

Abstract
Prototrophic bacteria grow on M-9 minimal salts medium supplemented with glucose (M-9 medium), which is used as a carbon and energy source. Auxotrophs can be generated using a transposome. The commercially available, Tn5-derived transposome used in this protocol consists of a linear segment of DNA containing an R6Kγ replication origin, a gene for kanamycin resistance and two mosaic sequence ends, which serve as transposase binding sites. The transposome, provided as a DNA/transposase protein complex, is introduced by electroporation into the prototrophic strain, Enterobacter sp. YSU, and randomly incorporates itself into this host’s genome. Transformants are replica plated onto Luria-Bertani agar plates containing kanamycin, (LB-kan) and onto M-9 medium agar plates containing kanamycin (M-9-kan). The transformants that grow on LB-kan plates but not on M-9-kan plates are considered to be auxotrophs. Purified genomic DNA from an auxotroph is partially digested, ligated and transformed into a pir+ Escherichia coli (E. coli) strain. The R6Kγ replication origin allows the plasmid to replicate in pir+ E. coli strains, and the kanamycin resistance marker allows for plasmid selection. Each transformant possesses a new plasmid containing the transposon flanked by the interrupted chromosomal region. Sanger sequencing and the Basic Local Alignment Search Tool (BLAST) suggest a putative identity of the interrupted gene. There are three advantages to using this transposome mutagenesis strategy. First, it does not rely on the expression of a transposase gene by the host. Second, the transposome is introduced into the target host by electroporation, rather than by conjugation or by transduction and therefore is more efficient. Third, the R6Kγ replication origin makes it easy to identify the mutated gene which is partially recovered in a recombinant plasmid. This technique can be used to investigate the genes involved in other characteristics of Enterobacter sp. YSU or of a wider variety of bacterial strains.
Keywords: Microbiology, Issue 92, Auxotroph, transposome, transposon, mutagenesis, replica plating, glucose minimal medium, complex medium, Enterobacter
Introduction
Prototrophic bacteria grow in M-9 minimal salts medium containing glucose (M-9 medium), converting glucose through central carbon metabolism pathways to generate precursors, such as amino acids, nucleic acids and vitamins, for biosynthesis1. M-9 medium contains ammonium chloride as a nitrogen source, sodium and potassium phosphate as a buffer and phosphorous source, magnesium sulfate as a sulfur source and glucose as a carbon and energy source. Luria-Bertani (LB) medium is rich in amino acids from tryptone and in vitamins and growth factors from yeast extract. It supports the growth of auxotrophs that cannot synthesize amino acids, vitamins and other growth factors required for growth on M-9 medium. Thus, prototrophs will grow in LB and M-9 medium, whereas auxotrophs will grow in LB medium but not in M-9 medium. By introducing mutations into a prototrophic population of bacteria and identifying mutated genes that cause auxotrophic phenotypes, it is possible to obtain a better understanding of the metabolism in a bacterial strain.
Transposon mutagenesis can be used to identify many of the genes required for growth on glucose in M-9 medium. Transposons insert themselves randomly into the host genome2. By spotting transposon transformants on LB agar plates and replica plating them onto M-9 medium agar plates, it is possible to screen for auxotrophs. Interrupted genes are identified through gene rescue. This study uses a commercially available Tn5-derived transposome which is shipped as a solution of a linear transposon DNA segment mixed with transposase protein. The DNA segment lacks a transposase gene but contains a gene for kanamycin resistance, an R6Kγ replication origin and two mosaic motifs which are DNA sequences for transposase binding at each end of the segment3,4. Since the transposase protein is added directly to the DNA, the DNA segment alone is defined as the transposon, and the DNA/transposase protein complex is defined as the transposome. The transposome is transformed by electroporation5 into a kanamycin sensitive host (Figure 1A). Colonies that grow on LB agar plates containing kanamycin (LB-kan) have transposon inserts (Figure 1B), and replica plated transformants that fail to grow on M-9 medium agar plates containing kanamycin (M-9-kan) are auxotrophs (Figire 1C). Genomic DNA from a mutant is purified and partially digested with the 4-base cutting restriction endonuclease, BfuC I (Figure 1D). The ligated DNA is transformed into a strain of Escherichia coli (E. coli) that contains the pir gene (Figure 1E). This gene allows the new plasmid containing the transposon and flanking host chromosomal region to replicate in E. coli6. The kanamycin resistance gene serves as a selectable marker for the new plasmid. Finally, sequencing using primers complementary to each end of the transposon and Basic Local Alignment Search Tool (BLAST) analysis7,8 of the resulting sequence are used to determine the identity of the interrupted genes.
This transposome mutagenesis strategy provides three advantages3. First, since the transposase protein is bound directly to the transposon, insertion does not depend on expression of the transposase gene within the host. Once the transposon incorporates itself into the host genome, the transposase is degraded, preventing additional movement of the transposon. However, additional movement cannot be prevented if the host possesses an endogenous Tn5 transpositional element. Second, introduction of the transposome by electroporation makes it possible to use it in a wide variety of hosts. It also eliminates the need to introduce the transposon by bacterial conjugation or by viral infection. Both processes require host susceptibility. Third, inclusion of the kanamycin resistance gene and the R6Kγ replication origin in the transposome makes it easier to identify the interrupted gene. Transposon-interrupted regions can be stored and sequenced as plasmids, eliminating the need to use the inverse polymerase chain reaction (PCR) technique for gene identification.
