present a robust theoretical model of the active form of ExoR protein, ExoRm, generated by means of the well-established approach of template-based modeling.11 We propose that the ExoR protein adopts an alpha–alpha superhelical fold, an inherently flexible fold designed for protein–protein interactions.12,13 In the absence of any structural representations of the ExoR family of proteins in the structural database, our study provides a first look into the structure of ExoR and its implications to ExoR function.
Results
S. meliloti Rm1021 ExoR protein is composed of six sel1-like repeats
In addition to the four Sel1-like repeats already identified in ExoR,14 our analysis of the ExoR sequence with the latest repeat detection methods reveal two additional repeats at the C terminus of ExoR, corroborated by secondary structure prediction (Supporting Information Table SI; Supporting Information Fig. S1; Fig. 1). The length of each of the six repeats ranges from 32 to 40 residues and consists of two α-helices; this repeat structure is in agreement with the expected configuration of Sel1-like repeats.12,14 The alignment of all six repeats reveals expected conserved alanine and glycine residues based on the Sel1-like repeat consensus except for ExoR6 that maintains only three out of seven conserved structural residues (Fig. 2).12,14
Figure 1.
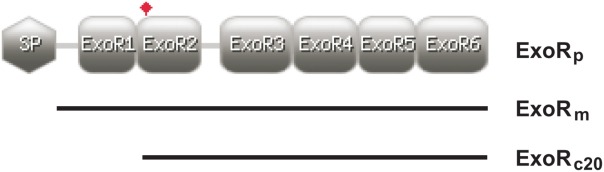
Schematic representation of the three forms of the ExoR protein (ExoRp, ExoRm, and ExoRc20). The cartoon shows the location of signal peptide (SP), six putative Sel1-like repeats (ExoR1–ExoR6), and the cleavage site of ExoR, A80 (flag-tagged).
Figure 2.

(a) Alignment of repeats found in the ExoR protein of Sinorhizobium meliloti Rm1021. Residues located in α-helices are shaded gray. The cleavage site (A80) is indicated in green. Proline residues in the ExoR3 loop 1 and helix A of ExoR6 are colored blue. (b) Sequence alignment of the ExoR C termini from 28 ExoR orthologs. The alignment is based on ExoR6 from S. meliloti Rm1021 and corresponds to residues 229–268 of the full-length ExoR that includes ExoR6 (black) and the ExoR Asp3 tag (gray). The following species are shown: S. meliloti Rm1021 (S.m.), Sinorhizobium medicae WSM419 (S.m.w.), Sinorhizobium fredii NGR234 (S.f.), Rhizobium leguminosarum bv. trifolii WSM1325 (R.l.1325), Rhizobium leguminosarum bv. trifolii WSM2304 (R.l.2304), Rhizobium etli CIAT 652 (R.e.c.), Rhizobium etli Kim 5 (R.e.k.), Agrobacterium tumefaciens str. C58 (A.t.), Agrobacterium vitis S4 (A.v.), Agrobacterium radiobacter K84 (A.r.), Bartonella grahamii as4aup (B.g.), Bartonella henselae str. Houston-1 (B.h.), Bartonella tribocorum CIP 105476 (B.t.), Bartonella schoenbuchensis R1 (B.s.), Ochrobactrum anthropi ATCC 49188 (O.a.), Ochrobactrum intermedium LMG 3301 (O.i.), Brucella abortus str. 2308 A (B.a.), Hoeflea phototrophica DFL-43 (H.p.), Chelativorans sp. BNC1 (C.sp.), Starkeya novella DSM 506 (S.n.), Beijerinckia indica subsp. indica ATCC 9039 (B.i.), Rhodopseudomonas palustris CGA009 (R.p.), Bradyrhizobiaceae bacterium SG-6C (B.b.), Bradyrhizobium japonicum USDA 110 (B.j.), Bradyrhizobium sp. BTAi1 (B.sp.), Mesorhizobium ciceri biovar biserrulae WSM1271 (M.c.), Mesorhizobium loti MAFF303099 (M.l.), and Mesorhizobium opportunistum WSM2075 (M.o.). The consensus sequences above the alignments reflect conservation of at least 80% of the first five repeats (ExoR1–ExoR5) (a) and of the 28 ExoR orthologs (b); numbering indicated above the consensus sequences follows the convention of Mittl and Schneider-Brachert.14 The numbering at the beginning and end of each repeat corresponds to the residue number of full length ExoR. The conserved structural residues common to the SLR consensus sequence12,14 are colored red. Residues are abbreviated as follows: s, small; b, big; h, hydrophobic; p, polar; c, charged; x, any residue.
