

Abstract
Regulation of protein synthesis represents a key control point in cellular response to stress. In particular, discreet RNA regulatory elements were shown to allow to selective translation of specific mRNAs, which typically encode for proteins required for a particular stress response. Identification of these mRNAs, as well as the characterization of regulatory mechanisms responsible for selective translation has been at the forefront of molecular biology for some time. Polysome profiling is a cornerstone method in these studies. The goal of polysome profiling is to capture mRNA translation by immobilizing actively translating ribosomes on different transcripts and separate the resulting polyribosomes by ultracentrifugation on a sucrose gradient, thus allowing for a distinction between highly translated transcripts and poorly translated ones. These can then be further characterized by traditional biochemical and molecular biology methods. Importantly, combining polysome profiling with high throughput genomic approaches allows for a large scale analysis of translational regulation.
Keywords: Cellular Biology, Issue 92, cellular stress, translation initiation, internal ribosome entry site, polysome, RT-qPCR, gradient
Introduction
Regulation of protein synthesis (translation) is a key cellular process which is intimately linked to cellular survival. Given that translation consumes more than 50% of cell’s energy, it is not surprising that translation is tightly regulated and that perturbations in translation are not well tolerated. Conversely, many aberrant cellular processes require that translation machinery and translation output (i.e. proteome) have to be modified. This is often the case in various stress response and disease states, and is probably best exemplified in cancer. A key advantage of translational control is the ability of cells to rapidly reprogram the protein output in response to various external and internal triggers, thereby fine tuning mechanism that tips the balance between cell survival and death1.
Two aspects of translational control are particularly interesting; understanding of the nature of the regulatory mechanism(s) and control elements that allow for specific translation, and identification of specific mRNAs and their protein products that participate in the cellular response to the particular trigger that elicited selective translation in the first place. Polysome profiling is a technique that allows effective study of both processes.
The overall goal of the polysome profiling technique is to study and quantify the translation state of specific mRNAs under different cellular conditions. The principle of the technique is to capture mRNA translation by ‘‘freezing’’ actively translating ribosomes on different transcripts and separate the resulting polyribosomes by ultracentrifugation on a sucrose gradient, thus allowing for a distinction between highly translated transcripts (bound by several ribosomes) and poorly translated ones (bound by one or two ribosomes). In this particular protocol, the antibiotic cycloheximide that binds to the 60S ribosomal subunit and blocks the release of deacetylated tRNA from the ribosome E site2, is used to inhibit ribosome translocation and stall ribosomes on translating mRNAs. However, other blocking agents such as emetine that also blocks translation elongation3, can be used.
Unlike the western blotting and 35S-Methionine labeling techniques that measure the final output of the translation process, polysome profiling has the advantage of measuring ribosomes association with actively-translated mRNAs, thus allowing for a more detailed study of translation mechanisms under different conditions. Hence, the technique can be easily adapted to specifically study different modes of translation4-6, proteins factors involved in translation regulation7-9, stress conditions affecting protein synthesis1,10,11 or the effects of mRNA structures such as IRES or upstream open reading frames on protein synthesis12-14. Nowadays, polysome profiling applications are even broader with the use of DNA microarrays 15 or next-generation sequencing 16,17 to analyze polysomes-associated mRNAs.
Protocol
NOTE: A note on working with RNA: Take standard precautions to protect against RNA degradation by RNases. Use gloves, barrier pipette tips, RNase-free plasticware and chemicals in all steps of the protocol. Use ultrapure or DEPC-treated water for all solutions. Decontaminate any suspected surfaces with RNase inactivation solution following the manufacturer’s protocol.
1. Preparation of Solutions.
Prepare 500 ml of Basic Solution (0.3 M NaCl; 15 mM MgCl2.6H2O; 15 mM Tris-HCl, pH 7.4). Mix using a stir bar until the solution is clear and pass through a 0.45 µm filter. Store at room temperature.
- Prepare 10 and 50% Sucrose Solutions and 60% Chase Solution as follows:
- For 10% Sucrose Solution (w/v), in a 50 ml Centrifuge tube add 5 g sucrose and fill to 50 ml with Basic Solution. For 50% Sucrose Solution (w/v), in a 50 ml centrifuge tube add 25 g sucrose and fill to 50 ml with Basic Solution.
- For 60% (w/v) Chase Solution, in a 50 ml centrifuge tube add 30 g sucrose and fill to 50 ml with Basic Solution. Add 100 µl of saturated bromophenol blue solution (add bromophenol blue to water until not more will dissolve; centrifuge to pellet the undissolved powder) to the 60% chase solution. Mix using a vortex and verify complete dissolution.
