

Abstract
INO80 chromatin remodeling complexes regulate nucleosome dynamics and DNA accessibility by catalyzing ATP-dependent nucleosome remodeling. Human INO80 complexes consist of 14 protein subunits including Ino80, a SNF2-like ATPase, which serves both as the catalytic subunit and the scaffold for assembly of the complexes. Functions of the other subunits and the mechanisms by which they contribute to the INO80 complex's chromatin remodeling activity remain poorly understood, in part due to the challenge of generating INO80 subassemblies in human cells or heterologous expression systems. This JOVE protocol describes a procedure that allows purification of human INO80 chromatin remodeling subcomplexes that are lacking a subunit or a subset of subunits. N-terminally FLAG epitope tagged Ino80 cDNA are stably introduced into human embryonic kidney (HEK) 293 cell lines using Flp-mediated recombination. In the event that a subset of subunits of the INO80 complex is to be deleted, one expresses instead mutant Ino80 proteins that lack the platform needed for assembly of those subunits. In the event an individual subunit is to be depleted, one transfects siRNAs targeting this subunit into an HEK 293 cell line stably expressing FLAG tagged Ino80 ATPase. Nuclear extracts are prepared, and FLAG immunoprecipitation is performed to enrich protein fractions containing Ino80 derivatives. The compositions of purified INO80 subcomplexes can then be analyzed using methods such as immunoblotting, silver staining, and mass spectrometry. The INO80 and INO80 subcomplexes generated according to this protocol can be further analyzed using various biochemical assays, which are described in the accompanying JOVE protocol. The methods described here can be adapted for studies of the structural and functional properties of any mammalian multi-subunit chromatin remodeling and modifying complexes.
Keywords: Biochemistry, Issue 92, chromatin remodeling, INO80, SNF2 family ATPase, structure-function, enzyme purification
Introduction
Evolutionarily conserved SNF2 family chromatin remodeling complexes are key regulators of chromatin organization and DNA accessibility1. These remodeling complexes always include a central SNF2-like ATPase subunit, which, in some cases, assembles with various accessory proteins and forms multi-subunit macro-molecular assemblies. To study the molecular details of the ATP-dependent chromatin remodeling process, it is important to understand the contributions of given subsets of subunits and/or domain structures to activities of the complexes. Such analyses require the ability to generate highly purified mutant complexes that lack particular protein subunits or domain structures.
Previous structure-function studies of ATP-dependent chromatin remodeling complexes have widely focused on the yeast model system due to the superior manipulability of the yeast genome (see, for example, refs1-4). Given the conservation of subunit composition and functionality among orthologous remodeling complexes, studies of the structure and function of yeast remodeling complexes have provided important insights into their counterparts in higher eukaryotes. Nonetheless, appreciable species-specific differences among remodeling complexes do exist, resulting from gain or loss of species-specific subunits, gain or loss of species-specific domains of conserved subunits, and sequence variability within conserved domains of conserved subunits. Such differences can in principle be driven by the need for higher eukaryotic cells to adapt to new molecular and cellular environments. Thus, understanding how subunits of higher eukaryotic remodeling complexes contribute to the nucleosome remodeling process is valuable, because it not only sheds light on basic mechanisms of the ATP-dependent chromatin remodeling process, but also can provide valuable insight into the mechanisms by which chromatin structure and gene expression in higher eukaryotes are regulated.
Thus far, there have been only limited structural and functional studies of multi-subunit mammalian chromatin remodeling complexes, due in part to the difficulties in obtaining biochemically defined chromatin remodeling complexes and subcomplexes. We have partially circumvented these difficulties with the procedures described below, in which immunoaffinity purification is used to prepare intact INO80 or INO80 subcomplexes from human cells stably expressing N-terminally FLAG epitope tagged wild type or mutant versions of Ino805-7 (Figure 1). To obtain intact INO80 complexes from human cells, Flp-mediated recombination is used to generate transgenic HEK293 cell lines stably expressing FLAG epitope tagged cDNAs encoding subunits of the INO80 complex8-10. Because over-expression of INO80 subunits can be somewhat toxic, it is necessary to isolate and maintain clonal cell lines under selective conditions to ensure stable transgene expression during the many passages needed for expansion of large-scale cell cultures. To obtain smaller INO80 subcomplexes that contain only a subset of subunits, we have successfully used two approaches (Figure 2A,B). In the first, we generate HEK293 Flp-In cell lines stably expressing mutant versions of Ino80 that lack domains required for interaction with specific subunits5. Alternatively, siRNA-mediated knockdown is used to deplete the desired subunit from cells expressing an appropriate FLAG-tagged INO80 subunit (unpublished data). Finally, to purify the human INO80 complexes, FLAG agarose based chromatography11 is used to enrich an INO80-containing fraction from nuclear extracts, thereby effectively reducing the presence of contaminating cytosolic proteins in the final fraction containing purified INO80 or INO80 subcomplexes.
