

Abstract
Understanding the mechanisms by which molecular motors coordinate their activities to transport vesicular cargoes within neurons requires the quantitative analysis of motor/cargo associations at the single vesicle level. The goal of this protocol is to use quantitative fluorescence microscopy to correlate (“map”) the position and directionality of movement of live cargo to the composition and relative amounts of motors associated with the same cargo. “Cargo mapping” consists of live imaging of fluorescently labeled cargoes moving in axons cultured on microfluidic devices, followed by chemical fixation during recording of live movement, and subsequent immunofluorescence (IF) staining of the exact same axonal regions with antibodies against motors. Colocalization between cargoes and their associated motors is assessed by assigning sub-pixel position coordinates to motor and cargo channels, by fitting Gaussian functions to the diffraction-limited point spread functions representing individual fluorescent point sources. Fixed cargo and motor images are subsequently superimposed to plots of cargo movement, to “map” them to their tracked trajectories. The strength of this protocol is the combination of live and IF data to record both the transport of vesicular cargoes in live cells and to determine the motors associated to these exact same vesicles. This technique overcomes previous challenges that use biochemical methods to determine the average motor composition of purified heterogeneous bulk vesicle populations, as these methods do not reveal compositions on single moving cargoes. Furthermore, this protocol can be adapted for the analysis of other transport and/or trafficking pathways in other cell types to correlate the movement of individual intracellular structures with their protein composition. Limitations of this protocol are the relatively low throughput due to low transfection efficiencies of cultured primary neurons and a limited field of view available for high-resolution imaging. Future applications could include methods to increase the number of neurons expressing fluorescently labeled cargoes.
Keywords: Neuroscience, Issue 92, kinesin, dynein, single vesicle, axonal transport, microfluidic devices, primary hippocampal neurons, quantitative fluorescence microscopy
Introduction
Intracellular transport is critical in all cell types for the delivery of proteins, membranes, organelles, and signaling molecules to various cellular domains1. Neurons are highly specialized cells with long, polarized projections that critically depend on intracellular transport of essential cargoes for their long-distance delivery to various axonal microdomains. This transport is mediated by kinesins and dyneins — two large families of molecular motor proteins — that bind to cargoes and track along polarized microtubules in anterograde and retrograde directions, respectively. While retrograde movement is mainly mediated by dynein, movement in the anterograde direction is facilitated by a large, functionally diverse family of kinesin motors. Consequently, anterograde transport of axonal cargoes could be mediated by various family members of the kinesin superfamily1-5. Though some cargoes move persistently in either direction, most cargoes move bidirectionally and reverse frequently on their way to their final destinations1,5-13. Furthermore, it has been shown that motors of opposing directionality associate simultaneously to cargoes, raising the question as to how regulated movement of cargoes is coordinated by opposite-polarity motors5-7. Together, transport of axonal cargoes is a concerted process that is regulated by the composition of motors and their specific biochemical activities, which in turn are dependent on various adaptors and regulatory binding partners14.
To faithfully describe the mechanism of axonal transport for a specific cargo and to uncover the underlying regulation of that transport, it is paramount to determine the composition of motor proteins and their regulatory binding partners associated with individual cargoes during their live transport. Other methods, for example biochemical approaches, provide estimates of average motor compositions on purified heterogeneous vesicle populations, but these estimates do not reveal the type or amounts of motors associated to single moving vesicles. Also, reconstitution of vesicle transport along preassembled microtubules in vitro enabled measuring the amount of one type of motor on a single vesicle level15. However, these experiments did not directly correlate the amount of motors with the transport characteristics of those vesicles, and measured transport in the absence of cellular regulatory factors.
A protocol is presented here, which determines the motor composition (type and relative amount of motors) of individual moving vesicles from immunofluorescence (IF) data measuring endogenously expressed motor proteins, and correlates these parameters to the live transport of the exact same vesicles in neurons16. This method entails precise mapping of IF-to-live cargo movement data. This is accomplished by growing hippocampal mouse neurons in microfluidic devices following established protocols17-19. These devices allow for the identification and correlation (“mapping”) of axons and single moving cargoes in fixed and live light microscopy modalities (Figure 1). Cultured neurons are transfected with fluorescently labeled cargo proteins whose transport is imaged at high spatial and temporal resolution to obtain detailed movement information that is plotted in kymographs. During the course of imaging, neurons are fixed with paraformaldehyde, and subsequently stained with antibodies against endogenous motor proteins. Fixed cargo and motor images are superimposed onto live movement kymographs to “map” (colocalize) them to the live cargo movement trajectories16. To correlate the live movement of cargoes with the association of motor proteins, colocalization is analyzed using a custom made MATLAB software package called “Motor Colocalization”16,20. Fluorescently labeled cargoes and motors generate diffraction-limited punctate features that can partially overlap. To resolve the position of overlapping puncta, the software first automatically fits Gaussian functions to each point spread function, representing individual fluorescent puncta, to determine their precise X-Y sub-pixel position coordinates and intensity amplitudes21-23. The positions of motors and cargoes are subsequently compared to each other to determine colocalization16,20. Therefore, this method more precisely assigns colocalization between fluorescent puncta as compared to other methods24.