Comparative modeling of S. meliloti ExoRm protein
The closest structural matches to the S. meliloti ExoR protein were found to be Helicobacter pylori cysteine-rich protein C (HcpC, PDB ID:1OUV)12 and a protein corresponding to locus C5321 of Escherichia coli strain Cft073 (PDB ID:4BWR),15 both of which are Sel1-like repeat containing proteins, using fold recognition algorithms (Supporting Information Table SII).
Out of approximately 200 ExoR models built, the structural ExoRm model generated by I-Tasser server16 was selected as the best structural representation from a group of top-ranked models (Supporting Information Table SIII). Energy profiles calculated using ProSA-web17 for the selected model showed low energies and a z-score of −6.99 comparable to solved structures. The evaluation via Verify3D18 shows high 1D–3D profile scores for almost the entire length of the model (Supporting Information Fig. S2). In addition, the model passed the checks of various stereochemical parameters implemented in WHAT_CHECK19 (Supporting Information Table SIV) and an overall favorable Ramachandran plot20 (Supporting Information Fig. S3).
Tertiary model of S. meliloti ExoRm protein shows a superhelical fold
The ExoRm sequence adopts a superhelical fold consisting of 12 α-helices forming six Sel1-like repeats preceded by a small 310 helix at its N terminus with high structural similarity to the 1OUV template12 (Supporting Information Table SV). Each repeat is formed by two antiparallel helices: A (N-terminal) and B (C-terminal), located at the inner (concave) and outer (convex) surfaces of the superhelix, respectively [Fig. 3(a)]. Individual repeats are structurally similar and superpose with low root-mean-square deviation (RMSD) values below 1 Å except ExoR3 (Table1). The marginally higher RMSD values (0.9–1.3 Å) for ExoR3 can be ascribed to its extended helix A and elongated loop region containing two proline residues absent from the other repeats (Supporting Information Fig. S1). The helical structure of the ExoR repeats follows the expected positioning of conserved structural residues characteristic of Sel1-like proteins: residues 14, 24, and 43 that dictate sharp turns are present in loop regions, whereas residues responsible for the tight packing of the repeats at positions 3, 8, 32, 39, and 40 are located within α-helices [Figs. 2(a) and 3(b)].12,14 The packing of the ExoR repeats is also conserved: the average inter-repeat helix-packing angle in the ExoR model is 42 (± 2.6)° and the average intra-repeat helix-packing angle is 18 (± 2.9)°.
Figure 3.
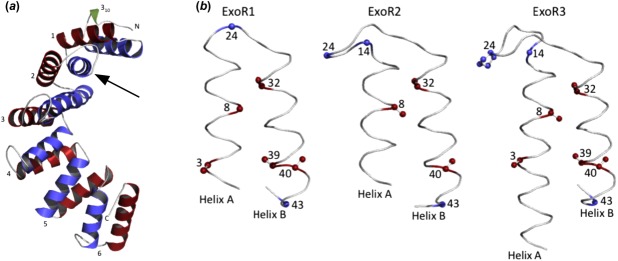
(a) Ribbon representation of the S. meliloti Rm1021 ExoRm model (residues 31–268). The repeats are numbered from 1 to 6 and helix A and helix B of each repeat are colored blue and red, respectively. The N-terminal 310 helix is colored green. The cleavage site is indicated with an arrow. (b) Backbone trace of the first three repeats of ExoR. Conserved structural residues responsible for tight packing of helices are colored red and those responsible for sharp turns in loop regions are colored blue. The numbers represent the positions of the amino acid residues in repeats.
Table 1.
RMSD Values for the Superposition of ExoR Repeats and Their Sequence Identity (%)
| ExoR1 43–74 | ExoR2 75–110 | ExoR3 114–160 | ExoR4 161–196 | ExoR5 197–228 | ExoR6 229–265 | |
|---|---|---|---|---|---|---|
| ExoR1 | — | 16 | 22 | 19 | 16 | 7 |
| ExoR2 | 0.7 | — | 28 | 25 | 9 | 17 |
| ExoR3 | 0.9 | 1.1 | — | 22 | 6 | 9 |
| ExoR4 | 0.5 | 0.6 | 1.1 | — | 9 | 9 |
| ExoR5 | 0.5 | 0.7 | 1.0 | 0.5 | — | 7 |
| ExoR6 | 0.7 | 0.7 | 1.3 | 0.6 | 0.7 | — |
The RMSD values (in bold) were calculated using TM-align server.36 The numbering corresponds to the residues of the full length ExoR, ExoRp.