Protocol
1. Generation and Culture of HEK293 Stable Cell Lines Expressing Full Length or Mutant Versions of FLAG Epitope-tagged Ino80 or Other INO80 Complex Subunits
Clone cDNA encoding full length or mutant human Ino80 ATPase or another INO80 subunit into the mammalian expression vector pcDNA5/FRT with an in-frame, N terminal FLAG epitope tag.
Confirm the sequence of the inserted cDNAs by DNA sequencing before proceeding.
To perform the transfection, grow Flp-In HEK293 cells in 10 cm tissue culture dishes in a medium containing DMEM (Dulbecco's Modified Eagle Medium), 5% glutamine, and 10% FBS (fetal bovine serum).
When cells reach ~70% confluency, add to each tissue culture dish a mixture of 40 µl of FuGENE6 transfection reagent, 0.5 µg of the appropriate pcDNA5/FRT expression plasmid, and 9.5 µg of pOG44, which encodes Flp recombinase, in a total volume of 800 μl.
48 hr later, trypsinize and split cells at a ratio of 1:30 into 10 cm dishes, and grow them in DMEM with 5% glutamine, 10% FBS, and 100 µg/ml hygromycin B for 3 - 4 weeks. Change the culture medium whenever it begins to turn yellow (typically every 3 - 5 days).
- To identify positive clones that express the highest level of FLAG-tagged protein, select individual hygromycin B-resistant colonies and transfer them to a single well of a 24-well plate.
- Once the cells reach 80% confluency, harvest cells from each well in ~1 ml PBS and pellet by centrifugation at 1,000 x g for 5 min.
- After removing the supernatant, resuspend the cell pellet in 60 µl of SDS-PAGE sample buffer.
- Subject half of the resuspended cell pellet to SDS page and western blotting to monitor expression of the FLAG tagged bait protein; save the other half for future analyses.
- If no FLAG-tagged human Ino80 protein is detected in cell lysates, expand the initial clonal cell population further by plating cells from single wells of a 24-well plate into 15 cm tissue culture dishes.
- To harvest cells from 15 cm dishes, resuspend near confluent cells in ice cold PBS and transfer to a 50 ml conical tube.
- Bring to a final volume of 50 ml by adding additional PBS.
- Pellet cells at 1,000 x g for 5 min, and remove as much as possible of the supernatant.
- Resuspend the cell pellet in 1 ml of Lys450 buffer (20 mM HEPES-NaOH pH 7.9, 450 mM NaCl, 0.5% TritonX-100, 10 mM KCl, 4 mM MgCl2, 0.2 mM EDTA, 10% glycerol, 1 mM DTT, 200 µM PMSF, and 1:1,000 Protease Inhibitor Cocktail). NOTE: here and elsewhere, always add DTT, PMSF, and protease inhibitor cocktail to buffers immediately before beginning an experiment.
- Immuno-precipitate FLAG-tagged proteins from the resulting whole cell lysate using 20 µl of anti-FLAG agarose gel, and analyze the FLAG eluates by western blotting. [For details of immunoprecipitation procedure, see step 5.]
Prepare frozen stocks of desired clonal cell lines12 and store them in liquid nitrogen until use.
2. Growing HEK293 Cell Lines in Roller Bottles
For large scale preparation of INO80 complexes, culture cells in 10 - 20 roller bottles; a typical yield from each roller bottle is ~1 ml of packed cells.
To each roller bottle, add 200 ml DMEM, 5% glutamine, and 10% calf serum, without hygromycin B.
Transfer all of the cells from a single near-confluent 15 cm dish into each roller bottle
Place roller bottles into a 37 °C roller bottle incubator and rotate at 0.2 rpm.
Once cells reach ~70% confluency, pour off and discard the medium.
Add ~50 ml of ice cold PBS to each bottle. Holding bottles horizontally, gently swirl to loosen the cell monolayer.
Transfer the resuspended cells to 250 ml plastic conical bottles and place on ice.
Rinse roller bottles with additional PBS, transferring it sequentially from bottle to bottle. When rinse solution is no longer clear (typically after being used to rinse about 5 bottles), add it to the cell suspension.
Pellet cells by centrifugation at 400 x g for 10 min.
Gently resuspend the cells in PBS, combine them into a single 250 ml conical bottle, and keep on ice until further processing.
3. siRNA-mediated Knockdown of INO80 Subunits in Cells Expressing Another FLAG-tagged INO80 Subunit
To obtain INO80 subcomplexes lacking a single subunit, use FLAG-immunopurification to purify INO80 complexes from siRNA treated cells or cells stably expressing shRNA. The “reverse” siRNA (small interfering RNA) transfection protocol described here is optimized for HEK293 cells growing in 15 cm dishes. The protocol is for a single 15 cm dish of cells and should be scaled up accordingly depending on the number of cells needed. To prepare biochemically useful amounts of INO80 complex from siRNA-treated cells, one should scale up to cultures grown in 40 15 cm dishes; these will yield approximately 2 - 4 ml of packed cell pellet.