The protocol presented in this video describes each step for transposon mutagenesis of Enterobacter sp. YSU9 using a transposome from its introduction into the bacterial cells to the identification of the putative gene it interrupted. In addition to previously published protocols3,4,10, detailed methods for using replica plating to screen for auxotrophs are presented. This mutagenesis technique may be used for investigating other phenotypes, such as antibiotic and metal resistances, in different types of bacteria, for identifying the minimal number of genes required for growth under defined culture conditions in synthetic biology studies, or for teaching a laboratory component of a genetics or microbial physiology course.
Protocol
1. Electroporation of Competent Cells5,11
Dilute an O/N LB culture of Enterobacter sp. YSU 1/20 into 50 ml of fresh LB medium and grow with shaking at 120 rpm and 30 °C to an O.D. (600 nm) between 0.4 and 0.6. Optionally, grow other bacterial strains at their optimal growth temperature.
Chill the cells on ice for 5 min and centrifuge at 4 °C and 7,000 x g for 5 min.
Discard the supernatant, resuspend the cells in 50 ml of sterile ice cold water and centrifuge at 4 °C and 7,000 x g for 5 min. Repeat this step.
Discard the supernatant and resuspend the cells in a volume of ice cold water equal to the cell pellet volume. For long term storage, wash the cells with 10 ml of ice cold 10% (v/v) glycerol, resuspend in an equal pellet volume of ice cold 10% (v/v) glycerol, store at -80 °C, and thaw on ice before electroporation.
Add 0.5 µl of the transposome to 40 µl of cells. The commercially available transposome solution is supplied at a DNA concentration of 0.33 µg/µl. Although 0.33 µg is recommended, 0.165 µg DNA is sufficient.
Place the cell/transposome mixture in an ice cold, 0.2 mm, electroporation cuvette and tap the mixture to the bottom of the cuvette. Shock the cells at 25 µF, 200 Ω, and 2.5 kV. To ensure that the cells remain chilled, store the electroporation cuvettes in a -20°C freezer before use.
Immediately, add 960 µl of filter sterilized super optimal broth with catabolite repression (SOC) medium. Mix by pipetting up and down and transfer the cells to a sterile 1.5 ml microfuge tube. NOTE: The cells begin to die after the shock, but the salt in the SOC medium allows them to recover. To economize, it is not necessary to add transposome or to shock the negative control cells. Just add 960 µl of sterile SOC medium to it.
Incubate the cells at 30 °C for 45-60 min with shaking at 120 rpm. This provides time for the transposome to recombine with the host genome and for the cells to express kanamycin resistance.
Spread 100 µl of cells on a LB-kan agar plate and incubate the plate O/N at 30 °C. Store the remaining transformation mix in the refrigerator at 4 °C. Spread additional amounts at a later date up to 2 weeks after the initial electroporation if required.
2. Gridding of Transformants
Tape a grid to the lid of an empty 100 x 15 mm petri dish. Copy a grid from “A Short Course in Bacterial Genetics”12.
Draw a line on the bottom of an LB-kan agar plate. Use a small piece of tape to anchor it to the gridded lid so that the grid is visible through the agar. Ensure that the line on the bottom of the plate is aligned with the top, center of the grid. Anchoring the plate with tape keeps it from sliding, allowing for even gridding. The line facilitates colony identification after replica plating.
Pick a single transformant with a sterile toothpick and spot it in the center of a square in the grid. Just touch the colony and the spot. Do not dig the toothpick into the agar. To avoid contaminating the plate, handle the toothpick only at one end.
Spot another transformant in an adjacent square as in step 2.3. When the plate is full, incubate it O/N at 30 °C. This is the master plate.
3. Replica Plating to Determine the Auxotroph Phenotype12
For each plate that was gridded, make a stack of three fresh agar plates numbered one through three: one for LB-kan, two for M-9-kan and three for LB-kan. Dry the plates upside down, O/N, at 37 °C before use.
Draw a line on the top of each plate as in step 2.2.
Wipe the replica plating tool with 70% ethanol, place a sterile velveteen square on top of the replica plating tool and clamp the velveteen square down. Hands are teeming with microbes even after washing them. Prevent the introduction of contaminating microbes by handling the velveteen square by its edge.
Align the line on the master plate with a line drawn on the clamp and place the plate on top of the velveteen square. Apply gentle pressure to transfer the bacteria from the plate to the velveteen square. Be careful not to smear the bacteria. Remove the master plate from the velveteen square.
Align the line drawn on plate number one with the line on the clamp and place it on top of the velveteen square. Apply gentle pressure to transfer the bacteria from the velveteen square to the plate. Remove plate number one.
Discard the velveteen square into a beaker of water and place a clean, sterile velveteen square on the replica plating tool as in step 3.3. This step prevents over-inoculation which causes breakthrough growth and false positive results.
Align the line on plate number one with the line on the clamp and place it on top of the new velveteen square. Apply gentle pressure to transfer the bacteria from plate number one to the velveteen square. Remove plate number one.
Align the line drawn on plate number two with the line on the clamp and place it on top of the velveteen square. Apply gentle pressure to transfer the bacteria from the velveteen square to plate number two. Remove plate number two.