The strength of this method is the ability to assess the colocalization of motors with individual cargo in fixed cells, for which live movement trajectories (e.g., the direction in which they were moving at the time of fixation) have been recorded. With this method, kinesins and dyneins were found to associate simultaneously to vesicles that carry the normal prion protein (PrPC-cellular), a neuronally enriched cargo that moves bidirectionally or remains stationary in axons16. This analysis allowed the formulation of a working model for the regulation of PrPC vesicle movement in which anterograde (kinesin) and retrograde (dynein) motors coordinate their activities in order to move the vesicles in either direction or to remain stationary while associated to the cargo. Another strength of this method is its potential broad applicability for characterizing colocalization/association of many fluorescently labeled cargoes that move in virtually any cell type, with any other protein(s) of interest. Thus, live/fixed correlation could potentially allow for the detection of transient protein-cargo interactions, as many individual fluorescently labeled moving particles can be analyzed over a desired period of time. Given the broad applicability and the type of questions that this method can address, this protocol will be of interest to a wide audience of cell biologists including those studying trafficking and transport in neurons or in other cell types.
Protocol
All experiments were conducted following approved protocols and according to institutional guidelines for the humane care of research animals. Neonate mice were euthanized by decapitation.
1. Preparation of Microfluidic Devices for Cell Culture
Prepare polydimethyl siloxane (PDMS) microfluidic devices for growth of hippocampal neurons as described by Harris and colleagues17-19. Below are some modifications that were adapted to the cargo mapping protocol. NOTE: Microfluidic devices are also commercially available (Materials List), thus access to a fabrication facility is not necessary.
Prepare No. 1½ cover glasses (24 x 40 mm) by washing them three times with acetone, followed by three washes with 100% ethanol and three washes of water. Store coverslips in water at 4 °C until use. These treatments help ensure that coverslips are clean from debris that might interfere with plating of cells, contamination, and survival. NOTE: Acetone and ethanol are flammable and hazardous in case of skin contact (irritant), of eye contact (irritant), of ingestion, of inhalation. Dispose according to official regulations.
When ready to use, place one cover glass in a 60 mm cell culture dish per microfluidic device, and let it air dry uncovered for 45 min inside a biosafety cabinet to avoid contamination.
After devices are cut out from the masters and punches have been cut to create the reservoirs (Figure 1A, B), place them with the microfluidic channels side-up inside a biosafety cabinet equipped with an UV lamp for 45 min for sterilization. NOTE: Leaving devices under UV treatment for longer than 2 hr might affect the integrity of the PDMS.
Assemble the devices by placing one device onto each cover glass inside the 60 mm plastic cell culture dish so that the microfluidic channels face down. Apply gentle pressure to the top of the device (avoid touching the microchannels) to provide a non-permanent seal of the device to the cover glass. Cover each 60 mm cell culture dish with a lid.
Using a micropipette, hydrate the devices by adding cell culture grade water to the top two microfluidic reservoirs (1 and 2 in Figure 1A), allowing water to flow through. Remove water with a micropipette and add 1 mg/ml poly-L-lysine into microfluidic reservoir 1 (200 µl) and 2 (100 µl). The volume differential will allow for the poly-L-lysine to flow through the microchannels (Figure 1A, B).
To ensure that the devices inside the 60 mm cell culture dishes do not topple over inside a 37 °C incubator, place three 60 mm cell culture dishes containing the devices into a 150 mm plastic cell culture dish and cover the dish with a lid. Incubate O/N in a 37 °C incubator with 5% CO2. Typically more than 90% of the channels are usable per device and do not contain any air bubbles or physical blockage.
On the next day, replace poly-L-lysine with water and leave device in incubator for at least 1 hr. Repeat this washing three times. Remove water and add Neurobasal-A growth medium (containing 2% B-27 supplement and 500 µM GlutaMAX) to each device. Leave O/N in a 37 °C incubator with 5% CO2.
On the next day, plate hippocampal cells as described below.
2. Plating of Mouse Hippocampal Primary Neurons in Microfluidic Devices
NOTE: The preparation of cultured hippocampal neurons from neonate rats has been previously published in JoVE25. Below are some modifications that were adapted to plate mouse hippocampal neurons in microfluidic devices, and that were optimal for transfections.
- Before mouse brain dissection, pre-warm Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS; without antibiotics), Mixture A (125 mg DL-cysteine, 125 mg bovine serum albumin (BSA) and 3.125 g of D-glucose in 500 ml of phosphate buffered saline (PBS); filter sterilize through a 0.22 µm filter), and deoxyribonuclease I solution (DNase I: 50 mg of DNase I and 0.5 ml of a 1.2 M MgSO4 solution in 100 ml Hank’s Balanced Salt Solution (HBSS) filter sterilize through a 0.22 µm filter) in a 37 °C water bath.
- Pre-warm Neurobasal-A growth medium inside a 37 °C incubator with 5% CO2 to equilibrate the pH.
Dissect hippocampi from 1-3 day old neonate mice according to established protocols26. Keep hippocampi in a 15 ml conical tube containing 10 ml dissection buffer (HBSS containing 10 mM HEPES pH 7.4, 50 mM glucose, 100 U/ml penicillin and 100 µg/ml streptomycin), on ice. Put up to 4 hippocampi per 15 ml conical tube.
Perform the remaining protocol is under sterile conditions inside a biosafety cabinet.