- Store solutions at 4 °C until needed.
Prepare RNA lysis buffer. Prepare this buffer fresh on the day of the experiment. Prepare 500 µl of RNA lysis buffer for each sample. To 1 ml of Basic Solution add 10 µl Triton X-100 (1% [v/v] final), 1 µl of 100 mg/ml cycloheximide in DMSO (CHX, 0.1 mg/ml final) and 3.5 µl RNasin (140 U/ml). Mix using a vortex. Keep on ice. NOTE: Cycloheximide is highly toxic and may cause mutations. Avoid skin contact and inhalation. To avoid handling the powder form too often, prepare a larger quantity of 100 mg/ml stock in DMSO, aliquot and keep at -80 °C for long term storage. Keep working stock at -20 °C.
The day of the experiment, prepare Sucrose+CHX Solutions by adding CHX (final concentration of 0.1 mg/ml) to the desired volume of 10% and 50% sucrose solution (~7 ml of each solution per gradient).
2. Preparation of Sucrose Gradient Using an Automated Gradient Maker
Turn the gradient maker ‘‘ON’’ and level the plate that will hold the gradients (criticalstep!) Click on ‘‘Done’’ when plate is leveled.
- Select on the menu the gradient program to be run:
- Press ‘‘GRAD’’ menu and select ‘‘LIST’’.
- Select “SW41”.
- Scroll through the list and select the ‘‘10 - 50% gradient - 11 steps’’ program by pressing the ‘‘RUN’’ button.
Place a conical ultracentrifuge tube (SW-41 tube, 14 x 89 mm) into the Marker Block and use the provided fine tube marker to trace a mark on the tube along the upper step of the Marker Block. Place tubes in a fitting rack on a steady surface. NOTE: This mark will be the filling guide for layering the 10 and 50% sucrose solutions.
Attach the two provided cannula to two 10 ml syringes and wash by pipetting up and down with warm distilled water containing RNase inactivation solution. Make sure to remove all traces of water afterwards.
Fill one of the syringes with 10% sucrose+CHX and insert the cannula at the bottom of the ultracentrifuge tube. Gently dispense 10% sucrose solution until it goes just past the mark made on the tube. NOTE: Avoid making bubbles. If bubbles form, gently tap tube to displace them.
Fill the other syringe with 50% sucrose+CHX, wipe extra liquid off with a wipe and gently insert cannula at the bottom of the ultracentrifuge tube. Gently dispense the 50% sucrose solution until it reaches exactly the mark. Quickly and smoothly remove the cannula. NOTE: The 10% sucrose should rise in the tube and form a meniscus at the top. If not, add more 10% sucrose solution at the surface.
Insert the provided cap by tilting the ultracentrifuge tube slightly and displacing all air bubbles. Remove excess liquid in the cap’s reservoir with a micropipet.
Wipe excess liquid along the tube wall and place on the gradient maker plate.
Press the ‘‘RUN’’ button. NOTE: The plate will tilt at the specified angle, pause for 5 sec and the rotations to form the gradient will begin.
When gradient formation is complete (about 2 min, the machine will beep), gently remove the ultracentrifuge(s), place on rack and swiftly remove cap in an upward movement to avoid disturbing the gradient.
Keep gradient(s) on ice. Don’t disturb! Alternatively, gradients keep overnight at 4 °C.
- Alternatively, use a two chamber gradient maker to make gradients as follows:
- Rinse tubing and gradient maker with RNase inactivation solution and RNase-free water.
- Add 5 ml of 50% sucrose+CHX to proximal chamber of gradient maker and ensure that any air between the two chambers is removed.
- Add 5 ml of 10% sucrose+CHX to distal chamber of gradient maker.
- Add 0.5 ml of 50% sucrose+CHX to bottom of a conical centrifuge tube (SW-41 tube, 14 x 89 mm) and place in tube holder.
- Turn on stirrer placed in proximal chamber, open stop-cocks and deposit gradient at speed set to “3” (about 1ml/min).
- Place gradient(s) on ice. Do not disturb. NOTE: Gradients can be kept overnight at 4 °C.
3. Cell Lysis and Ultracentrifugation.
Place SW-41 rotor and buckets at 4 °C and set centrifuge to 4 °C. NOTE: The cell lysis procedure is optimized for two 150 mm subconfluent (~70-80% confluent) plates of HEK293 cells per gradient and the volumes used may need to be adjusted for different cell lines.
Plate cells at the appropriate cell density in the growth media at least 24 hr prior to lysis.
Prepare an aliquot (20 ml per 150 mm plate) of growth media containing 0.1 mg/ml CHX.