Repeat step 3.8 for plate number three. The third plate is a control for complete transfer from the master plate to the other two plates. Discard the velveteen square into a beaker of water. To reuse the velveteen squares, autoclave the beaker containing the contaminated velveteen squares, wash them, dry them, wrap them in aluminum foil and re-autoclave them.
Incubate the plates at 30 °C O/N. Colonies that grow on both LB-kan agar plates but fail to grow on M-9-kan plates are considered to be auxotrophs (Figure 2).
Streak out auxotrophs from plate number one onto a fresh LB-kan agar plate, and incubate the plate at 30 °C O/N.
Streak out for isolation 3 colonies from the streak plate in step 3.11 onto a fresh LB-kan plate. Incubate the plate at 30 °C O/N. This ensures that the mutant is well isolated. Divide the plate into thirds and streak out each colony in one section.
Spot colonies from the streaked out colonies derived from step 3.12 onto a fresh LB-kan plate to form a grid as in steps 2.1-2.4. Each mutant is being re-tested in triplicate.
Replicate plate again as in steps 3.1-3.10 to confirm the auxotroph phenotype.
4. Gene Rescue
Grow a 3 ml O/N culture of an isolated mutant colony from step 3.13 in liquid LB-kan medium at 30 °C. Purify genomic DNA from 1 ml of culture using a commercially available genomic DNA purification kit.
Set up a mixture of 0.025 U BfuC I restriction endonuclease and 14 µl of 1 μg/μl genomic DNA in a 20 µl reaction on ice. Since BfuC I cuts the transposon at twelve different sites, it may be more efficient to use restriction endonuclease, Bfa I or Hae III, which cuts the transposon at two and three different sites, respectively.
Incubate the reaction at 37 °C for 25 min and then at 80 °C for 20 min to inactivate the enzyme. Analyze 3 µl of the partially digested DNA on a 0.8% agarose gel (Figure 3). A successful partial digestion results in a smear from above 10 kb down to the bottom of the gel.
Ligate 15 µl of partially digested genomic DNA using 800 units of T4 DNA ligase in a 500 µl reaction. Incubate the reaction O/N at 4 °C. The large reaction volume maximizes the ligation of single fragments to themselves and minimizes interactions between two or more DNA fragments.
Precipitate the ligated DNA by adding 50 µl of 3 M sodium acetate, pH 5.5, and 1 ml of 95% ethanol and incubating it at -20°C for 20 min.
Microfuge the DNA at 4 °C and 13,500 x g for 10 min, wash the pellet with 70% ethanol, dry it in a centrifugal vacuum concentrator, and resuspend it in 10 µl of nuclease free water. Alternatively, the DNA may be air dried at RT for 15 min.
Use 4 µl of resuspended DNA to transform E. coli strain ECD100D pir or ECD100D pir116 by electroporation as in section 1. The R6kγ replication origin requires a bacterial strain with a pir gene to replicate6. The pir strain results in low copy plasmids, and the pir116 strain contains a pir mutant gene that results in high copy plasmids. Each transformant contains a new plasmid with the transposon and a region of the interrupted chromosome (Figure 1E).
Inoculate 5 ml of LB-kan medium with a single colony, grow it O/N with shaking at 120 rpm and 37 °C, and purify plasmid DNA from the entire O/N culture using a commercial kit.
Digest 14 µl of the plasmid using the restriction endonuclease Xho I. Make sure the final volume of the reaction is 20 µl. This enzyme recognizes a site in the middle of the transposon at the 5’ end of the kanamycin resistance gene. Analyze 10 µl of the undigested and digested plasmid on a 0.8% agarose gel (Figure 3). The plasmid consists of the 2 kb transposon plus flanking host DNA.
5. DNA Sequencing
Sequence the plasmid using a commercially available sequencing kit, primers that are homologous to each end of the transposon, and a capillary sequencing analysis system. Alternatively, the plasmid may be shipped to an outside institution for sequencing.
Use the molecular weight standard (1 kb ladder) in the gel to estimate the size of the plasmid. Then, estimate the number of nanograms (ng) of plasmid that is required for 50 fmol.
Measure the concentration of the plasmid DNA using a spectrophotometer at wavelengths of 260 nm (A260) and 280 nm (A280). Ensure the light path length through the cuvette is 1 cm. Determine the concentration by multiplying the absorbance at 260 nm by 50 ng/µl. High quality plasmid DNA will have an A260/ A280 ratio between 1.8 and 2.0.
The volume of DNA required for each sequencing reaction is the number of ng required for 50 fmol divided by the concentration of the plasmid. Mix the volume of required plasmid with water to bring the total volume up to 10 µl.
Heat the plasmid DNA/water mixture at 96 °C for 1 min and then cool it to RT.
Add 8 µl of master mix from the sequencing kit and 2 µl of 1.6 μM of one of the sequencing primers.
Follow the protocol from the sequencing kit for the thermal cycler program and reaction cleanup.
Analyze each sample using a capillary DNA analysis system.
Use a commercially available software package to view the DNA sequence. Search for the sequence, “5’-GAGACAG-3’,” which is the last 7 bp of the transposon (Figure 4). The sequence before the 5’ end of this 7 bp segment belongs to the transposon. The sequence after the 3’ end of this 7 bp segment belongs to the interrupted gene.