Dilute 45 units of papain in 5 ml of Mixture A, vortex, and incubate for 5 min at 37 °C until papain powder is dissolved. Add 1 ml of DNase I solution. Filter the mix through a 0.22 µm syringe filter unit.
Carefully aspirate HBSS’ from 15 ml conical tube containing hippocampi. Add 10 ml cold (4 °C) HBSS to hippocampi and let them settle to the bottom of the tube. Carefully aspirate HBSS from 15 ml conical tube.
Add 1 ml papain/DNase I mix to up to 4 hippocampi and incubate for 20-30 min at 37 °C. Tilt conical tube every 5 min to bathe hippocampi with mix. Alternatively place hippocampi in a 37 °C water bath shaker with gentle shaking (~100 rpm).
Aspirate papain/DNase I mix and wash hippocampi twice with pre-warmed DMEM (37 °C) containing 10% FBS.
After second wash, add 2 ml of pre-warmed DMEM containing 10% FBS to hippocampi and manually triturate hippocampi by pipetting up and down ~10-12 times using a 1 ml pipette tip to dissociate neurons.
Let triturate settle for ~1 min as this will allow non-dissociated neurons to settle to the bottom of the conical tube. Transfer supernatant containing dissociated neurons to a new 15 ml conical tube avoiding transfer of larger tissue that has settled at the bottom of the tube.
Spin cells at 1,000 x g for 2 min and carefully remove supernatant without disturbing the cell pellet. Resuspend cells in 80 μl Neurobasal-A growth medium by pipetting gently.
Remove medium from microfluidic reservoir 1 and 1’. Apply 20 µl of cells (~275,000) to microfluidic reservoir 1, and allow them to flow through. Place device in 37 °C incubator with 5% CO2 for 20 min.
Look under microscope to ensure that cells have adhered to the cover glass. Add Neurobasal-A growth medium to top off all reservoirs. Return device with cells to a 37 °C incubator with 5% CO2.
Check devices every ~2-3 days and top off reservoirs with Neurobasal-A containing 2% B-27 supplement (without GlutaMAX) to avoid evaporation. Neurons start to extend their axons through microfluidic channels ~2-3 days after plating. Subject neurons to transfection typically 7-10 days after plating.
3. Transfection of Hippocampal Neurons Grown in Microfluidic Device
Per device, prepare a mix of 1.2 µl Lipofectamine 2000 in 30 µl Neurobasal-A, and a mix of 0.5 µg fluorescent cargo fusion plasmid in 30 µl Neurobasal-A and incubate 5 min at RT. Add Lipofectamine mix to DNA mix and incubate for 20 min at RT. Add 60 µl of Neurobasal-A containing 4% B-27 supplement to the DNA/Lipofectamine mix and mix by flicking the tube.
Remove all media from microfluidic reservoir 2 and 2’ and carefully fill up wells with Neurobasal-A containing 2% B-27 supplement without spilling over the medium into the other compartments.
Remove media from microfluidic reservoir 1 and 1’. Add 120 µl of DNA/Lipofectamine mix to microfluidic reservoir 1 and let it flow. Return device to 37 °C incubator with 5% CO2 for 3-4 hr.
Remove all media from microfluidic reservoir 1 and 1’ and replace with Neurobasal-A containing 2% B-27 supplement for 24 hr (or until ready to image). Typically, 5-7 axons that grow through microchannels are transfected under those conditions, which represent approximately 1-3% transfection efficiency consistent with efficiencies published for hippocampal cultured cells 26.
4. “Cargo Mapping” Analyses
- Live Imaging of cargo movement
- Perform the imaging on an inverted epifluorescence light microscope equipped with a 37 °C incubator and a CO2 control chamber.
- Mount the coverslip with hippocampal neurons grown in a microfluidic device onto the microscope stage. To avoid neuronal death, work expeditiously to maintain the samples at the microscope for no longer than 1 hr.
- Using a high-resolution objective (100X), find axons of transfected cells that extend through channels of microfluidic chambers and record the number of the channels in which the transfected axon was found. Count the number of channels using a hand tally counter.
- For optimal recording of live movement and subsequent colocalization analysis, choose axons that grow relatively flat through the channels so that imaging of most vesicles can be performed in focus.
- Remove most of the medium from reservoirs 2 and 2’ with a 1 ml plastic transfer pipette. To facilitate subsequent alignment of fixed images with movement trajectories on kymographs, align the right edge of the microchannels with the field of view during imaging (Figure 1B, C).
- To facilitate fixing of the samples during imaging, perform live imaging with two people. Alternatively, if live imaging is performed by a single person, use a microscope system equipped with drift correction.
- Start imaging live cargo movement with time-lapse specifications specific to the transport dynamics of the cargo being analyzed (microscope specifications and settings for imaging YFP-PrPC are specified in the legend of Figure 2 and Materials List).
- After sufficient live movement data have been collected, have one person fill up reservoirs 2 and 2’ with pre-warmed 4% paraformaldehyde in PBS containing 0.04 g/ml sucrose to fix the cells. Avoid touching the microfluidic device, as this will disrupt the focus of the live imaging. Have the other person adjust the focus while the fixative is added. Fixation is successful when immediate stop of movement is observed. Add fixative to reservoir 1 and 1' afterwards. NOTE: Temporary photobleaching of the cargo fluorescence might also be observed, but the fluorescence persists after IF staining for motor proteins is completed (next step). NOTE: Paraformaldehyde is harmful by inhalation, in contact with skin, and if swallowed. Irritating to eyes, respiratory system and skin. Dispose according to official regulations.