Incubate cells in CHX + media (20 ml per 150 mm plate) for 3 min at 37 °C/5% CO2 to arrest and stabilize polysomes.
Working quickly, aspirate media from plates, place plates on ice and wash cells once with 10 ml ice cold phosphate buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 , 1.8 mM KH2PO4) supplemented with CHX (final concentration of 0.1 mg/ml) being careful to pipette slowly down the side of the dish wall to minimize lifting of the cell monolayer.
Aspirate PBS and add an additional 10 ml PBS+CHX to each plate, scrape cells with a cell lifter and transfer the total volume from both plates to a 50 ml Centrifuge tube on ice. Retain 1 ml of cell suspension in a microfuge tube on ice for downstream protein analysis.
Centrifuge the 50 ml tube of cells at 300 x g at 4 °C for 10 min.
Remove all PBS and resuspend pellet in 500 µl of ice cold RNA lysis buffer.
Transfer cell suspension to a 2 ml microcentrifuge tube.
Pipette up and down to lyse cells and incubate on ice for 10 min with occasional vortexing.
Centrifuge at 2,000 x g at 4 °C for 5 min to pellet nuclei.
Transfer supernatant to a new 2 ml microcentrifuge tube.
Centrifuge at 17,000 x g at 4 °C for 5 min to pellet cell debris.
Transfer supernatant to a new 2 ml microcentrifuge tube and use 2 µl to measure optical density at 260 nm (use lysis buffer as blank).
Remove 50 µl (10% of total) from each sample for analysis of total RNA. NOTE Lysate can be stored at -80 °C until needed.
Gently load equal A260 units onto each sucrose gradients (min. 10 OD, optimal 25 OD, in a maximum volume of 500 µl; dilute concentrated lysate with additional lysis buffer if needed).
Ensure gradients are within 0.1 g of each other prior to ultracentrifugation.
Centrifuge gradients at 39,000 rpm (260,343 x g) for 1.5 hr at 4 °C.
During spindown keep rotor brake on and turn it off during deceleration when it reaches 1,000 rpm. NOTE: This step minimizes any disturbance of the gradients caused by abrupt changes in acceleration as the brush heads contact the rotor during braking.
4. Gradient Fractionation System Instrument Set-up.
- Set-up the instrument and blank the spectrophotometer with water as follows:
- Turn on the components and allow the lamp to warm-up for at least 15 min.
- Ensure fraction collector is set to the 'polysome' method (press folder icon twice; return arrow twice to return to home screen).
- Ensure the valve on the fraction collector is set to waste (arrow pointing to trash bin icon). Place a 250 ml Erlenmeyer flask containing distilled water under the waste collection tube.
- Select the following settings on the chart recorder: Set Sensitivity dial to 'SET LAMP AND OPTICS'; set Noise filter switch to '1.5'; set Peak separator dial to 'OFF'; set Chart speed dial to '30 cm/hr' (5 cm/10 min); set Baseline Adjust dial (on top of the spectrophotometer unit) to 'MAX OPEN'. NOTE: Optional: A digital recorder software can be used instead of the chart recorder. On the computer, click on 'start'. An acquisition window will open: under the Options menu, uncheck Pause graphics. Under Scaling, set the graph’s limits to -10 and 100 (can be changed on-the-fly during run). Click on the Save button to create a new file and record the run.
- Place the syringe and barrel in the pump and tighten into place (use provided screws and Allen key). NOTE: Do not overtighten.
- Fill the syringe with Chase Solution by using the ‘‘REV’’ command at RAPID speed. Place the pump in upright position and chase air through the tubing using the ‘‘FORWARD’’ command. Only use RAPID for this function and washes.
- Install an ultracentrifuge tube filled with RNase-free water.
- Pierce the ultracentrifuge tube with the cannula until the two black marks are visible (the dispensing hole is located between these two marks) and start syringe pump on ‘‘Manual’’ at 6.0 ml/min.
- As the water passes through the flow cell (when chase solution fills 1/3 of the tube), switch speed to variable and set to 1.0 ml/min.
- At a flow rate of 1.0 ml/min, adjust the Baseline Adjust dial on the spectrophotometer until the voltage on the chart is at zero (+/- 0.5 units). NOTE: Observe water exit from the waste tube into the Erlenmeyer flask.
- Set the desired Sensitivity to '0.5' or '1.0' AU. NOTE: 1.0 AU works best for 10 OD units in a 10 ml gradient. If OD is lower than 10, '0.5' sensitivity can be used to enhance signal.