Determine the possible function of the interrupted gene using the Basic Local Alignment Search Tool (BLAST) 7,8. Use the following link: http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&BLAST_PROGRAMS=megaBlast&PAGE_TYPE=BlastSearch. Paste the sequence of the interrupted gene into the query box, select “others” under the database and click on “BLAST” at the bottom of the page. When the result appears, scroll down the page to view the alignment results (Figure 5).
Representative Results
Transformation of Enterobacter sp. YSU by electroporation with the transposome initiated random genome insertion into the host genome (Figure 1A,B). A successful electroporation yielded several thousand transformants which grew on LB-kan plates. To obtain 300-400, well-spaced colonies per plate, the amount of transformation mixture spread on each LB-kan agar plate was optimized. Each transformant contained at least one transposon insert, but it was not clear if an important gene for growth on M-9 medium was interrupted without screening by replica plating. Transformants were transferred to fresh LB-kan plates in grids of 88 and replica plated onto M-9 medium. Note, to ensure that the transposon is maintained in the transformants, the Protocols section recommends adding kanamycin to the M-9 medium plates. The M-9 medium was not supplemented with kanamycin in this case, but it was still possible to obtain auxotrophs without it. In Figure 2, the black squares show two colonies that grew on the LB-kan plate (2A) but not on the M-9 minimal medium plate (2B). These auxotrophs probably contained interruptions in genes involved in the synthesis of an amino acid, a nucleic acid or a vitamin which are found in LB medium but not in M-9 minimal medium (Figure 1C). The other 86 colonies did not have an interruption in a gene that is required for growth on M-9 minimal medium.
Some colonies may have been contaminated with adjacent colonies during replica plating. To isolate a mutant as a pure culture, it was streaked from the LB-kan plate number one in Step 3.1 onto a fresh LB-kan agar plate and grown O/N at 30 °C. Three colonies that grew were streaked out onto a second fresh LB-kan plate and grown O/N at 30 °C. Then, one colony that arose from each of the three colonies was spotted onto a third fresh LB-kan plate. Replica plating back onto an M-9 minimal medium plate verified in triplicate the auxotrophic phenotype that was observed in Figure 2. Of the 1,760 transposon transformants screened in a 2013 Microbial Physiology course, 23 were auxotrophs.
The interrupted gene from an auxotroph was then identified using gene rescue. An auxotrophic colony from the second streak plate above (Step 3.13) was grown O/N in LB-kan medium, and genomic DNA was purified from the culture using a genomic DNA purification kit. Agarose gel electrophoresis showed that the purified DNA was larger than 10 kb in size (Figure 3, lane 2). To break the genomic DNA into smaller fragments that contained the transposon and flanking interrupted host regions (Figure 1D), it was partially digested with 0.025 units of BfuC I for 25 min at 37 °C. This restriction endonuclease cuts at 5’-GATC-3’ sites which occur approximately every 200 to 500 base pairs. Even though there are twelve 5’-GATC-3’ sites within the transposon, a large number of fragments containing the intact transposon and flanking host genome will still be present in the partially digested DNA. Lane 3 of Figure 3 shows partially digested genomic DNA with a smear of DNA beginning above 10 kb, all the way to the bottom of the gel to below 0.5 kb. If the DNA smear begins below 2 kb, the size of the transposon, then the DNA has been digested too much. It will be necessary to decrease the amount of enzyme added, the 37 °C incubation time, or both. If there is no smear and the lane with the digested DNA is the same size as the DNA with the undigested DNA, then no digestion occurred. It will be necessary to increase the amount of enzyme added, the 37°C incubation time or both. Instead of using BfuC I, it may be possible to use the restriction endonucleases Bfa I or Hae III, which cut the transposon at two and three different sites respectively. Use of one of these enzymes may increase the E. coli pir+ transformation yield during gene rescue.
The partially digested DNA was ligated in a 500 µl reaction. This increased volume minimized interactions between other DNA fragments and maximized the probability that each fragment ligated with itself to form a circular molecule. The ligated DNA was precipitated and transformed by electroporation into E. coli strain ECD100D pir116. The transformation mixture was spread on LB-kan plates. Colonies that grew contained a new plasmid consisting of the transposon and a region of the interrupted host gene (Figure 1E). The number of colony forming units ranged from 0 to 2,000 per ml of transformation mix. If the flanking chromosomal region encoded a protein that was toxic to the E. coli host, the transformation efficiency was low or only a small segment of the chromosome was associated with the new plasmid. Plasmid DNA was purified from a culture that was inoculated with a single transformant and digested with the restriction endonuclease, Xho I, which recognized a single site in the center of the transposon near the 5’ end of the kanamycin resistance gene. Lane 4 in Figure 3 contained the undigested plasmid and lane 5 contained the digested plasmid. Because purified plasmid DNA tends to be supercoiled, it migrated at a faster rate than the same strand of linear digested DNA. The correct size of this plasmid was 3 kb. It contained 2 kb of the transposon, plus about 1 kb of the interrupted host gene. If the flanking host DNA contains one or more Xho I recognition sites, two or more plasmid bands will be observed in the lane with Xho I digested DNA.