Grow HEK293 cells stably expressing the desired subunit of the INO80 complex to near confluency in 15 cm dishes.
Add a volume of of siRNA resuspension buffer (20 mM KCl, 6 mM HEPES-pH 7.5, and 0.2 mM MgCl2) sufficient to prepare a 50 µM stock solution of siRNA to a tube containing lyophilized siRNA. Pipette the solution up and down a few times and incubate on a nutator for 30 min at RT to ensure the siRNA is fully dissolved.
Prepare a transfection cocktail containing siRNAs and transfection reagent. Mix 10 μl of the 50 µM siRNA stock solution with 32 µl Lipofectamine RNAiMAX and add it to 4 ml of Opti-MEM Reduced Serum Medium with very gentle mixing; allow all reagents to equilibrate to RT before use.
Incubate the mixture at RT for 30 min.
- Prepare Flp-In HEK293 cells stably expressing the desired INO80 subunit for transfection.
- During the incubation of Step 3.4, wash cells in the 15 cm dishes once with RT PBS.
- After removing PBS, treat cells with 1 ml trypsin just until they begin to lift off the plate.
- Immediately add 10 ml of complete medium (DMEM + 5% glutamine + 10% FBS) to trypsinized cells and mix gently. Collect cells by centrifugation at 1,000 x g for 5 min at RT.
- Resuspend the cell pellet in ~4 ml complete medium, and count the cell resuspension using a haemocytometer.
- Dilute cells with complete medium to a concentration of ~5.4 x 106/ml.
To each 15 cm dish, add 15 ml complete culture medium followed by 4 ml transfection cocktail. Swirl gently to ensure the medium and transfection cocktail are thoroughly mixed. Finally, add one ml of cell suspension and again swirl gently to disperse cells uniformly.
After 60 hr of culture in a 37 °C, 5% CO2 incubator, gently remove medium, resuspend cells in ice cold PBS, and proceed immediately to prepare nuclear extracts.
4. Preparation of Nuclear Extracts
This procedure has been modified from the protocol of Dignam13 and can be scaled up or down depending on the size of starting cell pellets. Typically, 1 ml of packed cell pellet yields 1 ml of final nuclear extract. All buffers should be ice cold, and all steps should be performed in a cold room or on ice if a suitable cold room is not available.
- Isolation of nuclei:
- Gently transfer cells to a suitably sized (15 or 50 ml) graduated conical tube and spin at 1,000 x g for 10 min at 4 °C.
- Remove the supernatant, and measure the volume of the packed cell pellet. 1 ml of packed cells corresponds to ~3 x 108 HEK293 cells.
- Add 5 packed cell volumes of Buffer A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl and freshly added 1 mM DTT, 200 µM PMSF, and 1:1,000 Protease Inhibitor Cocktail). Resuspend the cell pellet by gentle pipetting.
- Incubate on ice for exactly 10 min.
- Pellet the cells at 1,000 x g for 10 min at 4 °C, and remove the supernatant.
- The size of the cell pellet should increase up to 2-fold during incubation in Buffer A.
- Resuspend the cells in two packed cell volumes of buffer A, and transfer the cell suspension to an appropriately sized Dounce tissue homogenizer. If starting with less than 2 ml of packed cells, use a 7 ml homogenizer; for 2 - 4 ml packed cells, use a 15 ml homogenizer; and for 10 or more ml of packed cells, use a 40 ml homogenizer.
- Homogenize the cell suspension with the LOOSE glass pestle of the Dounce homogenizer until 90% of the cells stain positively with 1% trypan blue.
- Transfer the suspension to a 45 ml high-speed centrifuge tube and spin at 25,000 x g for 20 min at 4 °C in a JA-17 or similar rotor (see Table of Materials/Equipment).
- From the nuclear pellet, remove the supernatant, which contains cytosolic proteins or proteins that leak out of the nucleus during fractionation.
- Extracting nuclei with salt:
- Add Buffer C (20mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA, and freshly added 1 mM DTT, 200 µM PMSF, and 1:1,000 Protease Inhibitor Cocktail) to the nuclear pellet; use 2.5 ml Buffer C for every 3 ml of starting packed cell volume (~1 x 109 cells).
- Using a glass rod or a pipette, dislodge the nuclear pellet from the wall of the tube and transfer the entire mixture to a Dounce homogenizer of an appropriate size.
- Resuspend the nuclei by homogenizing the mixture with two strokes of a LOOSE pestle.
- Transfer the resuspended nuclear fraction into a chilled beaker. Choose a beaker such that the suspension will fill the beaker to at least 0.5 cm deep.