- Push the Auto Baseline button and adjust the baseline setting to the 10% mark on the chart recorder by turning the Recorder offset dial (optional if using digital recorder).
- Once a stable baseline is achieved, turn the pump switch to 'OFF' and the Chart Speed dial to 60 cm/hr if using analogue chart recorder or 'OFF' if using digital recorder.
- Recover the Chase Solution by switching pump to 'REV' position (if needed one can increase flow rate back to 6.0 ml/min...DO NOT USE RAPID) until it reaches the first mark on the piercing cannula.
- Remove centrifuge tube and chase any air bubbles within the cannula by running a little bit of the Chase Solution through it. Wipe the piercing stage clean. NOTE: Some residual water may leak from the flow cell.
5. Gradient Fractionation and Collection of Samples.
Install and pierce the centrifuge tube containing the sucrose gradient.
Place eleven 2 ml microcentrifuge tubes in the fraction collector tray positions 1-11 such that the caps do not protrude upwards. Replace ‘‘tube #1’’ for now with a ‘‘waste tube’’ to collect any liquid left in the collector tubing.
Set the pump control dial to 'Manual' and flow rate on the syringe pump to '6.0 ml/min'
Press the 'Play' icon on the fraction collector. As soon as the arm reaches the first tube, press the ‘‘pause’’ button (this will position the arm and open the fraction-dispensing valve).
Start the pump by using the ‘‘FORWARD’’ command and monitor Chart recorder pen. When it starts deflecting upwards (indicating that sample is passing through the flow cell of the spectrophotometer), quickly replace ‘’waste tube’’ with ‘‘tube #1’’.
When the first drop is collected, quickly switch commands to 'Remote start/stop', ‘‘Variable (1.0 ml/min)’’ and press ‘‘Play’’ icon. NOTE: The pump is now controlled by the fraction collector interface and 1 ml fractions will be collected every min.
From this point on, A260 readings will appear on the digital recorder interface (or the analogue chart recorder). Make sure the ‘‘Record’’ button on the software is pressed!
After the blue Chase Solution enters the collecting tube or the last tube (usually tube 11), press the 'Stop' icon. NOTE: The dispensing valve should switch to the waste valve.
Keep fractions on ice until all gradients have been fractionated. At the end of the day fractions can be stored at -80 °C prior to protein or RNA isolation. If protein and RNA analysis is required, split each fraction in half (500 µl each for protein and RNA analysis).
6. Wash
NOTE: (this is an optional step to be performed only if multiple gradients are to be collected).
Submerge the waste tube into the Erlenmeyer flask containing ~50 ml of ultrapure water and reverse the pump using a 6 ml/min flow rate.
Stop the pump when the Chase Solution reaches the first mark on the piercing cannula.
Remove the centrifuge tube, chase any air out of the cannula and clean the piercing stage. NOTE: Some residual water may leak from the flow cell.
Repeat the fractionation procedure starting at step 5.1.
7. Final Clean up.
Disconnect the pump tubing from the bottom of the piercer and pump the Chase Solution into a 50 ml centrifuge tube using the rapid flow setting. Store the Chase Solution at 4 °C as it can be reused.
Connect pump tube to a 3 way tubing system and close the valve leading to the syringe. Connect one tube to a 50 ml syringe filled with distilled water and the other tube to the base of the piercer.
Install the ultracentrifuge tube used for the blanking step and pass at least 50 ml of distilled water through the spectrophotometer and collection tube (switch to collection menu on the interface by clicking the ‘‘Start/Pause’’ icon).
Remove tube, syringe (use Allen key to remove screws) and all other removable components and wash thoroughly using warm tap water and then rinse with ultrapure water. NOTE: Take special care when washing the plunger, barrel, tubing and piercing cannula. NOTE: Handle the glass syringe barrel with care! Store all components away from dust.
Wipe bottom of flow cell with a soft tissue moistened with distilled water. Wipe down the fraction collector tray and any other areas where the sucrose may have been spilled.
8. Isolation of RNA from Sucrose Fractions.
Prepare 50 µl of Proteinase K solution for every 1 ml of sucrose fraction (37.5 µl 10% SDS, 7.5 µl 0.5M EDTA, 1 µl Glycoblue, 4 µl of 20 mg/ml Proteinase K).
In 2 ml flat bottom tube, incubate 1 ml sucrose fraction with 50 µl of Proteinase K solution at 55 °C for 1 hr (if using 500 µl fractions adjust volume of Proteinase K solution accordingly).