Identification of the ExoRm protein–protein interaction sites
Analysis of the ExoR structural model predicted three non-overlapping binding sites: A, B, and C. Site A, composed of 36 residues, is located at the inner face of the N-terminal end of the protein and encompasses the ExoR proteolytic site. Site B that is found at the center of the inner face of the protein and extends to the C terminus is formed by 21 residues. The putative interaction site C is formed by eight residues located on the C-terminal convex surface of ExoRm ([Fig. 4(a–c)]; Supporting Information Table SVI).
Figure 4.

(a) Location of putative protein–protein interaction sites. The sites A, B, and C are colored red, blue, and green, respectively. (b, c) Putative functional sites mapped to the surface of ExoRm protein. Site A (residues identified by more than one prediction program in red; residues predicted using just one program in light red) and site C (green) in (b); and site B (residues identified by more than one prediction program in blue; residues predicted using just one program in light blue) in (c). Residues identified as hot spots are shown in black. (d) Superposition of the crystal contact II (orange) of HcpC protein12 and site A (red) of ExoR. The superposition is based on residues 28–116 from HcpC and residues 31–143 from ExoR. The side-chains of Asn66 (HcpC), Asn83 (ExoR), and Asn122 (ExoR) are shown. (e) Superposition of the crystal contact I (orange) of HcpC protein12 and site B (blue) of ExoR. The superposition is based on residues 225–292 from HcpC and residues 162–268 from ExoR.
Analysis of the electrostatic features of ExoRm
The surface electrostatic profile of the modeled ExoRm protein was examined to further analyze the biophysical features of ExoR that may drive interactions with its binding partners. The modeled ExoRm has a net charge of −3 at pH 7 and a dipole moment of 584 Debye oriented from the inside toward the outside of the superhelix. The concave face of the ExoR superhelix is highly negatively charged with acidic patches at the putative protein–protein interaction site A and the C terminus. The convex face of ExoR is predominantly hydrophobic with a prominent basic patch (Fig. 5).
Figure 5.
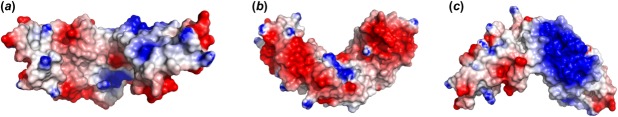
Electrostatic potentials mapped to the molecular surface of ExoRm. The ExoR protein is oriented to visualize the interfaces of the functional sites A and C in (a), site B in (b), and the basic surface patch at the outer surface of the super-helix in (c). In (a) and (b) the protein is oriented as in Figure 4. In all shown structures the N-terminal ends are placed on the left and the C-terminal ends on the right. The surface potentials are color-graded from −4 kT/e (red) to +4 kT/e (blue).
Analysis of the accessibility of the cleavage site
Because the proteolysis of the ExoRm protein has been implicated as a key step in regulating its function, the experimentally determined proteolysis site6 was mapped on the modeled structure of ExoR [Fig. 3(a)]. Surface accessibility of Ala80 and Leu81 in the modeled ExoRm protein corroborates the sequence-based surface accessibility predictions and suggests that both residues are not surface exposed (16% surface accessible area for Ala80 and 0% for Leu81).
Discussion
ExoR is a key player in regulating the ExoS/ChvI two-component system responsible for the successful establishment of the symbiotic relationship between S. meliloti and its leguminous host, alfalfa.2–7 Similarly, homologs of ExoR, ExoS, and ChvI in Agrobacterium tumefaciens and Brucella abortus are essential for host invasion by these plant and animal pathogens.21,22 Yet, a major gap in knowledge exists in understanding ExoR functionality because no solved ExoR structure exists. This study attempts to bridge this gap, providing a robust theoretical structural model of S. meliloti Rm1021 ExoRm, providing a first look into the details of the structural fold of this protein and predicting its implications to ExoS/ChvI two-component signaling.
