

Abstract
We previously described the bioconversion of Notoamide T into (+)-Stephacidin A and (−)-Notoamide B, which suggested that Versicolamide B (8) is biosynthesized from 6-epi-Notoamide T (10) via 6-epi-Stephacidin A. Here we report that [13C]2-10 was incorporated into isotopically enriched 8 and seven new metabolites, which were not produced under normal culture conditions. The results suggest that the addition of excess precursor activated the expression of dormant tailoring genes giving rise to these structurally unprecedented metabolites.
Keywords: Notoamide, Aspergillus sp, Bioconversion, Alkaloid
The Notoamides are a family of prenylated indole alkaloids produced by the marine-derived Aspergillus sp. MF297-2,1 and have also been detected as natural metabolites of A. versicolor NRRL 35660.2 These natural substances contain a unique bicyclo[2.2.2]diazaoctane core structure and have attracted significant attention from the synthetic community.3 Previously, Sammes4 and Birch5 suggested that the bicyclo[2.2.2]diazaoctane core of these alkaloids is likely to arise from a biosynthetic intramolecular hetero Diels-Alder (IMDA) reaction. Initially, we isolated Notoamides A–D (1–4) and Stephacidin A (5) from the fungal extract1a and proposed a biogenetic pathway,6 in which a putative biosynthetic precursor was postulated to be Notoamide E (6) (Scheme 1). The precursor incorporation experiment utilizing [13C]-6 afforded 3 and 4 but, metabolites containing the bicyclo[2.2.2]diazaoctane core, such as 1, 2, and 5, were not obtained.1c
Scheme 1.
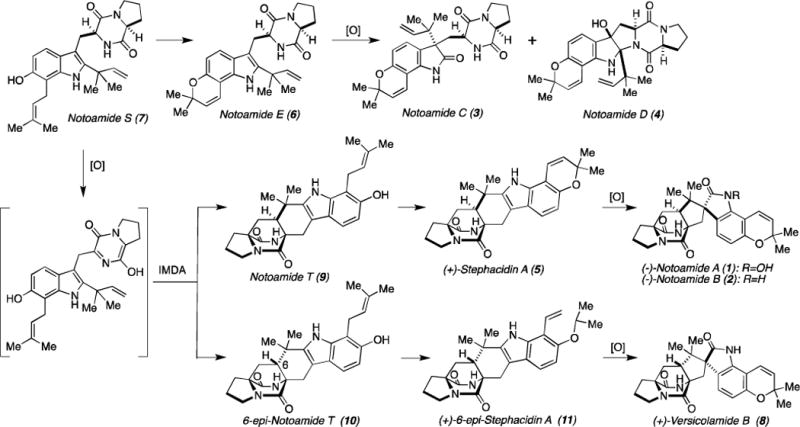
Biosynthetic Pathway for the Notoamides, Stephacidin A, and Versicolamide B
We then speculated that Notoamide S (7) would be the progenitor of 6 and converted to 5/2 and Versicolamide B (8) by an enzyme-catalyzed IMDA reaction through Notoamide T (9) and 6-epi-Notoamide T (10), respectively. Although we initially reported that 8 isolated from Aspergillus sp. MF297-2 was the (−)-enantiomer,1d careful, recent re-analysis of the natural metabolite revealed that the absolute configuration should be revised to the (+)-form. We have also carefully confirmed that the absolute configurations of Stephacidin A and Notoamide B are as originally assigned. Thus, the marine-derived Aspergillus sp. MF297-2 produces (+)-Stephacidin A, (−)-Notoamide B, and (+)-Versicolamide B and the terrestrial A. versicolor NRRL 356602 produces the enantiomers: (−)-Stephacidin A, (+)-Notoamide B, but the same enantiomer of (+)-Versicolamide B as that produced in the marine-derived fungus.7 These stereochemical facts present numerous fascinating biogenetic puzzles that have yet to be unraveled. In our ongoing efforts toward understanding the molecular bases for the biogenesis of the Stephacidins, Notoamides, and Versicolamide B we have previously described the bioconversions of 5 into 28 and of 9 into 5/2.9 These results clearly suggested that (+)-8 would be biosynthesized from 10 via 6-epi-Stephacidin A (11).10 In order to verify this hypothesis, we evaluated the detection of 11 in the culture of the marine-derived Aspergillus sp. MF297-2 and the bioconversion of 10 into 11 and 8.