Figure 4 shows a sequencing result for a plasmid that was isolated from an auxotroph. After removing the short transposon sequence in bold, the host DNA sequence was submitted for Basic Local Alignment Search Tool (BLAST) analysis7,8. One of the BLAST results suggested that the interrupted sequence is similar to an Enterobacter cloacae subsp. cloacae NCTC 9394 (emb|FP929040.1|) gene for chorismate mutase, which is involved in the biosynthesis of L-tyrosine and L-phenylalanine (Figure 5)13. Sometimes during the ligation of the partially digested genomic DNA, the linear transposon ligates to one or more other BfuC I fragments. This can make the BLAST results confusing because the identity of a gene changes abruptly. For example, the first 41 base pairs of another auxotroph matched to a gene for glutamate-5-semialdehyde dehydrogenase which is involved in proline biosynthesis1. Then, the next 219 base pairs matched to a gene for a ribonucleoside-diphosphate reductase subunit, followed by a 62 base pair segment for lipid-A-disaccharide kinase. The last 295 base pair segment matched to glutamate-5-semialdehyde dehydrogenase again. The presence of 5’-GATC-3’ sites for BfuC I suggested that two smaller DNA fragments ligated into the rescued plasmid. Thus, dilution of partially digested DNA for ligation reactions does not completely eliminate background caused by the ligation of smaller fragments. From these results, it was concluded that the transposome inserted itself into the gene for glutamate-5-semialdehyde dehydrogenase because the sequence for this gene was immediately adjacent to the transposon. Other auxotrophs contained interruptions in the gene for cystathione beta lyase involved in methionine biosynthesis; for histidinol dehydrogenase involved in histidine biosynthesis; for phosphoserine phosphatase involved in serine, glycine, and cysteine biosynthesis; and for ketol-acid reductoisomerase involved in leucine, isoleucine and valine biosynthesis1.
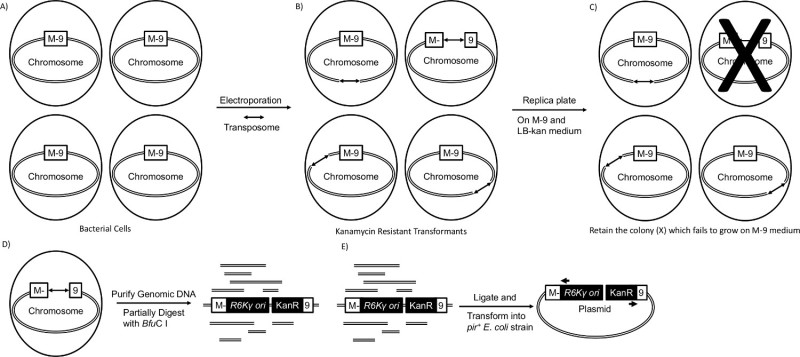 Figure 1. Transposon Mutagenesis. (A) Bacterial cells containing a gene that is required for growth on M-9 minimal salts medium containing glucose. The boxed M-9 represents an auxotrophic gene. (B) The transposome is introduced into the bacterial strain by electroporation, and the transposase protein bound to each mosaic end (arrows) randomly inserts the transposon into the host chromosome3,4. Only transposon containing transformants will grow on LB-kan medium plates. (C) The transformants are replica plated onto M-9 medium containing glucose and on LB-kan medium. The transformants that fail to grow (crossed-out) on M-9 medium containing glucose are auxotrophs. (D) Genomic DNA is purified from the auxotrophs and is partially digested with the 4-base cutter, BfuC I, which recognizes the DNA site, 5’-GATC-3’. (E) The partially digested DNA is treated with T4 DNA ligase and transformed by electroporation into E. coli strain ECD100D pir to form a new plasmid that contains the transposon and the interrupted chromosomal gene which is required for growth on M-9 medium. The pir gene allows for circular DNA fragments containing the R6Kγ replication origin in the transposon to serve as a replication origin in the new plasmid6. The kanamycin resistance gene serves as a selectable marker for the new plasmid. The plasmid is purified, and a ~500 bp sequence of the interrupted gene is determined using primers (arrows) that are homologous to each end of the transposon. Please click here to view a larger version of this figure.
Figure 1. Transposon Mutagenesis. (A) Bacterial cells containing a gene that is required for growth on M-9 minimal salts medium containing glucose. The boxed M-9 represents an auxotrophic gene. (B) The transposome is introduced into the bacterial strain by electroporation, and the transposase protein bound to each mosaic end (arrows) randomly inserts the transposon into the host chromosome3,4. Only transposon containing transformants will grow on LB-kan medium plates. (C) The transformants are replica plated onto M-9 medium containing glucose and on LB-kan medium. The transformants that fail to grow (crossed-out) on M-9 medium containing glucose are auxotrophs. (D) Genomic DNA is purified from the auxotrophs and is partially digested with the 4-base cutter, BfuC I, which recognizes the DNA site, 5’-GATC-3’. (E) The partially digested DNA is treated with T4 DNA ligase and transformed by electroporation into E. coli strain ECD100D pir to form a new plasmid that contains the transposon and the interrupted chromosomal gene which is required for growth on M-9 medium. The pir gene allows for circular DNA fragments containing the R6Kγ replication origin in the transposon to serve as a replication origin in the new plasmid6. The kanamycin resistance gene serves as a selectable marker for the new plasmid. The plasmid is purified, and a ~500 bp sequence of the interrupted gene is determined using primers (arrows) that are homologous to each end of the transposon. Please click here to view a larger version of this figure.