Our analysis of the ExoR sequence suggests that it houses six Sel1-like repeats, in contrast to previous studies that report only four.14 Of these, we classify ExoR6 as a nontraditional low conservation repeat. Sel1-like repeats with low sequence conservation have been found to play important roles in other solenoid proteins such as the last Sel1-like repeat of H. pylori HcpC that deviates from the SLR consensus but plays a key role in protein–protein interactions.12 The “succinoglycan overproduction” phenotype of the S. meliloti exoR95 mutant with a disrupted C terminus23 supports the importance of ExoR6 and suggests that the newly identified repeats are an integral and functional part of the complete ExoR structural fold and protein.
The modeled ExoRm adopts the typical superhelical fold observed for Sel1-like repeat proteins known to be a suitable scaffold for protein–protein interactions.12 A comparison of ExoR and HcpC reveals that the crystal contact II at the concave face of the HcpC N terminus12 matches the putative protein–protein interaction site A at the concave face of the ExoR N terminus [Fig. 4(d)]. Because asparagine residues are recognized to be important to peptide recognition in HcpC,12 Hsp70/Hsp90 organizing protein,24 and PEX5,25 we propose that two asparagine residues present in site A of ExoR (Asn83 and Asn122) play a role in mediating protein–protein interactions, although their role would have to be confirmed through site-directed mutagenesis. The putative site B identified in ExoR protein also partially overlaps with the identified crystal contact I at the HcpC C terminus [Fig. 4(e)].12 Because HcpC and ExoR belong to the same structural family and share similar structural features, we anticipate that the mode of protein–protein interactions in ExoR will be similar to that of HcpC.12 Although the prediction of Site C is not as reliable as Sites A and B, it is possible that it forms a novel interface site characteristic of the ExoR protein family. Further validation of the identified interaction sites comes from our ongoing modeling studies (data not shown) of the experimentally characterized ExoR reduced-function mutants, ExoRG76C and ExoRS156Y, which show loss of stabilizing interactions with the ExoS protein.5
In addition, ExoR is predicted to form unique 20 residues long extended helix A of ExoR3 that encompasses a part of the protein–protein interaction site A. A similar unusual extended conformation of a repeat has been observed in the third repeat of the crystallized Trypanosoma brucei PEX5 protein, a helical multirepeat protein.26 On the other hand, the crystallized human PEX5 does not show this unusual elongated conformation.25 Based on similarity in sequence and function of T. brucei and human PEX5, it has been suggested that the third repeat in T. brucei PEX5 can adopt the extended form and the standard conformation.26 We speculate a similar helix to random coil pliability,27 which may be important for ExoR function and/or regulation.
Electrostatic interactions are important for selective binding of interacting partners in multirepeat α-α proteins.14,28 Our analysis of the electrostatic profile of the modeled ExoRm protein shows asymmetry in the charge distribution on the surface of the ExoR protein with the inside of the protein being more negatively charged relative to the outer surface of the superhelix that may play a key role in driving interactions between ExoR and its binding partners.
Mapping the experimentally determined cleavage site6 on the modeled ExoRm [Fig. 3(a)] reveals that it is buried and therefore would not be accessible to periplasmic proteases without undergoing conformational changes. The requirement for a similar conformational change is observed for substrates of DegP, a periplasmic serine endoprotease in E. coli, which recognizes three residues of the substrate protein and cleaves after a hydrophobic residue (Val, Ala, and Ile) that is almost completely buried in most DegP substrates.29 Although the protease involved in ExoR proteolysis has not been identified, S. meliloti Rm1021 does have a homolog of E. coli DegP.30 The presence of a solvent inaccessible hydrophobic residue in the vicinity of the experimentally determined ExoR cleavage site falls in line with the proposed model of DegP cleavage6,29 and can be further investigated experimentally.
In conclusion, we present the first attempt to generate a 3D model of S. meliloti Rm1021 ExoR in the absence of its crystal structure revealing important insights toward understanding its function. The findings of our structural analyses make it possible to study the molecular mechanism of ExoR cleavage and ExoRm–ExoS interactions using rational hypotheses-driven approaches and facilitate studies of pathogenicities of animal and plant pathogens in general.