- To extract nuclear proteins from chromatin or other insoluble structures, gradually increase the salt concentration of the suspension to 0.42 M NaCl by dropwise addition of 5 M NaCl while gently stirring the suspension with a pipette or glass rod on ice; once all of the 5 M NaCl has been added, the solution should become very viscous or gel-like. Calculate the volume of 5 M NaCl needed to bring the solution to a final concentration of 0.42 M NaCl according to the following formula:
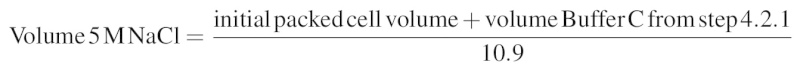
- Carefully transfer the viscous suspension into 10 ml polycarbonate tubes for a Type 70.1 Ti rotor (or equivalent) or 70 ml polycarbonate bottles for a Type 45 Ti or similar rotor (see Table of Materials/Equipment). Seal tightly with parafilm if using 10 ml tubes or with cap assembly if using 70 ml bottles.
- Slowly rock the sealed tubes at 4 °C for 30 min using a nutator.
- Spin the samples in a Type 45 Ti or 70.1 Ti rotor for 30 min at 120,000 x g at 4 °C.
- Transfer the supernatant to a single plastic tube or bottle. This supernatant is the nuclear extract, and the pellet contains chromatin and other nuclear debris.
- Divide the nuclear extract into conveniently sized aliquots, freeze it in liquid nitrogen, and store it at -80 °C.
5. Immunoaffinity Purification of the Human INO80 or INO80 Subcomplexes
To thaw frozen nuclear extract, place tubes containing the extract on the benchtop or roll tubes between hands until the frozen material becomes a slurry. Then place the tubes on ice or in the cold room until the extract is completely thawed.
Transfer the thawed nuclear extract to 10 ml polycarbonate ultracentrifuge tubes, and spin at 100,000 x g for 20 min at 4 °C in a Type 70.1 Ti rotor or equivalent to remove any precipitate that may have formed during the freeze-thaw cycle.
Transfer the supernatant to a 15 ml conical tube. Add fresh DTT, PMSF, and Protease Inhibitor Cocktail to final concentrations of 1 mM DTT, 200 µM PMSF, and 1:1,000 Protease Inhibitor Cocktail.
To prepare anti-FLAG agarose for the immunopurification, transfer 200 µl of 50% slurry of anti-FLAG agarose beads to a 1.5 ml microcentrifuge tube using a P200 or similar pipette with a tip from which the end has been cut off with a clean scalpel or razor blade.
Pellet the beads by centrifugation in a benchtop microcentrifuge at 8,000 x g for 30 sec. Remove the supernatant, and wash the beads by resuspending the beads in 1 ml of Lys450 buffer, and pellet the beads at 8,000 x g for 30 sec. Wash the beads two more times.
Resuspend the washed anti-FLAG agarose beads in about 100 µl of the nuclear extract using a P200 or similar adjustable volume pipette with a tip from which the end has been cut off with a clean scalpel or razor blade and, using the same tip, transfer the resuspended beads to the 15 ml conical tube containing the extract. Repeat a few times until all of the beads have been transferred to the 15 ml tube.
Incubate the extract/bead mixture for 4 hr at 4 °C with slow rotation on a laboratory rotator. OPTIONAL: Include benzonase at a concentration of 25 Units/ml to remove contaminating DNA.
Collect the FLAG agarose by centrifugation at 1,000 x g for 5 min at 4 °C.
Resuspend in 10 ml Lys450 to wash, incubate 5 min at 4 °C with gentle rocking on a nutator. Pellet the beads at 1,000 x g for 5 min at 4 °C.
Resuspend in 100 - 150 µl of Lys450 and transfer beads to a 1.5 ml microcentrifuge tube. Continue to rinse the 15 ml conical tube with 100 - 150 µl of Lys450 until all the beads have been transferred to the microcentrifuge tube.
Spin down the beads at 8,000 x g for 30 sec at 4 °C in a microcentrifuge. Wash three times more with 1 ml Lys450 and once with 1ml EB100 buffer (10 mM HEPES pH 7.9, 10% glycerol, 100 mM NaCl, 1.5 mM MgCl2, 0.05% TritonX-100, and freshly added 1 mM DTT, 200 µM PMSF, and 1:1,000 Protease Inhibitor Cocktail).
To elute bound proteins, add 200 µl EB100 buffer containing 0.25 mg/ml 1x FLAG peptide. Incubate 30 min at 4 °C each on a nutator.
Pellet the beads at 8,000 x g for 30 sec at 4 °C in a microcentrifuge. Transfer the supernatant, which contains the eluted INO80 complex, to a fresh microcentrifuge tube.
Repeat the elution four more times, and pool all the supernatants into a single tube.
To remove any residual FLAG-agarose beads from the eluted protein fraction, pass the eluate through an empty spin column.