- IF staining of motor proteins for colocalization analysis NOTE: For quantitation of relative levels of motor proteins (as determined by antibody staining), associated to individual moving cargoes, validate and titrate antibodies to ensure that the antibody-antigen binding is saturated. This is done by determining the specificity of the antibody using neurons in which the protein of interest has been either knocked-out or knocked-down, and by performing IF analysis with increasing antibody concentrations. For negative controls, neurons are stained with secondary antibodies in the absence of primary antibodies. More detailed description of antibody validation can be found in Encalada et al.16, and Szpankowski et al.20.
- Remove the coverslip with hippocampal neurons grown in microfluidic device from the microscope stage. Do not detach the microfluidic device from the coverslip, as the microchannels need to remain intact in order to superimpose the live and fixed images. Perform the following steps in a humidity chamber.
- To ensure the flow of solutions into the microchannels, maintain a buffer volume differential between reservoirs 1, 1’, and 2, 2’ of at least 30 µl. in all subsequent steps. For example, add 100 µl media volume to microfluidic reservoir 2, 2’, and 70 µl media volume to microfluidic reservoir 1, 1’.
- Remove fixative from chambers of the microfluidic device and wash cells three times with PBS waiting 10 min between washes.
- Permeabilize cells by adding 0.1% Triton X-100 diluted in PBS for 5 min to all reservoirs.
- Remove solution from all reservoirs and wash with PBS. Repeat washing three times waiting 10 min between washes.
- Remove solution from all reservoirs and apply blocking buffer containing 10% Normal Donkey Serum and 3% protease free and immunoglobulin G (IgG)-free BSA in PBS and incubate for at least 30 min at RT.
- Spin primary antibody stock solution at 20,000 x g for 5 min to remove precipitates. Dilute antibody to an appropriate concentration in blocking buffer. Replace the blocking buffer from microfluidic device with antibody solution. Incubate samples for 2 hr at RT or O/N at 4 °C.
- Remove solution from all reservoirs and wash three times with PBS, waiting 10 min between washes.
- Spin secondary antibody stock solution at 20,000 x g for 5 min to remove precipitates. Dilute antibody to an appropriate concentration in blocking buffer. Replace PBS with secondary antibody solution. Incubate samples for 1 hr at RT.
- Remove solution from all reservoirs and wash three times with PBS, waiting 10 min between washes.
- Replace PBS with ~10-20 µl mounting medium by directly pipetting mounting medium into the opening of the channels connecting the reservoirs. Let mounting media dry for ~20 min – 1 hr at RT.
- Store devices at 4 °C protected from light to avoid photobleaching, and image axons within two days (maximum one week) of staining.
- After staining is completed, mount coverslip with hippocampal neurons grown in microfluidic device onto the microscope stage. Locate the region of the transfected axon imaged in step 4.1.1-4.1.7.
- Acquire Z-stack images (300 nm steps) for each of the three channels: cargo, motor 1, motor 2, using a high-resolution objective (e.g., a high numerical aperture oil or water objective). NOTE: Acquisition of Z-stacks is important as axons often do not grow perfectly flat in microchannels and therefore not all fluorescent puncta are in the same focal plane.
- Cargo Mapping
- Generate a kymograph of cargo movement using image analysis software. The ImageJ plugin developed by Rietdorf and Seitz (EMBL) is recommended for generating kymographs (for download seeMaterials List). NOTE: ImageJ is a public domain, Java-based image processing program developed at the National Institutes of Health27,28 (for download see Materials List).
- Align the fixed images of the fluorescent cargo (generated in step 4.2.14) manually by superimposing them onto the position of the trajectories in the kymograph at the point of fixation (Figure 2A, B), using a commercially available graphing software program (Materials List). Manually align by first superimposing the fixed cargo image onto the kymograph and manually choosing the cargo puncta that corresponds to a specific trajectory on the kymograph. For best alignment results, use the Z slice that is in best focus for the cargo mapped. Several Z slices may be used.
- For each image in the Z-stack (cargo, motor 1 and motor 2; generated in step 4.2.14) determine the X-Y coordinates and intensity amplitudes for each fluorescent puncta using an established algorithm to fit 2D Gaussians to the point spread function that represents each point source in each of the three channels16,20,21 (Figure 2D). NOTE: To obtain the X-Y coordinates, a custom built MATLAB software package called “Motor Colocalization” was developed16,20, which incorporates a Gaussian fitting algorithm developed by Jaqaman, Danuser and colleagues21-23. The software package and detailed instructions for its use can be obtained upon request.
- For each of the individually cargo puncta that were mapped onto the mobile or stationary trajectories on the kymograph, select the X-Y coordinates and intensity amplitudes from the results of the “Motor Colocalization” program (these cargoes are individually labeled in Figure 2B). Use several Z-stack images acquired in step 4.2.14) to map more cargoes that are found on different focal planes.
- Compare the individual X-Y coordinates for the mapped cargo to those obtained for each of the motor channels in the corresponding Z slice (Figure 2D). Determine and select the X-Y motor coordinates that are within a 300 nm radius of the cargo. For cargoes and motors that have signal within the same focal plane, use the “Motor Colocalization” software package to determine the puncta that are located within a 300 nm radius.