Add an equal volume of phenol:chloroform:Isoamyl alcohol (125:24:1, preferably acidic (pH 4.5) to minimize DNA contamination) to the sucrose fractions. NOTE: Add an extra 200 µl of chloroform to fractions 7-10 because they are heavier and phases could get inverted. NOTE: phenol/chloroform can cause burns on contact and inhalation. Wear lab coat, safety goggles, and use a fumehood.
Vortex ~30 sec and centrifuge at maximum speed for 5 min at room temperature.
Remove ~80-90% of aqueous phase (do not touch interphase which could contain genomic DNA) and place in new tube.
Add an equal volume of chloroform and repeat step 8.4.
Remove aqueous phase being careful to avoid interphase.
Add 1:10 volume of 3 M sodium acetate, pH 5.2 (alternatively 5 M ammonium acetate can be used).
Add 1.5 volumes of chilled, absolute ethanol and vortex for 15 sec.
Precipitate overnight at -20 °C (or 1 hr at -80 °C if in a rush). NOTE: The samples can be left at -20°C for longer if needed.
Centrifuge at maximum speed for 30 min at 4 °C.
Wash the pellet with 1 ml of chilled RNase-free 70% ethanol.
Air dry the pellet and resuspend in 20 µl of RNase-free water.
Quantify total RNA by spectrophotometry to ensure adequate yield and purity (based on A260/280 ratio) and proceed to standard RT-qPCR analysis14 using equal volumes of each fraction and PCR primers specific to the mRNA(s) of interest.
9. Isolation of Proteins from Sucrose Fractions.
In addition to RNA, proteins can be isolated and identified in individual polysome fractions as well. Use one of many excellent protocols available for protein isolation, e.g. 18.
Representative Results
We have recently described a novel role for the tumour suppressor PDCD4 as a selective translation inhibitor of IRES-harboring mRNAs encoding apoptotic inhibitors XIAP and Bcl-xL12. Polysome profiling was a key technique to demonstrate the specific involvement of PDCD4 in IRES-mediated translation. HEK293 cells were transiently transfected to deplete endogenous levels of PDCD4 (Figure 1A) and then subjected to polysome profile analysis. We observed that reducing levels of PDCD4 did not impair global cellular translation, as judged by the lack of changes in the polysome profile when comparing siCTRL and siPDCD4 treated cells (Figure 1B). Similarly, polysome distribution of non-IRES variant of XIAP 19 was unchanged between treated and untreated cells (Figure 1C). In contrast, polysome distribution of the two IRES-harboring mRNAs, XIAP and Bcl-xL, was significantly altered. In the absence of PDCD4 the distribution of these two mRNAs is shifted into heavy ribopolysomes (Figure 1D, E). Since this is not accompanied by changes in steady-state levels of XIAP and Bcl-xL mRNA (Figure 1F) these results demonstrate enhanced translation of XIAP and Bcl-xL in cells with reduced PDCD4 levels, which was further confirmed by Western blotting (Figure 1G).
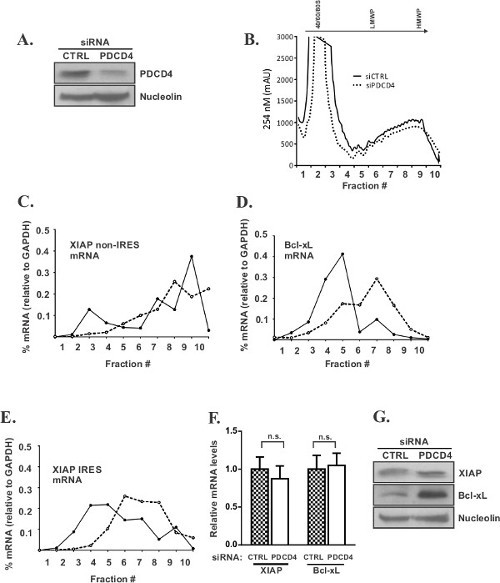 Figure 1: Polysome profiling identifies PDCD4 as selective inhibitor of XIAP and Bcl-xL translation.(A) HEK293 cells were treated with PDCD4 siRNA or control (CTRL), non-targeting siRNA, lysed and subjected to Western blot analysis with anti-PDCD4 and anti-Nucleolin antibodies to verify the extent of PDCD4 knock-down. (B) PDCD4 was knocked down as in (A) and cell lysates were subjected to polysome profiling. Representative polysome profile is shown in the top panel; distribution of the XIAP non-IRES (C), Bcl-xL (D), and XIAP IRES (E) mRNAs relative to that of GAPDH is shown as the percent of total mRNA (% mRNA) in each fraction. (F) Steady-state mRNA levels were measured by RT-qPCR in PDCD4 siRNA or control (CTRL) siRNA treated cells. (G) The lysates from (A) were also subjected to western blot analysis with anti-XIAP, anti-Bcl-xL, and anti-Nucleolin antibodies. (NB. Data presented in Figure 1 were originally published in 12) Please click here to view a larger version of this figure.