Since 6-epi-Stephacidin A (11) was expected to be a short-lived precursor, the time-course of the metabolite profile was analyzed by HPLC. The fungus was cultured on agar medium in petri dishes and the extract was semi-purified by ODS column chromatography. To monitor the presence of 11, the 1H NMR spectrum of the fraction, of which retention time corresponded to authentic, synthetic 11, was measured. The spectra revealed that 11 existed only in the culture incubated for six days and subsequently disappeared. The CD spectrum of 11 revealed that this substance possesses the 11S,17S-configuration (Supplementary data, Figure S1).11 The experimental data clearly indicated that 11 was produced by the fungus in the early phase of growth and was then rapidly converted to other downstream metabolites. We have previously demonstrated that Notoamide E (6) was also a short-lived endogenous precursor and was rapidly converted into Notoamide C (3) and Notoamide D (4).1c
Next, we conducted a precursor incorporation experiment using synthetic, [13C]2-(±)-6-epi-Notoamide T (10).9 The fungal cells of Aspergillus sp. MF297-2 were incubated in a trace element solution with [13C]2-(±)-10. After fourteen days, the culture was extracted with n-BuOH and the extract was purified by ODS column chromatography followed by ODS HPLC to afford [13C]2-Versicolamide B (8) and very surprisingly, seven new metabolites, [13C]2-6-epi-Notoamides T3–T8 (12–17) and [13C]2-6-epi-Notoamide I (18) (Scheme 2).
Scheme 2.
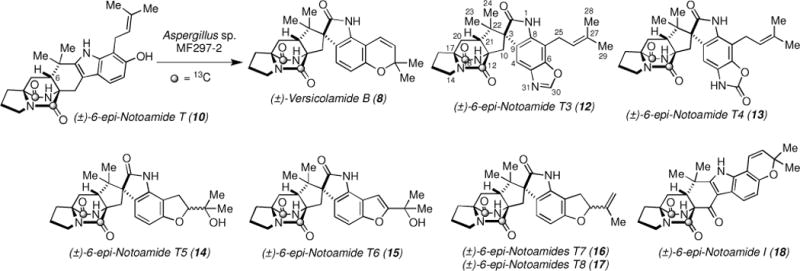
Bioconversion of 6-epi-Notomide T into Versicolamide B and seven new metabolites
A molecular formula of [13C]2-6-epi-Notoamide T3 (12) was determined to be C2513C2H30N4O4 on the basis of the HRESIMS, which indicated the presence of additional carbon and nitrogen atoms in [13C]2-12. Analysis of NMR spectra in DMSO-d6 (Supplementary data, Table S1) indicated that [13C]2-12 possessed a spiro-oxindole structure, similar to that which constitutes Versicolamide B (8). The isoprene unit still remained at C-7 yet, the pyran ring was absent in [13C]2-12. In addition, instead of ortho-coupled aromatic protons, δ 7.11 (H-4) and δ 6.37 (H-5), observed in 8, a single aromatic proton at δ 7.75 (H-4, singlet) and a quaternary carbon at δ 133.8 (C-5) were observed in [13C]2-12. These data clearly suggested that the remaining CHNO unit was attached to C-5. The HSQC spectrum of this substance revealed that a single proton (δ 8.60, H-30, singlet) was directly attached to a carbon at δ 152.0 (C-30), and the carbon was attached to hetero atoms as judged by the chemical shift. HMBC cross peaks from δH 8.60 to δC 133.8 (C-5) and 148.0 (C-6) and the chemical shifts of these carbons suggested that an oxazole ring was fused to the tryptophan moiety as shown in Figure 1. The presence of an oxygen atom bonded to C-6 is firmly explained by the biosynthetic relationship with the precursor, [13C]2-10.
Figure 1.
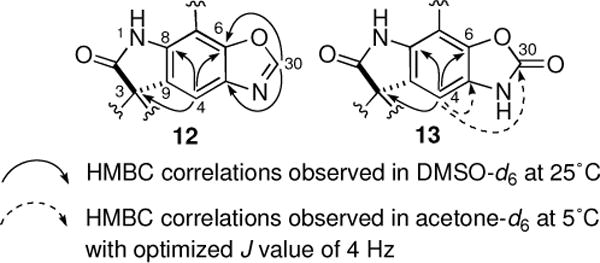
HMBC correlations observed for 12 and 13.