Representative Results
Figure 1 shows an overview of the microfluidic device used to grow hippocampal neurons (Figure 1A, B). Neurons are plated in reservoir 1. The size of the microchannels prevents the diffusion of cell bodies (soma) into the axonal compartment while the length of the channels prevents dendritic projections from crossing all the way to the axonal compartment. After ~2-3 days in culture, neurons start extending their axons across microchannels into the axonal compartment (Figure 1B, C). Transfected axons expressing normal prion protein labeled with yellow fluorescent protein (YFP-PrPC) can be identified by fluorescence microscopy (Figure 1C). Expression of fluorescently labeled proteins using plasmid transfection should be checked for proper subcellular localization and function as overexpression might introduce experimental artifacts due to altered protein conformations. Thus, colocalization experiments ought to be complimented both with biochemical co-precipitation studies, as well as with IF staining for endogenously expressed proteins. The behavior of the YFP-PrPC used here was tested previously16. Expression of YFP-PrPC in transiently transfected neuroblastoma cells (N2a) was low, suggesting that transient transfection does not result in highly overexpressed YFP-PrPC levels16. Moreover, IF of N2a cells transfected with YFP-PrPC using antibodies against PrPC indicated that YFP-PrPC highly colocalized with endogenous PrPC, suggesting that both transfected and endogenous PrPC behaved similarly. To ascertain that PrPC associated with motors, vesicle immunoisolations were performed16.
For optimal cargo mapping studies, the right edge of the microchannels is aligned with the field of view during live and fixed imaging in order to image the same region of the axon (indicated by red rectangle in Figure 1B, C). Figure 2 and Figure 3 show representative results for the cargo mapping analyses of neurons transiently transfected with YFP-PrPC. Vesicles carrying YFP-PrPC are transported in mammalian axons by members of the Kinesin-1 family and Dynein Heavy Chain 1 (DYNC1H1)16, therefore YFP-PrPC vesicles were mapped to the Kinesin-1 subunit Kinesin Light Chain 1 (KLC1), and to DYNC1H1 motors. YFP-PrPC vesicle movement was imaged and plotted in a kymograph (Figure 2A). In kymographs, anterograde and retrograde movement of YFP-PrPC vesicles is represented by trajectories with negative and positive slopes, respectively, and stationary vesicles are depicted by vertical trajectories. At fixation, all movement stopped indicated by the absence of diagonal lines in the kymograph (Figure 2A). The IF signal of molecular motors and cargoes in fixed, permeabilized axons is punctate16 (Figure 2B, C). The goal of “cargo mapping” is to determine if puncta in the cargo channel (Figure 2B) colocalize with motor puncta (Figure 2C) in the other two channels. To do this, fluorescent images of fixed YFP-PrPC vesicles, and the IF images of corresponding KLC1 and DYNC1H1 associated to vesicular puncta were aligned to the kymograph (Figure 2B,C). Gaussian functions were fitted to the point spread functions of fluorescent point sources in all three channels in order to map the precise X-Y positions of motors to cargo trajectories obtained by live imaging (Figure 2D). Endogenously expressed KLC1 and DYNC1H1 motors appear as punctate staining and colocalized differentially with YFP-PrPC vesicles (Figure 2D). Colocalization was defined by the presence of YFP-PrPC and motor puncta within a distance of 300 nm and quantified by the level of colocalization of the Gaussian X-Y coordinate positions between the YFP-PrPC and each of the KLC1 and DYNC1H1 puncta (Figure 3A). This distance was chosen based on the optics, diffraction limit of light, and the relevant physical size of the cargo and motor subunits. The relative amount of KLC1 and DYNC1H1 associated with YFP-PrPC vesicles was obtained from the Gaussian amplitudes, which represent the intensities of each puncta (Figure 3B). These data can be further analyzed as desired. For example, intensities of motors colocalizing with YFP-PrPC puncta between control (wild-type - WT) neurons and those lacking other Kinesin-1 subunits (KIF5C -/- neurons in Figure 3A, KIF5C is a member of the Kinesin-1 family), can be compared to determine if the absence of KIF5C disrupts association of KLC1 and/or DYNC1H1 to YFP-PrPC vesicles (KLC1 and DYNC1H1 intensities were unchanged in our experiments16). Furthermore, KLC1 and DYNC1H1 intensities can be plotted to scrutinize possible correlations between relative motor amounts and directionality of movement (Figure 3B). For example, YFP-PrPC vesicles that are moving and/or stationary associated with both KLC1 and/or DYNC1H1 (Figure 3B).