Figure 1: Polysome profiling identifies PDCD4 as selective inhibitor of XIAP and Bcl-xL translation.(A) HEK293 cells were treated with PDCD4 siRNA or control (CTRL), non-targeting siRNA, lysed and subjected to Western blot analysis with anti-PDCD4 and anti-Nucleolin antibodies to verify the extent of PDCD4 knock-down. (B) PDCD4 was knocked down as in (A) and cell lysates were subjected to polysome profiling. Representative polysome profile is shown in the top panel; distribution of the XIAP non-IRES (C), Bcl-xL (D), and XIAP IRES (E) mRNAs relative to that of GAPDH is shown as the percent of total mRNA (% mRNA) in each fraction. (F) Steady-state mRNA levels were measured by RT-qPCR in PDCD4 siRNA or control (CTRL) siRNA treated cells. (G) The lysates from (A) were also subjected to western blot analysis with anti-XIAP, anti-Bcl-xL, and anti-Nucleolin antibodies. (NB. Data presented in Figure 1 were originally published in 12) Please click here to view a larger version of this figure.
Discussion
Polysomal profiling is a powerful technique that allows for the quantification of the state of translation of specific transcripts under different treatment conditions. It is a very simple technique in principle yet it requires several steps of execution, some of which are critical for producing good polysome profiles. These key steps are: 1) using RNase-free chemicals and plasticware, 2) preparing good sucrose gradients (in our experience a 10-50% sucrose gradients work best for efficiently resolving 40/60S subunits and mRNAs with bound ribosomes), and 3) using cells that are exponentially growing and no more than 80% confluent in order to capture the highest rates of translation. This latter step should be taken into consideration when doing long cell treatments, such as gene knockdown or overexpression, so that the amount of cells to plate will be a function of how much the cells will be confluent at endpoint.
In the results presented here, polysome profile analysis was an important tool for assessing PDCD4 role in the translation regulation of the IRES-harboring mRNAs XIAP and Bcl-xL12. The fact that we did not observe any change in the general polysome profiles upon PDCD4 knock-down (Figure 1B) allowed us to conclude that PDCD4 did not impair global cellular translation under the conditions of the experiment. We next used RT-qPCR analysis to determine the distribution of the XIAP and Bcl-xL mRNAs in cells treated with PDCD4 or control siRNAs. qPCR is a method of choice for analyzing polysome profiles, as opposed to standard RT-PCR or Northern blotting, because it allows for a quantitative rather than semi-quantitative analysis of mRNA distribution and for the detection of subtle shifts in translational efficiency between treatments. Using this technique, we were able to show that the distribution of the XIAP and Bcl-xL mRNAs (expressed as a percent of total target mRNA across the gradient) was significantly shifted into heavy polyribosomes (mRNAs with a large number of ribosomes associated with them) after PDCD4 knock-down (Figure 1D, E), thus indicating an increase in translation of these mRNAs. This was further confirmed by an increase in XIAP and Bcl-xL protein levels but not in their steady-state RNA levels (Figure 1F, G). Importantly, the polysome distribution of the non-IRES variant of XIAP was unchanged between treated and untreated cells (Figure 1C). Alternatively, translational efficiency could be presented as a ratio of mRNA content in polysomes (usually fractions 5 to 10) versus monosomes (usually fractions 2 to 4)14.
The use of controls throughout the protocol is important in validating any polysome profiling result, as peaks observed from absorbance readings at 254 nm cannot on their own be attributed to ribosomal RNA (detergents, such as Triton X-100 strongly absorb in this region of the spectrum and may obscure 40/60S peaks at the top of the gradient). Blank gradients without lysate and/or with lysate buffer need to be run to determine the relative contribution of the buffers to the absorbance at 254 nm. Artefactual higher order structures can also lead to erroneous interpretations of polysomes where there are in fact none (e.g. “pseudo-polysomes”20). Sequestering magnesium (used throughout the procedure to stabilize 80S ribosomes) with the divalent cation chelator EDTA added to lysates and to the gradient buffer (20 mM for 15 min) will cause separation of the ribosome into its component small (40S) and large (60S) subunits. Thus, if absorbance peaks recorded at 260 nm are indeed polysomes, EDTA treatment will collapse the profile such that a single maximum will be observed near the top of gradient that corresponds to free mRNA and ribosomal units (data not shown). Coincident with EDTA-induced collapse of the profile, all of the specific targets analyzed by qPCR should now be found at the top of the gradient.