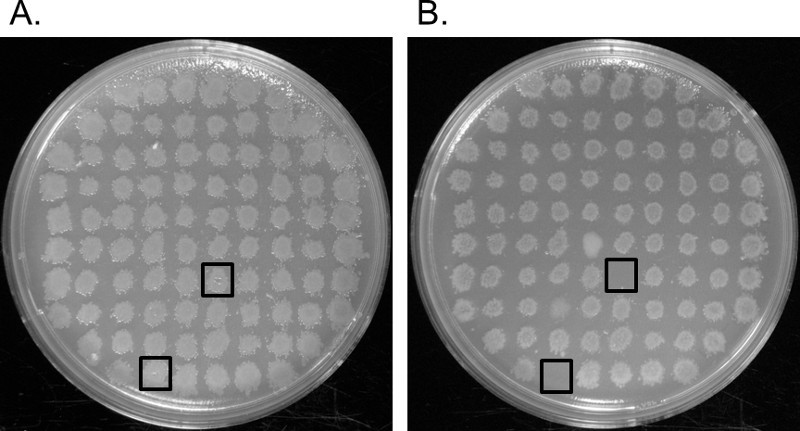 Figure 2. Replica Plating. (A) Grid of transformants that were transferred to an LB-kan agar plate. (B) Grid of the same transformants that were transferred to an M-9 medium plate. Auxotrophs grew on the LB-kan plates but not on the M-9 medium plates. These are identified by the black squares. Please click here to view a larger version of this figure.
Figure 2. Replica Plating. (A) Grid of transformants that were transferred to an LB-kan agar plate. (B) Grid of the same transformants that were transferred to an M-9 medium plate. Auxotrophs grew on the LB-kan plates but not on the M-9 medium plates. These are identified by the black squares. Please click here to view a larger version of this figure.
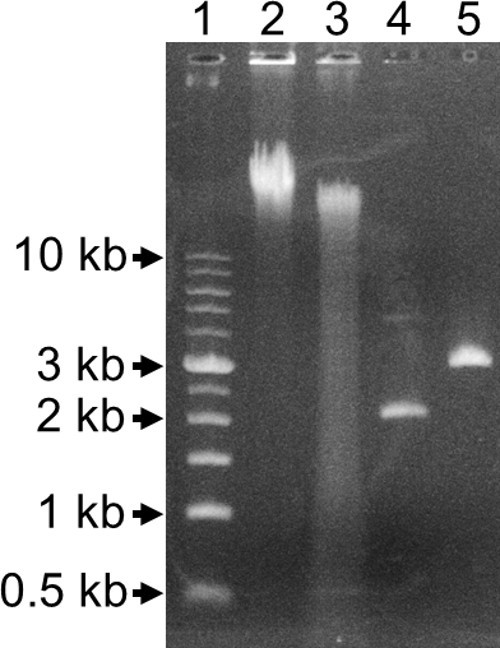 Figure 3. 0.8% Agarose Gel. Lane 1: 1 kb ladder. Lane 2: Undigested genomic DNA. Lane 3: BfuC I partially digested genomic DNA. Lane 4: Undigested rescued plasmid. Lane 5: Xho I digested rescued plasmid.
Figure 3. 0.8% Agarose Gel. Lane 1: 1 kb ladder. Lane 2: Undigested genomic DNA. Lane 3: BfuC I partially digested genomic DNA. Lane 4: Undigested rescued plasmid. Lane 5: Xho I digested rescued plasmid.
 Figure 4. Partial DNA Sequence of a Plasmid. The sequence in bold is part of the transposon and was not submitted for BLAST analysis. The rest of the sequence is a segment of the rescued genes from the host.
Figure 4. Partial DNA Sequence of a Plasmid. The sequence in bold is part of the transposon and was not submitted for BLAST analysis. The rest of the sequence is a segment of the rescued genes from the host.
 Figure 5. BLAST Analysis7,8. Starting from the first base (Query), the interrupted gene appears to encode an Enterobacter cloacae chorismate mutase, which is involved in phenylalanine and tyrosine biosynthesis.
Figure 5. BLAST Analysis7,8. Starting from the first base (Query), the interrupted gene appears to encode an Enterobacter cloacae chorismate mutase, which is involved in phenylalanine and tyrosine biosynthesis.
Discussion
Transposon mutagenesis using a transposome is an efficient tool for generating auxotrophs in Enterobacter sp. YSU and other types of Gram negative and Gram positive bacteria3,4. The process was initiated by introducing the Tn5-derived transposome into the host by electroporation. To identify colonies with inserts, the untransformed target strain had to be sensitive to kanamycin in order to select for the resistance marker carried by the transposon. Some hosts produce restriction endonucleases which degrade the transposon DNA before it can recombine with the host genome. Addition of a restriction endonuclease inhibitor to the electroporation mix may minimize degradation and increase the transformation yield3. While preparing electroporation competent bacteria, it was important to remove as much salt as possible to prevent an electric arc or spark that kills the bacteria caused by the presence of salt. Even in the absence of an electric arc, the cells began to die rapidly after shocking them. Adding the salt containing SOC growth medium immediately restored the osmotic balance, minimized cell death and increased transformation efficiency5. Kanamycin resistant transformants were then spotted onto a fresh LB-kan plate and replica plated on to M-9 minimal medium agar. To prevent over-inoculation which can cause false positive results that miss possible auxotrophs, velveteen squares were switched at step 3.6 before the rest of the plates were inoculated. Then, during replica plating, just enough pressure was applied to transfer the colonies without causing smearing or contamination between adjacent colonies.