Concentrate the eluted protein fraction ~10-fold using an ultra centrifugal filter device (50,000 molecular weight cutoff).
To remove the FLAG peptide, pass the concentrated protein fraction sequentially through two desalting columns.
Divide the purified, desalted protein fraction into 20 µl aliquots, frozen in liquid nitrogen, and stored at -80 °C.
Analyze the subunit composition of INO80 or INO80 subcomplexes on silver-stained gels or by western blotting, and estimate their concentrations by semi-quantitative western blotting using preparations of recombinant INO80 subunits of known concentration as standards.
Representative Results
Figure 1 shows a flow chart summarizing the procedures used to generate, purify, and characterize human INO80 ATP-dependent chromatin remodeling complexes.
As illustrated in Figures 2 and 3, these procedures enable the generation of both wild type INO80 and INO80 subcomplexes that lack various subunits, thereby enabling subsequent biochemical analyses of the contribution of these missing subunits to INO80's enzymatic activities. Figure 2 describes two strategies we have used to define the architecture of the INO80 complex and to generate INO80 subcomplexes. In the first, shown in Figure 2A, intact INO80 complexes are purified through FLAG-tagged versions of wild type subunits (Ies2 or INO80E). Subcomplexes can be purified through epitope-tagged versions of mutant Ino80 proteins that lack individual domains on which different sets of subunits assemble. As shown in Figure 3A, the purity of the resulting complexes is assessed using silver stained SDS-polyacrylamide gels, while the composition of the complexes is assessed using western blotting with antibodies against various INO80 subunits (Figure 3B) or by mass spectrometry (data not shown). Using this approach, we found that the subunits shown in red in Figure 2A were lost upon deletion of the Ino80 NTD (N-terminal domain); subunits shown in blue were lost upon deletion of the Ino80 HSA (Helicase SANT Associated) domain; and that the SNF2 ATPase domain, composed of SNF2N and HelicC regions (purple) separated by a long insertion region (white), was necessary and sufficient for binding to subunits shown in purple. Thus, INO80 subcomplexes (INO80ΔN.com or INO80ΔNΔHSA.com) that lack either the subunits shown in red or subunits shown in red and blue can be purified through FLAG-Ino80ΔN or FLAG-INO80ΔNΔHSA, respectively5.
It is also possible to generate subcomplexes by depleting individual subunits from cells expressing an appropriate FLAG-tagged INO80 subunit (unpublished results). In the first example shown in Figure 2B, siRNA-mediated knockdown of subunit X depletes only X from the INO80 complex, suggesting X is not required for assembly of any other subunits into INO80. In the second and third examples, siRNA knockdown of either Y or Z leads to co-depletion of both the Y and Z subunits, suggesting Y and Z assemble into the INO80 complex in a mutually dependent manner.
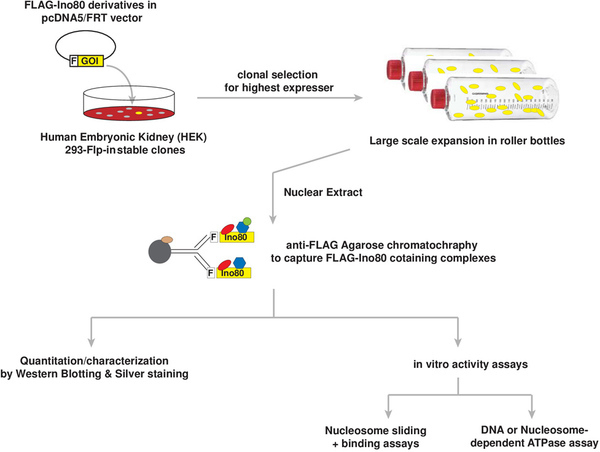 Figure 1. Flow chart describing procedures used to generate, purify and characterize human INO80 ATP-dependent chromatin remodeling complexes. F, an N-terminal in-frame FLAG epitope tag; GOI, Gene-of-interest. Please click here to view a larger version of this figure.
Figure 1. Flow chart describing procedures used to generate, purify and characterize human INO80 ATP-dependent chromatin remodeling complexes. F, an N-terminal in-frame FLAG epitope tag; GOI, Gene-of-interest. Please click here to view a larger version of this figure.