The number of ribosomes associated with a mRNA is not always an indicator of how efficiently that particular mRNA is being translated. Indeed, there are specific contexts in which active translation on polysomes becomes stalled 21,22 which the reader should be aware of. Determining whether or not transcripts are actively engaged with the translation machinery can be done by a) treating the sample with puromycin, which unlike EDTA, causes 'run-off' of ribosomes that are actively translocating across a mRNA, or b) treating the sample with homoharringtonine which inhibits translocation of only the first ribosome at the start codon, again causing 'run-off' of any downstream translocating ribosomes.
The use of control transcripts for qPCR analysis is also critical. The selection of transcripts that are not affected by different treatment conditions such as some housekeeping genes (GAPDH, Actin, Tubulin, Nucleolin), ribosomal mRNAs (18S, RPL13A) or in this case the non-IRES variant of the XIAP IRES, is important in verifying if the treatment’s effect on translation is specific or generalized. These control transcripts can be used to normalize the distribution of the analyzed mRNAs as was done here (XIAP and Bcl-xL mRNAs were normalized to GAPDH mRNA and expressed as a percentage of the total mRNA content represented by the sum of mRNA content in every fraction) or alternatively, target and control mRNAs can be expressed as absolute values and shown separately. qPCR analysis of the input lysates (usually 10% of total volume) is another step that will control for the distribution of specific mRNAs. Ideally, the mRNA content of a specific transcript in the input lysate should be comparable to the sum of mRNA content in all 10 fractions analyzed. Finally, an optional step to control for the phenol:chloroform extraction step is to spike each polysome fraction with an in vitro transcribed reporter mRNA (such as CAT, chloramphenicol acetyl transferase) that will be subsequently quantified by qPCR and can even be used to normalize the data 14.
As with any technique, the polysome profiling presents some limitations. The fact that usually a minimum of 10 fractions (more subtle shifts in translation efficiency may require 20 or more) need to be collected in order to have nice polysome distributions makes this step limiting, in the sense that only a maximum of 4 samples can be analyzed at a time to be able to comfortably handle RNA extraction and qPCR analysis of 40 samples and 4 input controls at once. The sample size can easily add up with how many transcripts are to be analyzed and how many qPCR replicates to do, and this can be costly. Another limitation of a qPCR-based polysomal profiling analysis is that it can only be done on a candidate-based approach (where the transcripts to be analyzed are known beforehand) and cannot be applied for genome-wide analysis of translation. However, at the beginning of the 21st century, investigators started to interrogate their polysomal RNA at a genomic level using DNA microarrays 15 and more recently with next-generation sequencing 16,17. In this powerful approach, polysomal profiling is performed as described in this protocol except that rather than performing qPCR analysis of individual fractions, monosomes fractions (usually fraction 2-4) and polysomes fractions (usually fractions) are pooled together and variations in global translation analyzed by DNA microarray or whole-genome RNA sequencing. Hence with this approach, it is possible to assess all the mRNAs whose distribution shifts from polysomes to monosomes (decrease in translation) or vice-versa (increase in translation) upon a specific treatment. Validation and quantification of specific mRNA polysomes distribution can then be done by qPCR. An even more powerful use of the polysomal profiling principle is ribosome profiling. Further high throughput extension of this technique was reported recently that allows for simultaneous monitoring of hundreds of polysome fractions 23. This provides a clear advantage when probing translational efficiencies of mutant collections or in a large-scale RNAi screens. Ribosome profiling is a technique that takes advantage of next generation sequencing to map RNA fragments protected by ribosomes (ribosome footprints) engaged in protein synthesis24,25. In this technique, ribosome translocation can be inhibited with cycloheximide (reversible) or emetine (irreversible) followed by cell lysis and RNase digestion to yield a population of ribosome footprints. Ribosomes are enriched, total RNA is isolated, and contaminating ribosomal and mitochondrial ribosomal RNA is removed. RNA footprints of approximately 26-28 nucleotides are isolated, reverse transcribed, converted into a cDNA library and sequenced26. Unlike standard polysome profiling, this approach not only identifies specific mRNAs that are translationally regulated but also yields mRNA-specific information at codon resolution such as the exact occupation and density of ribosomes along a specific transcript. This is particularly of interest for mRNAs with specific modes of translation such as IRES-harboring mRNAs, as it could give more information on where on the transcript ribosome-recruitment is occurring.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are grateful to the past and present members of the laboratory, in particular Urszula Liwak, for critical discussion regarding polysome profiling protocols and sharing of data. This work was supported by operating grants from the Canadian institutes of Health Research, Cancer Research Society, and the National Science and Engineering Research Council of Canada to MH. MDF was supported by the Vanier Canada Graduate Scholarship Doctoral Award and the Ontario Graduate Scholarship, and this work forms part of her Ph.D. dissertation.