[13C]2-6-epi-Notoamide T4 (13) has a molecular formula of C2513C2H30N4O5 as established by HRESIMS, and possesses one oxygen atom more than [13C]2-12. The NMR spectra of [13C]2-13 in DMSO-d6 was almost superimposable on those of [13C]2-12 except for the high-field shift of H-4 (δ 7.03 in [13C]2-13; δ 7.75 in [13C]2-12) (Supplementary data, Table S1). Two carbon signals at C-5 and C-30 of 13 were not detected in the HMBC spectrum in DMSO-d6, suggesting the possibility of two rapidly equilibrating isomers; an HMBC spectrum was measured in acetone-d6 at 5 °C with optimized J value of 4 Hz. Under this condition, cross peaks from H-4 (δ 7.20) to C-5 (δ 135.2) and C-30 (δ 156.8) were observed (Figure 1). These data suggested that [13C]2-13 possess an 2-oxazolone unit instead of the oxazole unit present in [13C]2-12.
Analyses of NMR spectra of [13C]2-6-epi-Notoamides T5-T8 (14–17) (Supplementary data, Tables S2 and S3) readily indicated the presence of spiro-oxindole structures with various biosynthetic modifications derived from the isoprene unit at C-7 and the hydroxyl group at C-6. [13C]2-6-epi-Notoamide T5 (14) has a formula of C2413C2H31N3O5, suggesting that the attached portion at C-6 and C-7 was composed of C5H10O2. The COSY spectrum showed a -CH2CH- unit and the methylene signals (H2-25) showed HMBC correlation to C-7 (δ 108.0) (Figure 2). An exchangeable proton at δ 4.58 (27-OH) showed HMBC correlations with carbons at δ 24.6 (CH3, C-28), 26.1 (CH3, C-29), 69.6 (C, C-27), and 89.2 (CH, C-26). A singlet methyl signal at δ 1.12 (H3-29) showed HMBC correlations with carbons at δ 24.6, 69.6 and 89.2. These data suggest that [13C]2-14 contains a substituted dihydrofuran ring instead of the pyran ring present in 8. Although H-26 was observed as a single signal, [13C]2-14 might be a mixture of 26-epimers. [13C]2-6-epi-Notoamide T6 (15) has a formula of C2413C2H29N3O5; one H2 unit less than [13C]2-14. An olefin proton at δ 6.59 (H-25) showed HMBC correlations with three quaternary carbons at δ 113.7 (C-7), 155.0 (C-6), and 164.6 (C-26) (Figure 2). Therefore, the structure of [13C]2-15 corresponds to a dehydrogenated derivative of [13C]2-14. [13C]2-6-epi-Notoamides T7 (16) and T8 (17) were afforded in the ratio of 1:1 and they possess an identical formula C2413C2H29N3O4, an oxygen less than [13C]2-15. Their NMR spectra were almost superimposable, and differences were observed at proton signals at δ 10.40/10.42 (H-1, 16/17), 5.04/5.05 (H-28), and 1.70/1.72 (H3-29). Interpretation of 2D NMR spectra implicated that they corresponded to dehydro derivatives of [13C]2-14 and are C-26-epimers of each other. The structure of [13C]2-6-epi-Notoamide I (18) was deduced by the comparison of NMR data of Notoamide I1b and a formula C2413C2H27N3O4 shown by HRESIMS. The diversity of structures involving prenyl group ring closures on a terminal benzyl group is reminiscent of a recent report of novel Aspulivinones, where similar open ring, pyran, and furan ring systems were described.13
Figure 2.
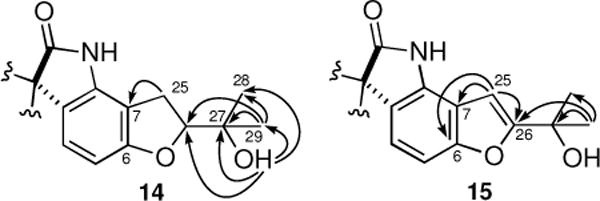
HMBC correlations observed for 14 and 15.
Although the precursor incorporation experiment was performed with a synthetic, racemic precursor [13C]2-(±)-10, metabolites [13C]2-12–18 and [13C]2-8 were found to be optically inactive, as revealed by the CD spectra of these substances (Supplementary data, Figures S47–S54) compared with a natural sample of (+)-Versicolamide B (Supplementary data, Figure S55). These results definitively suggest that the fungus converted both enantiomeric precursors, [13C]2-(±)-10, which clearly suggested that enzymes involved in these biotransformations were effective for these exogenic substrates.