Materials and Methods
Protein sequence analyses
The amino acid sequence of the ExoR protein from S. meliloti Rm1021 was retrieved from the NCBI Protein database (GenBank: AAA26260.1).31 The mature ExoR protein, ExoRm, that is, without its 30-residue signal sequence,4 was used for modeling the protein and sequence and structure analyses. SMART32 was used to identify and validate boundaries of the four previously predicted Sel1-like repeats of ExoR14 and cross-checked with other programs (Supporting Information Table SI). Previously unidentified Sel1-like repeats were detected with TPRpred,33 HHrepID,34 and REPRO35 and validated for correspondence with secondary structure prediction (Supporting Information Fig. S1). Structure-based sequence alignment of ExoR repeats was performed using the TM-align algorithm36 and manually refined by anchoring key conserved structural residues14 (Fig. 2). The sequences of putative ExoR orthologs were retrieved using NCBI-BLASTP31 against the nonredundant protein sequence database and aligned using T-COFFEE server v.9.01.37
Modeling methodology
The fold of the ExoR protein was identified using BLAST31 and threading algorithms (Supporting Information Table SII). To generate high-quality representations of the 3D structure of ExoRm, a large number of theoretical models were generated based on various alternative alignments for each template and also multitemplate modeling using comparative modeling methodology (detailed in Supporting Information Fig. S4). MODELLER versions 9.8 and 9.938 were used for model-building using alignments generated from T-COFFEE v9.02,37 MAFFT v6.864,39 FUGUE,40 and HHpred.41 I-TASSER16 and other modeling servers (Supporting Information Table SII) were explored for automated model building. The final model was generated with I-TASSER16 using the templates 1OUV12 and 3E4B.42 I-Tasser is currently ranked as the top server for automated protein structure prediction43 and its superior performance stems from the use of multiple threading alignments, iterative template fragment assembly simulation and ab initio modeling of unaligned regions.16 The prediction of side-chain conformations was implemented through the program SCWRL444 via the AS2TS system.45 The generated models were evaluated using ProSA-web,17 Verify3D,18 WHAT_CHECK,19 and RAMPAGE.20
Analyses of the 3D ExoRm model
The visualization and analyses of the shape, structural alignments, solvent-accessible molecular surfaces, and the electrostatic potential profile of the generated models were performed using the surface property analyses tools in PyMOL.46 All cartoons and diagrams were constructed with PyMOL46 and Prosite: MyDomains-Image Creator.47 STRIDE server48 was used to ascertain the location of secondary structure elements in the modeled ExoRm protein. Individual repeats were structurally aligned using the TM-align algorithm.36 The helical packing angles of the ExoRm models were computed using the PyMOL script to calculate angles between helices.49
Identification of protein–protein interaction sites
The putative protein–protein interaction sites were identified with PIER,50 SPPIDER v2,51 and cons-PPISP.52 Interaction hot spot residues were detected using the ISIS method.53
Electrostatics
The distribution of surface electrostatic potential for the ExoRm model was calculated using the Poisson-Boltzmann solver, DelPhi v.4 release 1.1.54 The net charge and the dipole moment of the modeled ExoRm protein were computed using the Protein Dipole Moments Server.55
Solvent accessibility analysis of the cleavage site
The solvent-accessible surface area of the ExoR cleavage site based on primary structure of ExoR was determined using several programs, including SABLE56 and RVP-NET.57 To determine the solvent accessibility of these residues in the generated theoretical models, the GETAREA58 server was used.
Acknowledgments
The authors thank the members of the Singh Laboratory and the Cheng Laboratory, specifically Mary Ellen Heavner, for helpful discussions and critical comments.