References
- Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6(4):318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- Obrig TG, Culp WJ, McKeehan WL, Hardesty B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem. 1971;246(1):174–181. [PubMed] [Google Scholar]
- Tscherne JS, Pestka S. Inhibition of protein synthesis in intact HeLa cells. Antimicrob Agents Chemother. 1975;8(4):479–487. doi: 10.1128/aac.8.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard CK, Lykke-Andersen J. Translational coregulation of 5'TOP mRNAs by TIA-1 and TIAR. Genes Dev. 2011;25(19):2057–2068. doi: 10.1101/gad.17355911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakor N, Holcik M. IRES-mediated translation of cellular messenger RNA operates in eIF2alpha- independent manner during stress. Nucleic Acids Res. 2012;40(2):541–552. doi: 10.1093/nar/gkr701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney C, et al. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep. 2014;6(1):9–17. doi: 10.1016/j.celrep.2013.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jivotovskaya AV, Valasek L, Hinnebusch AG, Nielsen KH. Eukaryotic translation initiation factor 3 (eIF3) and eIF2 can promote mRNA binding to 40S subunits independently of eIF4G in yeast. Mol Cell Biol. 2006;26(4):1355–1372. doi: 10.1128/MCB.26.4.1355-1372.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross JD, et al. Ribosome Loading onto the mRNA Cap Is Driven by Conformational Coupling between eIF4G and eIF4E. Cell. 2003;115(6):739–750. doi: 10.1016/s0092-8674(03)00975-9. [DOI] [PubMed] [Google Scholar]
- Graber TE, Baird SD, Kao PN, Mathews MB, Holcik M. NF45 functions as an IRES trans-acting factor that is required for translation of cIAP1 during the unfolded protein response. Cell Death Differ. 2010;17(4):719–729. doi: 10.1038/cdd.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs KA, Stoneley M, Bushell M, Willis AE. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol Cell. 2008;100(1):27–38. doi: 10.1042/BC20070098. [DOI] [PubMed] [Google Scholar]
- Uniacke J, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature. 2012;486(7401):126–129. doi: 10.1038/nature11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liwak U, et al. Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol Cell Biol. 2012;32(10):1818–1829. doi: 10.1128/MCB.06317-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Occhi G, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. 2013;9(3) doi: 10.1371/journal.pgen.1003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faye MD, et al. Nucleotide composition of cellular internal ribosome entry sites defines dependence on NF45 and predicts a posttranscriptional mitotic regulon. Mol Cell Biol. 2013;33(2):307–318. doi: 10.1128/MCB.00546-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Q, Schummer M, Hood L, Messenger Morris DR. RNA translation state: the second dimension of high-throughput expression screening. Proc Natl Acad Sci U S A. 1999;96(19):10632–10636. doi: 10.1073/pnas.96.19.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnik EM, et al. Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum. Genome Biol. 2013;14(11) doi: 10.1186/gb-2013-14-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri A, Maitra U, Evans T. Translation initiation factor eIF3h targets specific transcripts to polysomes during embryogenesis. Proc Natl Acad Sci U S A. 2013;110(24):9818–9823. doi: 10.1073/pnas.1302934110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin F, et al. The fragile X mental retardation protein is associated with poly(A)+ mRNA in actively translating polyribosomes. Hum Mol Genet. 1997;6(9):1465–1472. doi: 10.1093/hmg/6.9.1465. [DOI] [PubMed] [Google Scholar]
- Riley A, Jordan LE, Holcik M. Distinct 5' UTRs regulate XIAP expression under normal growth conditions and during cellular stress. Nucleic Acids Res. 2010;38(14):4665–4674. doi: 10.1093/nar/gkq241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R, Hentze MW. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature. 2007;447(7146):875–878. doi: 10.1038/nature05878. [DOI] [PubMed] [Google Scholar]
- Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber TE, et al. Reactivation of stalled polyribosomes in synaptic plasticity. Proc Natl Acad Sci U S A. 2013;110(40):16205–16210. doi: 10.1073/pnas.1307747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ringquist S, Cho AH, Rondeau G, Welsh J. High-throughput polyribosome fractionation. Nucleic Acids Res. 2004;32(10) doi: 10.1093/nar/gnh077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15(3):205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7(8):1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]