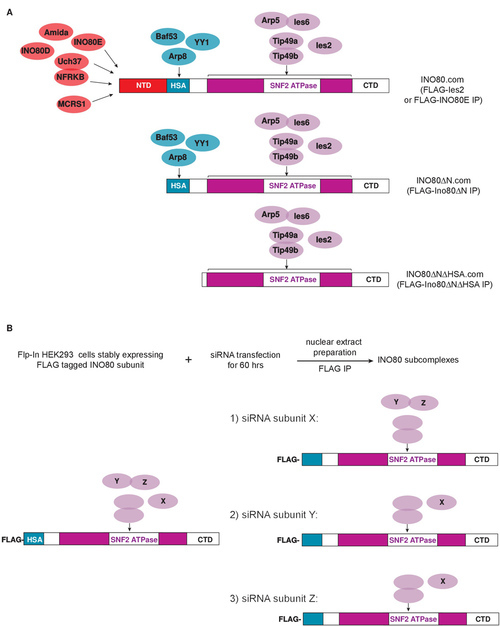 Figure 2. Diagram showing the two strategies used to generate INO80 subcomplexes that contain a subset of subunits. (A) The Ino80 ATPase contains regions that function as modular scaffolds on which the other INO80 subunits assemble. (B) siRNA-mediated knockdown can be used to deplete the desired subunit (X or Y or Z) from cells expressing an appropriate FLAG-tagged INO80 subunit (e.g., FLAG-Ino80ΔN). Please click here to view a larger version of this figure.
Figure 2. Diagram showing the two strategies used to generate INO80 subcomplexes that contain a subset of subunits. (A) The Ino80 ATPase contains regions that function as modular scaffolds on which the other INO80 subunits assemble. (B) siRNA-mediated knockdown can be used to deplete the desired subunit (X or Y or Z) from cells expressing an appropriate FLAG-tagged INO80 subunit (e.g., FLAG-Ino80ΔN). Please click here to view a larger version of this figure.
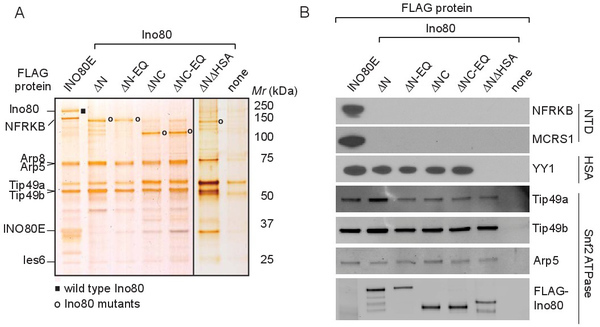 Figure 3. Analysis of the composition of immunopurified INO80 complexes and subcomplexes. (A) INO80 complexes or subcomplexes were purified from cells stably expressing the indicated FLAG-tagged proteins, subjected to SDS-polyacrylamide gel electrophoresis, and visualized by silver staining. The sample in the right-most lane was prepared from parental 293 cells that express no FLAG-tagged protein; proteins that appear in this control lane are contaminants that bind non-specifically to FLAG-agarose. Labels on the left indicate bands corresponding to several INO80 subunits, and those on the right the mobilities of molecular weight markers. The position of wild type Ino80 is indicated by the black rectangle, and positions of Ino80 mutants are indicated by open circles. The lanes separated by the black line are from separate gels. (B) INO80 complexes and subcomplexes were analyzed by western blotting using antibodies against representative subunits of the NTD, HSA, and SNF2 modules. This research was originally published in The Journal of Biological Chemistry. Chen et al, Subunit Organization of the Human INO80 Chromatin Remodeling Complex. An Evolutionarily Conserved Core Complex Catalyzes ATP-dependent Nucleosome Remodeling. The Journal of Biological Chemistry Vol 286: pp. 11283-11289. © 2011, by the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
Figure 3. Analysis of the composition of immunopurified INO80 complexes and subcomplexes. (A) INO80 complexes or subcomplexes were purified from cells stably expressing the indicated FLAG-tagged proteins, subjected to SDS-polyacrylamide gel electrophoresis, and visualized by silver staining. The sample in the right-most lane was prepared from parental 293 cells that express no FLAG-tagged protein; proteins that appear in this control lane are contaminants that bind non-specifically to FLAG-agarose. Labels on the left indicate bands corresponding to several INO80 subunits, and those on the right the mobilities of molecular weight markers. The position of wild type Ino80 is indicated by the black rectangle, and positions of Ino80 mutants are indicated by open circles. The lanes separated by the black line are from separate gels. (B) INO80 complexes and subcomplexes were analyzed by western blotting using antibodies against representative subunits of the NTD, HSA, and SNF2 modules. This research was originally published in The Journal of Biological Chemistry. Chen et al, Subunit Organization of the Human INO80 Chromatin Remodeling Complex. An Evolutionarily Conserved Core Complex Catalyzes ATP-dependent Nucleosome Remodeling. The Journal of Biological Chemistry Vol 286: pp. 11283-11289. © 2011, by the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
Discussion
Structural and functional studies of multi-subunit mammalian chromatin remodeling complexes from higher eukaryotes have been hampered by the difficulty of preparing biochemically useful amounts of such complexes containing mutant subunits or lacking certain subunits altogether. There are a number of technical hurdles: First, genetic manipulation in mammalian cells has been technically challenging and time-consuming. Unlike yeast cells, whose genome can be readily edited and targeted using recombineering techniques, the mammalian genome is more structurally complex and less susceptible to recombineering interventions. Thus, deletion or modification of mammalian genes in cultured cells or animals is more time consuming and requires more specialized expertise. As a consequence, the generation of mutant mammalian complexes carrying structural mutations or lacking subsets of subunit(s) has been rate-limiting. Second, biochemical reconstitution approaches using heterologous protein expression systems are often used to generate defined multi-protein assemblies. However, such approaches become technically challenging for very large complexes, whose subunits may need to be simultaneously expressed in proper stoichiometry and/or to be assembled in a particular order, with help from specific chaperones or cofactors. These problems are especially severe in the case of the INO80 complex, because its Ino80 ATPase subunit is particularly difficult to express in insect or E. coli cells in biochemically useful amounts. In studies of the INO80 chromatin remodeling complex, it has been possible to circumvent some of these challenges using the strategies described in this protocol.