Glossary
Abbreviations:
- RMSD
root-mean-square deviation
- RSI
ExoR-ExoS/ChvI signal transduction pathway
- SLR
sel1-like repeat
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- Stock JB, Ninfa AJ, Stock AM. Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol Rev. 1989;53:450–490. doi: 10.1128/mr.53.4.450-490.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao SY, Luo L, Har KJ, Becker A, Ruberg S, Yu GQ, Zhu JB, Cheng HP. Sinorhizobium meliloti ExoR and ExoS proteins regulate both succinoglycan and flagellum production. J Bacteriol. 2004;186:6042–6049. doi: 10.1128/JB.186.18.6042-6049.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HP, Walker GC. Succinoglycan production by Rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J Bacteriol. 1998;180:20–26. doi: 10.1128/jb.180.1.20-26.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DH, Chen EJ, Fisher RF, Long SR. ExoR is genetically coupled to the ExoS-ChvI two-component system and located in the periplasm of Sinorhizobium meliloti. Mol Microbiol. 2007;64:647–664. doi: 10.1111/j.1365-2958.2007.05680.x. [DOI] [PubMed] [Google Scholar]
- Chen EJ, Sabio EA, Long SR. The periplasmic regulator ExoR inhibits ExoS/ChvI two-component signaling in Sinorhizobium meliloti. Mol Microbiol. 2008;69:1290–1303. doi: 10.1111/j.1365-2958.2008.06362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HY, Luo L, Yang MH, Cheng HP. Sinorhizobium meliloti ExoR is the target of periplasmic proteolysis. J Bacteriol. 2012;194:4029–4040. doi: 10.1128/JB.00313-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HY, Cheng HP. Autoregulation of Sinorhizobium meliloti ExoR gene expression. Microbiology. 2010;156:2092–2101. doi: 10.1099/mic.0.038547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajava AV, Gorbea C, Ortega J, Rechsteiner M, Steven AC. New HEAT-like repeat motifs in proteins regulating proteasome structure and function. J Struct Biol. 2004;146:425–430. doi: 10.1016/j.jsb.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Bradley P. Structural modeling of TAL effector–DNA interactions. Protein Sci. 2012;21:471–474. doi: 10.1002/pro.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335:716–719. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biom. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Lüthy L, Grütter MG, Mittl PRE. The crystal structure of Helicobacter Cysteine-rich protein C at 2.0 A resolution: similar peptide-binding sites in TPR and SEL1-like repeat proteins. J Mol Biol. 2004;340:829–841. doi: 10.1016/j.jmb.2004.04.055. [DOI] [PubMed] [Google Scholar]
- Forwood JK, Lange A, Zachariae U, Marfori M, Preast C, Grubmuller H, Stewart M, Corbett AH, Kobe B. Quantitative structural analysis of importin-beta flexibility: paradigm for solenoid protein structures. Structure. 2010;18:1171–1183. doi: 10.1016/j.str.2010.06.015. [DOI] [PubMed] [Google Scholar]
- Mittl PRE, Schneider-Brachert W. Sel1-like repeat proteins in signal transduction. Cell Signal. 2007;19:20–31. doi: 10.1016/j.cellsig.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Urosev D, Ferrer-Navarro M, Pastorello I, Cartocci E, Costenaro L, Zhulenkovs D, Marechal JD, Leonchiks A, Reverter D, Serino L, Soriani M, Daura X. Crystal structure of c5321: a protective antigen present in uropathogenic Escherichia coli strains displaying an SLR fold. BMC Struct Biol. 2013;13:19. doi: 10.1186/1472-6807-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- Hooft RWW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272–272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Cα geometry: ϕ/Ψ and Cβ deviation. Proteins. 2002;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Guzman-Verri C, Manterola L, Sola-Landa A, Parra A, Cloeckaert A, Garin J, Gorvel JP, Moriyon I, Moreno E, Lopez-Goni I. The two-component system BvrR/BvrS essential for Brucella abortus virulence regulates the expression of outer membrane proteins with counterparts in members of the Rhizobiaceae. Proc Natl Acad Sci USA. 2002;99:12375–12380. doi: 10.1073/pnas.192439399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CF, Lin JS, Shaw GC, Lai EM. Acid-induced type VI secretion system is regulated by ExoR-ChvG/ChvI signaling cascade in Agrobacterium tumefaciens. PLoS Pathog. 2012;8:e1002938. doi: 10.1371/journal.ppat.1002938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty D, Leigh JA, Glazebrook J, Walker GC. Rhizobium meliloti mutants that overproduce the R. meliloti acidic calcofluor-binding exopolysaccharide. J Bacteriol. 1988;170:4249–4256. doi: 10.1128/jb.170.9.4249-4256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain–peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- Gatto GJ, Geisbrecht BV, Gould SJ, Berg JM. Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat Struct Biol. 2000;7:1091–1095. doi: 10.1038/81930. [DOI] [PubMed] [Google Scholar]