The mutated genes in the auxotrophs were identified through gene rescue. Genomic DNA from each auxotroph was sheared using a partial digestion with the frequent-base cutter BfuC I. Partial digestions using Bfa I or Hae III, which cut the transposon less frequently, may improve transposon transformation efficiency. It is also possible to use a complete digestion with other restriction endonucleases, 6-base and 8-base cutters, which lack sites within the transposon. The digested DNA was then ligated and transformed into E. coli strain ECD100D pir or ECD100D pir1166. Although the E. colipir116 strain was transformed by electroporation in step 4.8, it is also possible to use chemically competent cells. In this case, the whole transformation should be spread to obtain a large selection of rescued plasmids. The pir gene allows for the circularized transposon to replicate as a low copy number plasmid, and the pir116 mutant gene allows it to replicate as a high copy plasmid. Low transformation yields using the pir116 strain may be due to the expression of toxic gene products from the original host. Toxicity may be reduced by using the low copy pir strain. However, low plasmid yield from the pir strain can make sequencing more challenging.
Genomic sequencing10 and PCR sequencing14 are alternative methods to gene rescue. The genomic sequencing method sequences the flanking chromosomal regions directly from purified genomic DNA. This strategy is useful when the genome of the target organism has already been sequenced. Once the mutated region has been identified by genomic sequencing and BLAST, the whole region can be amplified by PCR and cloned for further analysis. The PCR method uses two PCR reactions and four different primers. The first reaction pairs a transposon homologous primer (primer 1) with a degenerate primer that contains a short DNA, linker sequence (primer 2). The second PCR reaction is a nested PCR reaction, pairing another transposon homologous primer (primer 3) with a homologous linker sequence primer (primer 4). The priming site for primer 3 is located on the 3’ side of the priming site for primer 1. Then, the amplicand from second PCR reaction is cleaned up and sequenced. This automated sequencing method amplifies DNA directly from the mutant cells, eliminating the need for both genomic DNA purification and plasmid rescue.
Instead of identifying genes involved in auxotrophy, transposon mutagenesis can also be used to study other easily assayed phenotypes. Enterobacter sp. YSU is a multi-metal resistant bacterial strain that is also resistant 100 µg/ml ampicillin (unpublished data). Its minimal inhibitory concentration (MIC) for HgCl2 is 70 µM, ZnCl2 is 800 µM, AgNO3 is 80 µM and NaAsO2 is 14 mM9. After introducing the transposome into this strain and gridding transformants onto LB-kan plates, the gridded colonies can be replica plated onto medium containing metals at concentrations below the MIC of this bacterial strain. Rescuing and sequencing of an interrupted gene from a mutant with a lowered MIC may reveal a gene that confers resistance to one of these metals.
In addition to generating mutants, the transposome can be used as a vector in vitro or in vivo to identify gain of function genes, such as antibiotic or metal resistance. For the in vitro strategy, genomic DNA from Enterobacter sp. YSU is digested with a restriction endonuclease that recognizes sites outside of the transposon. The DNA is ligated and mixed with the transposome in a buffer containing Mg2+. After recombination between the transposon and random genomic fragments, E. coli strain ECD100D pir is transformed with the DNA and spread on plates containing ampicillin or a metal salt. Colonies that grow will possess a new plasmid that contains the transposon located next to the host’s resistance gene. For the in vivo strategy, Enterobacter sp. YSU is transformed with the transposome. Then, the whole transformation reaction is grown in LB-kan liquid medium to select for a large population with random transposon inserts. Genomic DNA is digested with a restriction endonuclease that recognizes sites outside the transposome, ligated and transformed into E. coli strain ECD100D pir. As in the in vitro method, transformants are spread on ampicillin or metal plates. Again, the resulting plasmid will consist of the transposon inserted next to a resistance gene. These two strategies will be successful only if the target resistance gene can be expressed and is active in the E. coli strain.
Transposon mutagenesis can be used to identify the minimal number of genes a bacterial strain requires for growth15,16. Two transposomes containing loxP sites and different antibiotic resistance markers are required for this strategy in a bacterial strain with a known genome sequence. After the two transposons integrate, expressed cre recombinase catalyzes a recombination reaction between the two loxP sites, eliminating chromosomal regions between the transposon insertions. Knowledge of the genome sequence makes it possible to identify which genes are eliminated. The ability to grow demonstrates that the eliminated genes are not required for growth. This technique is labor intensive, and a minimal genome for growth using this strategy has not yet been completed. However, an automated transposon mutagenesis study of Pseudomonas aeruginosa using PCR sequencing estimated that 300-400 genes were required for the growth of this bacterium in rich medium14. This study did not use the loxP/cre recombinase system.