Generating human cell lines stably expressing epitope tagged subunits of the INO80 complex is a key step in this procedure, as it enables the purification of well-defined chromatin remodeling complexes that can be tested using various biochemical assays. Although it is in principle more time consuming to generate stable cell lines than to use transient transfection to deliver engineered cDNAs into mammalian cell lines for the production of recombinant protein, transient transfection suffers from several drawbacks. First, DNA―in the form of a transiently transfected extra-chromosomal vector―is short-lived and usually cannot self-propagate during the cell cycle. Second, transfection efficiency varies greatly depending on cell type and growth conditions. Heterogeneous transfection can cause a mosaic expression pattern that confers a selective growth advantage or disadvantage within the cell population. Thus, transiently introduced transgenes tend to get lost during the many cell passages required to generate the large amount of cells needed for purification of biochemically useful amounts of protein. Stable cell lines are most commonly generated using methods that result in random integration of cDNAs encoding a protein of interest. However, randomly integrated cDNAs can disrupt expression of endogenous genes and are subject to gene silencing with multiple cell passages. For these reasons, we typically introduce Ino80 cDNAs into cells using Flp-mediated recombination technology, in which the cDNA is stably incorporated into a specific chromosomal location via Flp recombinase-mediated insertion into a single FRT site stably integrated into the genome.
During selection of stable cell lines, it is essential to be able to detect exogenously expressed, FLAG-tagged, wild type or mutant Ino80 proteins. Because FLAG-tagged Ino80 protein is often expressed at very low levels and is therefore difficult or impossible to detect by western blotting in crude lysates of cells grown in a single well of a 24-well plate, it is often necessary to expand the initial clonal cell population prior to screening for expression of Ino80 protein. FLAG-tagged Ino80 protein can then be purified and concentrated by FLAG immunopurification prior to analysis by Western blotting.
The efficiency of siRNA-mediated knockdown is quite sensitive to cell density at the time of transfection; we found that knockdown efficiency decreased when transfections were performed with cells at densities other than that recommended in the protocol. The optimal length of time for siRNA transfection is variable and needs to be determined empirically for each target protein, as it depends on the stability of the target protein, the turnover rate of the targeted protein in protein complexes, and the degree to which the protein is essential for cell viability. As an alternative to the use of transient transfection of siRNAs to deplete individual subunits, one may wish to consider generating cell lines stably expressing shRNAs under selective pressure as it may be easier and more cost-effective to grow biochemically useful amounts of such cell lines. However, it can prove difficult to obtain sufficient knockdown using shRNA-mediated knockdown in stable cell lines.
Also key to the success of our procedure is the use of nuclear extracts as the starting material for purification of INO80 complexes. We have found that immunopurified INO80 complexes from whole cell extracts are often contaminated with ATPase and/or nucleosome remodeling activities that are independent of the Ino80 ATPase; such contamination is largely avoided when complexes are purified from nuclear extracts.
Preparation of nuclear extracts and isolation of intact INO80 complexes depends on a number of critical factors. First, the cells used to prepare nuclear extracts should be intact and healthy. The washed cell pellet is expected to swell up to two-fold following incubation in the hypotonic Buffer A. If cells do not swell and/or the supernatant becomes turbid at this step, the starting population of cells may have been unhealthy. Alternatively, cells may have been handled too roughly during harvest or resuspension steps, or they may have been incubated too long in Buffer A. Second, it is important not to over-homogenize the cells or the nuclear pellet, as this will shear the chromatin and release DNA into the soluble fraction. DNA in the soluble fraction can interfere with subsequent purification and assay of INO80 complexes. In particular, contaminating DNA can lead to increased formation of precipitates during incubation of the extract with anti-FLAG agarose, thereby reducing the efficiency of washing and elution steps and/or may increase the abundance of contaminating DNA-binding proteins in the final purified fraction. In addition, downstream biochemical assays measure activities that are dependent on DNA, thus the potential carry-over of DNA from the extract may complicate interpretation of these assays. To avoid over-homogenization, during cell lysis it is advisable to check the percentage of trypan blue-positive cells after each stroke of the homogenizer; for HEK293 cells, near complete cell lysis typically requires 4 - 6 strokes of homogenization. It is also is important to avoid introducing air bubbles while homogenizing. The supernatant from the 120,000 x g spin in Step 4.2.8 should be a clear, non-viscous solution, with only a very minimal amount of cloudy or viscous material near the chromatin pellet or floating on top. When collecting the supernatant, one should take care not to collect any of the cloudy or viscous material, as it may include chromatin and/or DNA. Finally, it is better to avoid freezing cells before preparing nuclear extracts, since complexes prepared from frozen cells tend to exhibit lower specific activity and to include more contaminating DNA and proteins.