Since 11 was proposed to be a precursor of 8, we expected to detect [13C]2-(±)-11 in precursor incorporation experiments of [13C]2-(±)-10 but we did not observe this species. We then performed precursor incorporation experiments of non-labeled-(±)-10 on minimal media agar plates. After sixteen days, the culture was extracted with n-BuOH and purification of the extract yielded 11 and four new metabolites, 6-epi-Notoamides T9–T12 (19–22) (Scheme 3; Supplementary data, Table S4). The CD spectra revealed that the metabolites were all racemates (Supplementary data, Figures S56–S60).
Scheme 3.
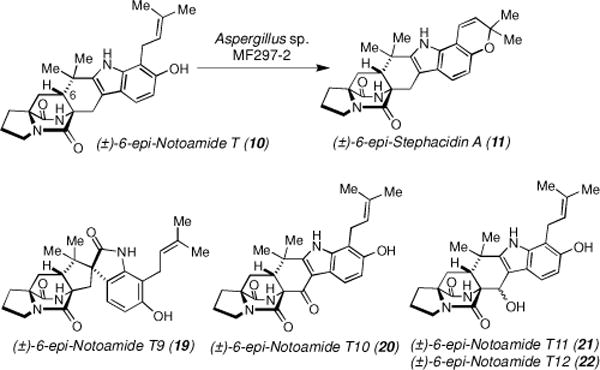
Bioconversion of (±)-6-epi-Notoamide T into (±)-6-epi-Stephacidin A and four new metabolites on the agar minimal media.
Biosynthetically, [13C]2-12 would be derived from [13C]2-10 by oxygenation at C-2 and successive pinacol rearrangement followed by an attachment of a CHN unit (Scheme 4). Glycine is one possible donor unit of both the new nitrogen and carbon in the oxazolidine and oxazolidinone units of 12 and 13. It is also possible that the nitrogen and carbon atoms are derived separately from distinct biosynthetic donors. While beyond the scope of this communication, we are seeking to elucidate the genes responsible for the production of these novel metabolites, the primary metabolic donor units and the mechanism for the formation of the new heterocyclic units of 12 and 13. At present, however, the origin of this unit, and particularly, the origin and mechanism for the addition of the new nitrogen atom remains unclear. To the best of our knowledge, the oxidative amination of an aromatic ring is an unprecedented biochemical transformation. Based on our previous work to obtain the complete sequence for the Aspergillus sp. MF297-2 fungal genome,12 we anticipate a thorough transcriptome analysis will enable initial identification of gene products expressed following introduction of [13C]2-(±)-6-epi-Notoamide T (10) into the culture medium leading to the reported series of novel alkaloids.
Scheme 4.
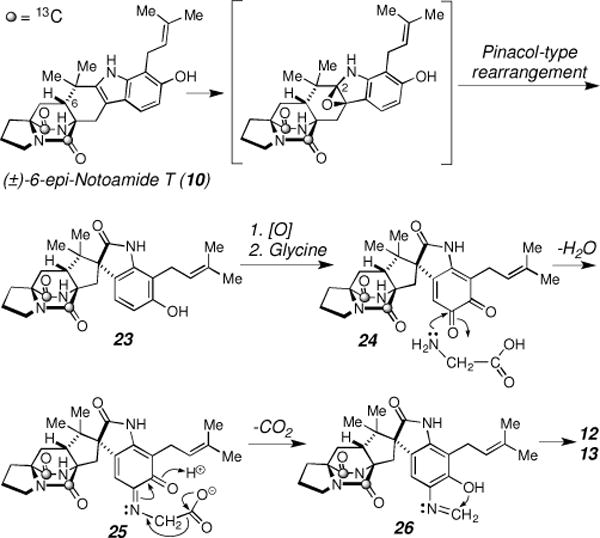
Possible biosynthetic pathway of 12 and 13 from 10.
In conclusion, we have demonstrated the bioconversion of 6-epi-Notoamide T (10) to 6-epi-Stephacidin A (11) and Versicolamide B (8). In the precursor incorporation experiment of [13C]2-(±)-10 in liquid media, seven new metabolites were unexpectedly obtained, [13C]2-12–18, which are not produced by the fungus (500 petri dishes) under normal culture conditions to any detectable extent. Among the metabolites, [13C]2-12 and [13C]2-13 contain unprecedented structures, including the oxazole and 2-oxazolone units, respectively, derived from the oxidative amination of the tryptophan moiety. On the other hand, the precursor incorporation experiment of (±)-10 on agar medium, four new oxidized metabolites 19–22 were obtained. These data suggest that the exogenous substrate, 6-epi-Notoamide T (10), selectively activates the expression of dormant tailoring genes that significantly expands the metabolome of the producing organism. Our laboratories are actively investigating the origins of the carbon and nitrogen atoms of the oxazole and 2-oxazolone units. We are also pursuing identification of the unique tailoring enzymes that catalyze construction of these unprecedented substances and how the exogenous substrate, [13C]2-(±)-10, has triggered gene expression. This study supports the growing body of evidence that the secondary metabolome of fungi may be considerably more plastic and subject to considerable chemical diversification through addition of both endogenous, as well as exogenous substances.14 Efforts to capitalize on these finding is under intensive investigation in our laboratories and will be reported in due course.