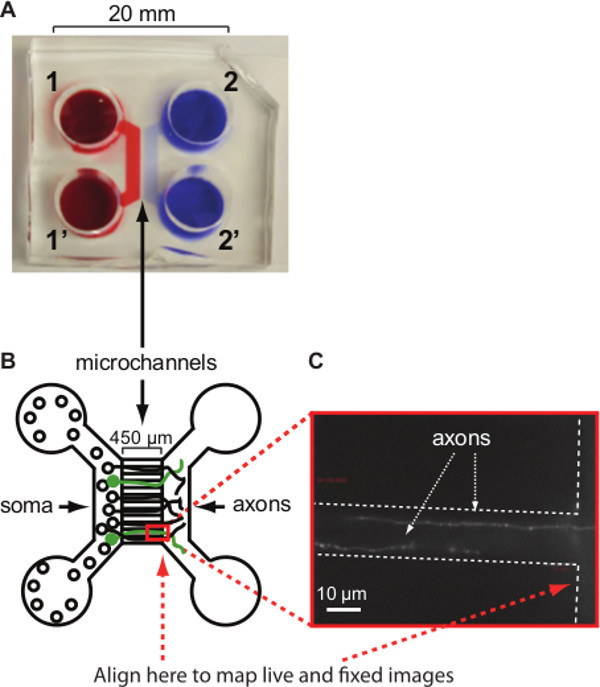 Figure 1. Hippocampal neurons cultured in microfluidic devices. (A) Picture of a microfluidic device. The outlines of reservoirs are visualized by red and blue dyes. Numbering refers to the number of microfluidic reservoirs used throughout the protocol. (B) Diagram of a microfluidic device. Numbers correspond to reservoirs in which media and neurons are added. Transfected neurons are shown in green. Red rectangle and red dotted arrows indicate the region recommended for live and fixed fluorescence imaging. (C) Image of two axons transfected with YFP-PrPC growing through a single microchannel (one microchannel and edge of the device is outlined by dotted white lines). Individually moving YFP-PrPC vesicles can be distinguished as brighter puncta. Scale bar is 10 µm. Figure is adapted from: Encalada et al.16. Please click here to view a larger version of this figure.
Figure 1. Hippocampal neurons cultured in microfluidic devices. (A) Picture of a microfluidic device. The outlines of reservoirs are visualized by red and blue dyes. Numbering refers to the number of microfluidic reservoirs used throughout the protocol. (B) Diagram of a microfluidic device. Numbers correspond to reservoirs in which media and neurons are added. Transfected neurons are shown in green. Red rectangle and red dotted arrows indicate the region recommended for live and fixed fluorescence imaging. (C) Image of two axons transfected with YFP-PrPC growing through a single microchannel (one microchannel and edge of the device is outlined by dotted white lines). Individually moving YFP-PrPC vesicles can be distinguished as brighter puncta. Scale bar is 10 µm. Figure is adapted from: Encalada et al.16. Please click here to view a larger version of this figure.
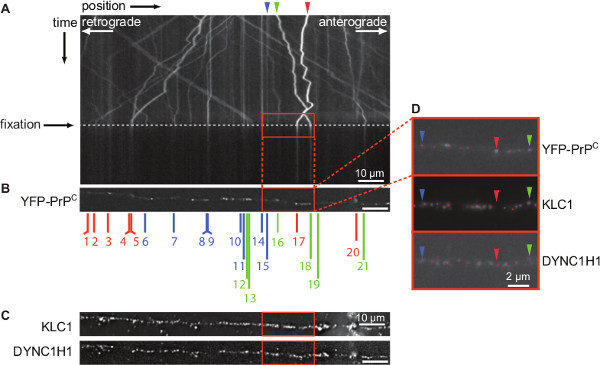 Figure 2. “Cargo mapping” to correlate YFP-PrPC vesicle transport with relative amounts of kinesin and dynein motors. Wide-field fluorescence images of live movement and of fixed cargoes were taken on an inverted microscope equipped with a motorized stage and a CCD camera and a 100X/1.4 NA oil objective. (A) Kymograph of YFP-PrPC vesicle movement of axons shown in Figure 1C. Live movement was recorded for a total time ranging from 30 to 60 sec at 100 ms exposure (10 Hz). Cells were fixed after imaging cargo movement for at least 15 sec. Dotted line indicates the time of fixation during live imaging. Red box shows region of vesicles highlighted in (B, C) and enlarged in (D). Arrowheads point to three vesicles shown in (D) corresponding to stationary (blue), retrograde (red), and anterograde (green) trajectories. (B) Image of fixed YFP-PrPC vesicles aligned to the corresponding kymograph. Numbers indicate the individual YFP-PrPC vesicles that were mapped to identifiable puncta on the kymograph. Anterograde vesicles are green, retrograde are red, and stationary ones are blue. (C) Fixed IF images of KLC1 and DYNC1H1 puncta corresponding to the same region shown in (B). Scale bar (A-C) is 10 µm. (D) Enlargement of fixed IF signals of each of the three channels with 2D Gaussian function assignments. Blue dots represent local intensity maxima pixels, red dots represent Gaussian fits, and pink dots are the overlap between the two. Arrowheads indicate examples of YFP-PrPC vesicles shown in (A), and their differential colocalization with KLC1 and DYNC1H1 motor proteins. Scale bar is 2 µm. Figure is adapted from: Encalada et al.16. Please click here to view a larger version of this figure.