Microbes with minimal genomes are useful for synthetic biology17. A bacterial strain with minimal a genome is especially useful for eliminating the production of undesired byproducts. For example, Clostridium acetobutylicum (C. acetobutylicum) is used to produce the biofuel, butanol18. During its growth, this strain produces butanol at the end of its exponential growth phase just before it enters stationary phase. At this point, it becomes sensitive to butanol and other fermentation products and forms dormant endospores which cease to synthesize product. Since transposomes have been used to study Clostridium perfringens3, it may be possible to use a similar transposome/loxP/cre recombinase system to construct a strain of C.acetobutylicum that lacks the genes for undesired fermentation products and sporulation, but contains genes for greater butanol tolerance. This strain would increase the product yield with minimal contaminants.
In summary, this video journal article provides a complete step by step description of the procedures used to create auxotrophs by transposon mutagenesis. It covers introducing the transposome by electroporation, screening for mutations by replica plating, rescuing mutated genes by cloning techniques and identification of interrupted genes by DNA sequencing. This technique can be used for identifying the function of unknown genes or for possibly constructing a bacterial strain that can be used in an industrial process.
Disclosures
The author has nothing to disclose.
Acknowledgments
The author would like to thank all of my undergraduate Independent Research Students and all of my Microbial Physiology graduate students who tested my transposon mutagenesis ideas during the 2010-2014 spring semesters. This work was funded by the Department of Biological Sciences at Youngstown State University.
References
- Kim BH, Gadd GM. Bacterial physiology and metabolism. Cambridge: Cambridge University Press; 2008. [Google Scholar]
- Hayes F. Transposon-based strategies for microbial functional genomics and proteomics. Annual review of genetics. 2003;37:3–29. doi: 10.1146/annurev.genet.37.110801.142807. [DOI] [PubMed] [Google Scholar]
- Hoffman LM. Methods in molecular biology. Vol. 765. Clifton, N.J: 2011. Random chromosomal gene disruption in vivo using transposomes; pp. 55–70. [DOI] [PubMed] [Google Scholar]
- Goryshin IY, Jendrisak J, Hoffman LM, Meis R, Reznikoff WS. Insertional transposon mutagenesis by electroporation of released Tn5 transposition complexes. Nature biotechnology. 2000;18(1):97–100. doi: 10.1038/72017. [DOI] [PubMed] [Google Scholar]
- Dower WJ, Miller JF, Ragsdale CW. High Efficiency Transformation of E. coli by High Voltage Electroporation. Nucleic Acids Research. 1988;16(13):6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf WW, Jiang W, Wanner BL. Use of the rep technique for allele replacement to construct new Escherichia coli hosts for maintenance of R6Kγ origin plasmids at different copy numbers. Gene. 1994;138(1-2):1–7. doi: 10.1016/0378-1119(94)90776-5. [DOI] [PubMed] [Google Scholar]
- Morgulis A, Coulouris G, Raytselis Y, Madden TL, Agarwala R, Schäffer A. Bioinformatics. 16. Vol. 24. Oxford, England: 2008. a Database indexing for production MegaBLAST searches; pp. 1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. Journal of computational biology a journal of computational molecular cell biology. 2000;7(1-2):203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- Holmes A, Vinayak A, et al. Comparison of two multimetal resistant bacterial strains: Enterobacter sp. YSU and Stenotrophomonas maltophilia OR02. Current microbiology. 2009;59(5):526–531. doi: 10.1007/s00284-009-9471-2. [DOI] [PubMed] [Google Scholar]
- Hoffman LM, Jendrisak JJ, Meis RJ, Goryshin IY, Reznikoff WS. Transposome insertional mutagenesis and direct sequencing of microbial genomes. Genetica. 2000;108(1):19–24. doi: 10.1023/a:1004083307819. [DOI] [PubMed] [Google Scholar]
- Ausubel F, Brent R, et al. Short Protocols in Molecular Biology. New York, NY: John Wile., & Sons, Inc; 1997. [Google Scholar]
- Miller JH. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Vol. 2. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Gibson MI, Gibson F. Preliminary studies on the isolation and metabolism of an intermediate in aromatic biosynthesis: chorismic acid. The Biochemical journal. 1964;90(2):248–256. doi: 10.1042/bj0900248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MA, Alwood A, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24) doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikoff W, Winterberg K. Transposon-Based Strategies for the Identification of Essential Bacterial Genes. Microbial Gene Essentiality: Protocols and Bioinformatics SE. 2008;2:13–26. doi: 10.1007/978-1-59745-321-9_2. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Inui M, Yukawa H. Random genome deletion methods applicable to prokaryotes. Applied Microbiology and Biotechnology. 2008;79(4):519–526. doi: 10.1007/s00253-008-1512-4. [DOI] [PubMed] [Google Scholar]
- Malley MA, Powell A, Davies JF, Calvert J. Knowledge-making distinctions in synthetic biology. BioEssays news and reviews in molecular, cellular and developmental biology. 2008;30(1):57–65. doi: 10.1002/bies.20664. [DOI] [PubMed] [Google Scholar]
- Zheng Y-N, Li L-Z, et al. Problems with the microbial production of butanol. Journal of industrial microbiolog., & biotechnology. 2009;36(9):1127–1138. doi: 10.1007/s10295-009-0609-9. [DOI] [PubMed] [Google Scholar]