The optimal ratio between the amount of starting extract and anti-FLAG agarose depends on the concentration of the FLAG bait protein present in the extracts and the accessibility of the FLAG epitope and needs to be determined empirically. We typically begin immunopurification with 100 µl bed volume of anti-FLAG agarose beads and 3 - 14 ml of nuclear extract; the amount of extract used depends on the goals of the experiment and availability of extract. A single step immunopurification using anti-FLAG agarose is usually sufficient to purify INO80 or INO80 subcomplexes to a degree adequate for reliable assays of their activities; however, additional purification step(s), such as gel filtration, density gradient sedimentation, or ion exchange chromatography may be needed for purification of other complexes or to remove contaminating activities that could complicate interpretation of biochemical assays.
The strategies described here should be more generally useful for studies of other chromatin remodeling enzymes as well as other large multiprotein complexes. Successful application of these strategies to the analysis of other multiprotein complexes depends on (i) identification of an individual subunit or subunit(s) that serve as scaffold(s) on which other subunits assemble and (ii) definition of specific domains or regions with which specific subsets of subunits assemble. The definition of such domains can be facilitated by analyzing the primary sequence of the core scaffold subunit(s) for evolutionarily conserved regions or regions that correspond to known structural domains.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
Work in the authors' laboratory is supported by a grant from the National Institute of General Medical Sciences (GM41628) and by a grant to the Stowers Institute for Medical Research from the Helen Nelson Medical Research Fund at the Greater Kansas City Community Foundation.
References
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes, Annual Review of Biochemistry. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- Szerlong H, Hinada K, Viswanathan R, Erdjument-Bromage H, Tempst P, Cairns BR. The HSA domain binds nuclear actin-related proteins to regulate chromatin-remodeling ATPases. Nature Struct. Mol. Biol. 2008;15(5):469–476. doi: 10.1038/nsmb.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Mizuguchi G, Hamiche A, Wu C. A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000;406(6795):541–544. doi: 10.1038/35020123. [DOI] [PubMed] [Google Scholar]
- Shen X, Ranallo R, Choi E, Wu C. Involvement of actin-related proteins in ATP-dependent chromatin remodelling. Mol. Cell. 2003;12:147–155. doi: 10.1016/s1097-2765(03)00264-8. [DOI] [PubMed] [Google Scholar]
- Chen L, et al. Subunit organization of the human INO80 chromatin remodeling complex: an evolutionarily conserved core complex catalyzes ATP-dependent nucleosome remodeling. Journal of Biological Chemistry. 2011;286(13):11283–11289. doi: 10.1074/jbc.M111.222505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer B. Site-specific recombination: developments and applications. 1994;5(5):521–527. doi: 10.1016/0958-1669(94)90068-x. [DOI] [PubMed] [Google Scholar]
- Gorman S, Fox DT, Wahl GM. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science. 1991;251(4999):1351–1355. doi: 10.1126/science.1900642. [DOI] [PubMed] [Google Scholar]
- Jin J, et al. A mammalian chromatin remodeling complex with similarities to the yeast INO80 complex. Journal of Biological Chemistry. 2005;280(50):41207–41212. doi: 10.1074/jbc.M509128200. [DOI] [PubMed] [Google Scholar]
- Cai Y, et al. YY1 functions with INO80 to activate transcription. Nature Structural and Molecular Biology. 2007;14(9):872–874. doi: 10.1038/nsmb1276. [DOI] [PubMed] [Google Scholar]
- Yao T, et al. Distinct Modes of Regulation of the Uch37 Deubiquitinating Enzyme in the Proteasome and in the Ino80 Chromatin-Remodeling Complex. Mol.Cell. 2008;31(6):909–917. doi: 10.1016/j.molcel.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzard BL, Chubet RG, Vizard DL. Immunoaffinity purification of FLAG epitope-tagged bacterial alkaline phosphatase using a novel monoclonal antibody and peptide elution, Biotechniques. 1994;16(4):730–735. [PubMed] [Google Scholar]
- Barker K. At the Bench: A Laboratory Navigator. Vol. 10. Cold Spring Harbor Laboratory Press; 2005. pp. 207–245. [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian cell nuclei. Nucleic. Acids. Res. 1983;11(5):1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]