Figure 2. “Cargo mapping” to correlate YFP-PrPC vesicle transport with relative amounts of kinesin and dynein motors. Wide-field fluorescence images of live movement and of fixed cargoes were taken on an inverted microscope equipped with a motorized stage and a CCD camera and a 100X/1.4 NA oil objective. (A) Kymograph of YFP-PrPC vesicle movement of axons shown in Figure 1C. Live movement was recorded for a total time ranging from 30 to 60 sec at 100 ms exposure (10 Hz). Cells were fixed after imaging cargo movement for at least 15 sec. Dotted line indicates the time of fixation during live imaging. Red box shows region of vesicles highlighted in (B, C) and enlarged in (D). Arrowheads point to three vesicles shown in (D) corresponding to stationary (blue), retrograde (red), and anterograde (green) trajectories. (B) Image of fixed YFP-PrPC vesicles aligned to the corresponding kymograph. Numbers indicate the individual YFP-PrPC vesicles that were mapped to identifiable puncta on the kymograph. Anterograde vesicles are green, retrograde are red, and stationary ones are blue. (C) Fixed IF images of KLC1 and DYNC1H1 puncta corresponding to the same region shown in (B). Scale bar (A-C) is 10 µm. (D) Enlargement of fixed IF signals of each of the three channels with 2D Gaussian function assignments. Blue dots represent local intensity maxima pixels, red dots represent Gaussian fits, and pink dots are the overlap between the two. Arrowheads indicate examples of YFP-PrPC vesicles shown in (A), and their differential colocalization with KLC1 and DYNC1H1 motor proteins. Scale bar is 2 µm. Figure is adapted from: Encalada et al.16. Please click here to view a larger version of this figure.
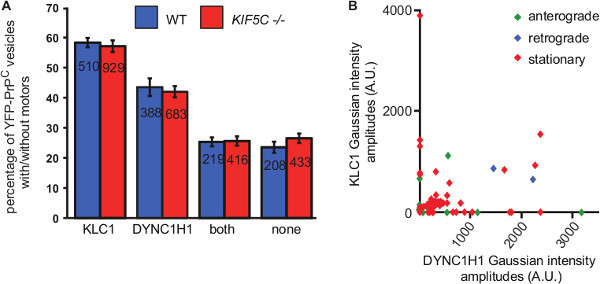 Figure 3. Quantitative analyses of “cargo mapping” of YFP-PrPC vesicles. (A) Quantification of colocalization between YFP-PrPC vesicles, KLC1 and DYNC1H1 in WT and KIF5C knockout neurons (KIF5C -/-). Total number of puncta analyzed was NWT= 887 and NKIF5C-/-= 162916. Numbers inside bars represent the number of vesicles per category. Shown are averages ± SEM. Non-parametric permutation t test was used for comparison between genotypes29. (B) Correlation of relative motor composition and directionality of movement in WT neurons of data analyzed in (A). Figure is adapted from: Encalada et al.16. Please click here to view a larger version of this figure.
Figure 3. Quantitative analyses of “cargo mapping” of YFP-PrPC vesicles. (A) Quantification of colocalization between YFP-PrPC vesicles, KLC1 and DYNC1H1 in WT and KIF5C knockout neurons (KIF5C -/-). Total number of puncta analyzed was NWT= 887 and NKIF5C-/-= 162916. Numbers inside bars represent the number of vesicles per category. Shown are averages ± SEM. Non-parametric permutation t test was used for comparison between genotypes29. (B) Correlation of relative motor composition and directionality of movement in WT neurons of data analyzed in (A). Figure is adapted from: Encalada et al.16. Please click here to view a larger version of this figure.
Discussion
The protocol presented here enables the correlation of directionality of movement of individual fluorescent microtubule-based moving cargo particles with the relative type and amount of associated motor proteins in live neurons. Previously, the total motor composition of axonal vesicular cargoes was assayed on heterogeneous populations of biochemically purified vesicles and organelles9,15. However, characterizing motor composition for a single type of cargo inside cells has been challenging because of the difficulty in purifying homogeneous vesicle populations. Moreover, while measurements of motor stall-forces provided estimates of active motor numbers in Drosophila embryos, it was unclear whether directionality of movement correlated to motors being stably bound to cargoes or to the rapid association/dissociation of active motors to/from cargo12. Therefore the “cargo mapping” protocol described here was developed to further investigate the correlation between the type and relative number of motors associated to individually moving cargo in live neurons.
To successfully obtain data from “cargo mapping” it is critical to 1) perform imaging of cargo movement and fixed IFs at a high temporal and spatial resolution, 2) ensure immediate fixing of the sample during the course of imaging, 3) align the region of interest in live and fixed images and map fixed cargo and motors to movement kymographs for the same images, and 4) precisely determine the X-Y coordinates of cargo and motor fluorescent puncta in order to determine the amount of colocalization between cargo and motor proteins.
While this protocol is uniquely suited to analyze transport characteristics in uniformly organized axons of neurons grown in microfluidic devices, it could be adapted to a broad range of research questions. For example, this application could be used to characterize association of vesicles with their adaptors involved in endo/exocytosis (e.g., trafficking assays in fibroblasts), or the growth of microtubules labeled with microtubule +tip binding proteins. For these, microfluidic devices could be substituted by gridded coverslips to facilitate the localization of imaged cells necessary for correlative live and fixed imaging studies. This protocol could also be used to characterize cellular events for which a parameter changes in time depending on the specific cellular environment. For example, the release of a fluorescently labeled viral protein from individual endosomal structures into the cytoplasm could be correlated with the expression and/or association of endosomal proteins.