Supplementary Material
Acknowledgments
This work was financially supported in part by Grants-in-Aid for Scientific Research (Nos. 23108518 and 25108719 to S.T. and 24710252 to H.K.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and a grant from the Nagase Science and Technology Foundation (to S.T.). Financial support from the National Institutes of Health (Grant #R01 CA070375 to R.M.W.) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S. Angew Chem Int Ed. 2007;46:2254–2256. doi: 10.1002/anie.200604381. [DOI] [PubMed] [Google Scholar]; (b) Tsukamoto S, Kato H, Samizo M, Nojiri Y, Onuki H, Hirota H, Ohta T. J Nat Prod. 2008;71:2064–2067. doi: 10.1021/np800471y. [DOI] [PubMed] [Google Scholar]; (c) Tsukamoto S, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. J Am Chem Soc. 2009;131:3834–3835. doi: 10.1021/ja810029b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tsukamoto S, Kawabata T, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. Org Lett. 2009;11:1297–1300. doi: 10.1021/ol900071c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tsukamoto S, Umaoka H, Yoshikawa K, Ikeda T, Hirota H. J Nat Prod. 2010;73:1438–1440. doi: 10.1021/np1002498. [DOI] [PubMed] [Google Scholar]
- 2.Greshock TJ, Grubbs AW, Jiao P, Wicklow DT, Gloer JB, Williams RM. Angew Chem Int Ed. 2008;47:3573–3577. doi: 10.1002/anie.200800106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller KA, Williams RM. Chem Soc Rev. 2009;38:3160–3174. doi: 10.1039/b816705m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter AEA, Sammes PG. J Chem Soc Chem Commun. 1970:1103. [Google Scholar]
- 5.Baldas J, Birch AJ, Russel RA. J Chem Soc Perkin Trans. 1974;1:50–52. [Google Scholar]
- 6.(a) Grubbs AW, Artman GD, III, Tsukamoto S, Williams RM. Angew Chem Int Ed. 2007;46:2257–2261. doi: 10.1002/anie.200604377. [DOI] [PubMed] [Google Scholar]; (b) Greshock TJ, Grubbs AW, Tsukamoto S, Williams RM. Angew Chem Int Ed. 2007;46:2262–2265. doi: 10.1002/anie.200604378. [DOI] [PubMed] [Google Scholar]
- 7.Li S, Anand K, Tran H, Yu F, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH. Med Chem Comm. 2012;3:987–996. doi: 10.1039/C2MD20029E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finefield JM, Kato H, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM. Org Lett. 2011;13:3802–3805. doi: 10.1021/ol201284y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sunderhaus JD, McAfoos TJ, Finefield JM, Kato H, Li S, Tsukamoto S, Sherman DH, Williams RM. Org Lett. 2013;15:22–25. doi: 10.1021/ol302901p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai S, Luan Y, Kong X, Zhu T, Gu Q, Li D. Org Lett. 2013;15:2168–2171. doi: 10.1021/ol400694h. [DOI] [PubMed] [Google Scholar]
- 11.Williams RM, Kwast E, Coffman H, Glinka T. J Am Chem Soc. 1989;111:3064–3065. [Google Scholar]
- 12.Ding Y, de Wet JR, Cavalcoli J, Li S, Greshock TJ, Miller KA, Finefield JM, Sunderhaus JD, McAfoos T, Tsukamoto S, Williams RM, Sherman DH. J Am Chem Soc. 2010;32:12733–12740. doi: 10.1021/ja1049302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz PG, Auld DS, Schultz PJ, Lovell S, Battaile KP, MacArthur R, Shen M, Tamayo G, Inglese J, Sherman DH. Chem Biol. 2011;18:1442–1452. doi: 10.1016/j.chembiol.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cichewicz RH. Nat Prod Rep. 2010;27:11–22. doi: 10.1039/b920860g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.