- Kumar A, Roach C, Hirsh IS, Turley S, deWalque S, Michels PA, Hol WG. An unexpected extended conformation for the third TPR motif of the peroxin PEX5 from Trypanosoma brucei. J Mol Biol. 2001;307:271–282. doi: 10.1006/jmbi.2000.4465. [DOI] [PubMed] [Google Scholar]
- Hite KC, Kalashnikova AA, Hansen JC. Coil-to-helix transitions in intrinsically disordered methyl CpG binding protein 2 and its isolated domains. Protein Sci. 2012;21:531–538. doi: 10.1002/pro.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortajarena AL, Kajander T, Pan W, Cocco MJ, Regan L. Protein design to understand peptide ligand recognition by tetratricopeptide repeat proteins. Protein Eng Des Sel. 2004;17:399–409. doi: 10.1093/protein/gzh047. [DOI] [PubMed] [Google Scholar]
- Kolmar H, Waller PRH, Sauer RT. The DegP and DegQ periplasmic endoproteases of Escherichia coli: specificity for cleavage sites and substrate conformation. J Bacteriol. 1996;178:5925–5929. doi: 10.1128/jb.178.20.5925-5929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galibert F, Finan TM, Long SR, Puehler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J. The composite genome of the legume symbiont Sinorhizobium meliloti. Science. 2001;293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting C. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpenahalli MR, Lupas AN, Söding J. TPRpred: a tool for prediction of TPR-, PPR- and SEL1-like repeats from protein sequences. BMC Bioinformatics. 2007;8:2. doi: 10.1186/1471-2105-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegert A, Söding J. De novo identification of highly diverged protein repeats by probabilistic consistency. Bioinformatics. 2008;24:807–814. doi: 10.1093/bioinformatics/btn039. [DOI] [PubMed] [Google Scholar]
- George RA, Heringa J. The REPRO server: finding protein internal sequence repeats through the Web. Trends Biochem Sci. 2000;25:515–517. doi: 10.1016/s0968-0004(00)01643-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Šali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiski CL, Harwich M, Jain S, Neculai AM, Yip P, Robinson H, Whitney JC, Riley L, Burrows LL, Ohman DE, Howell PL. AlgK is a TPR-containing protein and the periplasmic component of a novel exopolysaccharide secretin. Structure. 2010;18:265–273. doi: 10.1016/j.str.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Mao B, Aramini JM, Montelione GT. Assessment of template-based protein structure predictions in CASP10. Proteins 82(Suppl. 2014;2):S43–S56. doi: 10.1002/prot.24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemla A, Zhou CE, Slezak T, Kuczmarski T, Rama D, Torres C, Sawicka D, Barsky D. AS2TS system for protein structure modeling and analysis. Nucleic Acids Res. 2005;33:W111–W115. doi: 10.1093/nar/gki457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.3 Schrödinger, LLC.
- Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJA. The 20 years of PROSITE. Nucleic Acids Res. 2008;36:D245–D249. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder T. 2012. . Angle between helices, PyMOLWiki; 2010. Available at: http://pymolwiki.org/index.php/AngleBetweenHelices . Retrieved March 9,
- Kufareva I, Budagyan L, Raush E, Totrov M, Abagyan R. PIER: protein interface recognition for structural proteomics. Proteins. 2007;67:400–417. doi: 10.1002/prot.21233. [DOI] [PubMed] [Google Scholar]
- Porollo A, Meller J. Prediction-based fingerprints of protein–protein interactions. Proteins. 2007;66:630–645. doi: 10.1002/prot.21248. [DOI] [PubMed] [Google Scholar]
- Zhou H, Shan Y. Prediction of protein interaction sites from sequence profile and residue neighbor list. Proteins. 2001;44:336–343. doi: 10.1002/prot.1099. [DOI] [PubMed] [Google Scholar]
- Ofran Y, Rost B. ISIS: interaction sites identified from sequence. Bioinformatics. 2007;23:e13–e16. doi: 10.1093/bioinformatics/btl303. [DOI] [PubMed] [Google Scholar]
- Nicholls A, Honig B. A rapid finite difference algorithm, utilizing successive over-relaxation to solve the poisson-boltzmann equation. J Comp Chem. 1991;12:435–445. [Google Scholar]
- Felder CE, Prilusky J, Silman I, Sussman JL. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007;35:W512–W521. doi: 10.1093/nar/gkm307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczak R, Porollo A, Meller J. Accurate prediction of solvent accessibility using neural networks based regression. Proteins. 2004;56:753–767. doi: 10.1002/prot.20176. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Gromiha MM, Sarai A. RVP-net: online prediction of real valued accessible surface area of proteins from single sequences. Bioinformatics. 2003;19:1849–1851. doi: 10.1093/bioinformatics/btg249. [DOI] [PubMed] [Google Scholar]
- Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comp Chem. 1998;19:319–333. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information