One possible limitation of this protocol is that mapping of high density cargoes might be difficult. However, the assignment of Gaussian functions circumvents this issue to a great extent by allowing distinction of positions between fluorescent puncta at the sub-pixel level. Furthermore, the mapping of motors to axonal cargoes is low-throughput: currently, mapping can be done for 2 axons per microfluidic device, given the low rate of transfection of neurons and the limited field of view when using the high magnification necessary for high-resolution imaging with our camera. Future developments of this method might include the use of lentiviral systems to transduce neurons with higher efficiency, isolation of neurons from transgenic mice expressing fluorescently tagged proteins or the labeling of organelles with small fluorescent dyes such as Lysotracker or Mitotracker, all of which will increase the number of axons labeled with fluorescent markers. Furthermore, the size of the recorded area is limited by the magnification, the pixel size and the size of the pixel array of the camera used for imaging. Ongoing developments in the field of imaging techniques will allow for larger areas to be imaged in the future and therefore increase the amount of data obtained per experimental condition.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Ge Yang, Gaudenz Danuser, Khuloud Jaqaman, and Daniel Whisler for assistance with the adaptation and development of the software to quantitate cargo mapping analyses, and Emily Niederst for help in making the microfluidic devices. This work was supported in part by NIH-NIA grant AG032180 to L.S.B.G., and the Howard Hughes Medical Institute. L.S. was supported in part by a NIH Bioinformatics Training Grant T32 GM008806, S.E.E. was supported by a Damon Runyon Cancer Research Foundation Fellowship, NIH Neuroplasticity Training Grant AG000216, and by a grant from The Ellison Medical Foundation New Scholar in Aging Award, G.E.C was supported by an NIH/NCATS 1 TL1 award TR001114 and by the Achievement Rewards for College Scientists foundation.
References
- Goldstein AYN, Wang X, Schwarz TL. Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 2008;18:495–503. doi: 10.1016/j.conb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell. Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Welte MA. Bidirectional transport along microtubules. Curr. Biol. 2004;14:525–537. doi: 10.1016/j.cub.2004.06.045. [DOI] [PubMed] [Google Scholar]
- Bryantseva SA, Zhapparova ON. Bidirectional transport of organelles: unity and struggle of opposing motors. Cell. Biol. Int. 2011;36:1–6. doi: 10.1042/CBI20110413. [DOI] [PubMed] [Google Scholar]
- Gross SP. Hither and yon: a review of bi-directional microtubule-based transport. Phys. Biol. 2004;1:1–11. doi: 10.1088/1478-3967/1/2/R01. [DOI] [PubMed] [Google Scholar]
- Gross SP, et al. Interactions and regulation of molecular motors in Xenopus melanophores. J. Cell. Biol. 2002;156:855–865. doi: 10.1083/jcb.200105055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross SP, Welte MA, Block SM, Wieschaus EF. Coordination of opposite-polarity microtubule motors. J. Cell. Biol. 2002;156:715–724. doi: 10.1083/jcb.200109047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kural C, et al. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement. Science. 2005;308:1469–1472. doi: 10.1126/science.1108408. [DOI] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol. Biol. Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubeita GT, et al. Consequences of motor copy number on the intracellular transport of kinesin-1-driven lipid droplets. Cell. 2008;135:1098–1107. doi: 10.1016/j.cell.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppina V, Rai AK, Ramaiya AJ, Barak P, Mallik R. Tug-of-war between dissimilar teams of microtubule motors regulates transport and fission of endosomes. Proc. Natl. Acad. Sci. USA. 2009;106:19381–19386. doi: 10.1073/pnas.0906524106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev. Mol. Cell. Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Hendricks AG, et al. Motor Coordination via a Tug-of-War Mechanism Drives Bidirectional Vesicle Transport. Current Biol. 2010;20:697–702. doi: 10.1016/j.cub.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encalada SE, Szpankowski L, Xia C-h, Goldstein LSB. Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell. 2011;144:551–565. doi: 10.1016/j.cell.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J, et al. Non-plasma Bonding of PDMS for Inexpensive Fabrication of Microfluidic Devices. J. Vis. Exp. 2007;9(410) doi: 10.3791/410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J, et al. Fabrication of a Microfluidic Device for the Compartmentalization of Neuron Soma and Axons. J Vis. Exp. 2007;7(261) doi: 10.3791/261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, et al. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpankowski L, Encalada SE, Goldstein LSB. Subpixel colocalization reveals amyloid precursor protein-dependent kinesin-1 and dynein association with axonal vesicles. Proc. Natl. Acad. Sci. USA. 2012;109:8582–8587. doi: 10.1073/pnas.1120510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, et al. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomann D, Dorn J, Sorger PK, Danuser G. Automatic fluorescent tag localization II: Improvement in super-resolution by relative tracking. J. Microsc. 2003;211:230–248. doi: 10.1046/j.1365-2818.2003.01223.x. [DOI] [PubMed] [Google Scholar]
- Thomann D, Rines DR, Sorger PK, Danuser G. Automatic fluorescent tag detection in 3D with super-resolution: application to the analysis of chromosome movement. J. Microsc. 2002;208:49–64. doi: 10.1046/j.1365-2818.2002.01066.x. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Nunez J. Primary Culture of Hippocampal Neurons from P0 Newborn Rats. J. Vis. Exp. 2008;29(19) doi: 10.3791/895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat. Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DS, McCabe GP. Introduction to the Practice of Statistics. 5th edition. New York, NY: W.H. Freeman; 2005. [Google Scholar]