

Abstract
The presence of aromatic clusters has been found to be an integral feature of many proteins isolated from thermophilic microorganisms. Residues found in aromatic cluster interact via π-π or C-H···π bonds between the phenyl rings, which are among the weakest interactions involved in protein stability. The lone aromatic cluster in human carbonic anhydrase II (HCA II) is centered on F226 with the surrounding aromatics F66, F95 and W97 located 12 Å posterior the active site; a location which could facilitate proper protein folding and active site construction. The role of F226 in the structure, catalytic activity and thermostability of HCA II was investigated via site-directed mutagenesis of three variants (F226I/L/W) into this position. The measured catalytic rates of the F226 variants via 18O-mass spectrometry were identical to the native enzyme, but differential scanning calorimetry studies revealed a 3-4 K decrease in their denaturing temperature. X-ray crystallographic analysis suggests that the structural basis of this destabilization is via disruption and/or removal of weak C-H···π interactions between F226 to F66, F95 and W97. This study emphasizes the importance of the delicate arrangement of these weak interactions among aromatic clusters in overall protein stability.
Keywords: human carbonic anhydrase II, aromatic cluster, thermostability
Introduction
The carbonic anhydrases (CAs, EC 4.2.1.1) are a family of mostly zinc metalloenzymes found in all animals, photosynthetic organisms, and some non-photosynthetic bacteria (1-3). There are three evolutionarily and structurally unrelated CA families: the α-CAs, which are found primarily in animals and some plants; the β-CAs, which are found in photosynthetic organisms; and γ-CAs, which have been identified in several species of archae. Human CA isoform II (HCA II) is a ~30 kDa monomeric, globular protein that is ubiquitously expressed and is one of the fastest enzymes known, with a maximum turnover rate of 1 μs−1 with a catalytic efficiency approaching the diffusion limit at 100 M−1 μs−1 (4-8).
HCA II catalyzes the reversible interconversion between carbon dioxide and bicarbonate via a two-step ping-pong mechanism:
| (Reaction 1) |
| (Reaction 2) |
In the first reaction, the hydration reaction, a nucloephilic attack on carbon dioxide by the zinc-bound hydroxide converts carbon dioxide into bicarbonate. A water molecule then displaces the bicarbonate product, which diffuses away (Reaction 1). In the second reaction, the zinc-bound hydroxide is regenerated through transfer of a proton from the zinc-bound water to the proton shuttle resiude, H64, via a series of well-ordered, hydrogen-bonded water molecules that extends the length of the active site (9-13). The proton is then transferred to buffer in solution.
The X-ray crystallographic structure of CO2 bound in the hydrophobic pocket (V121, V143, L198, V207 and W209) of active site of HCA II (14) revealed that the CO2 molecule displaces a well-ordered water, termed ‘deep water’, and makes a potential hydrogen bond with backbone amide of T199. This coordination of CO2 allows for the nucleophilic attack by the Zn-OH− to form bicarbonate (Reaction 1). In addition to the catalytic binding site of CO2 in, a secondary site was observed ~11 Å away, buried in a hydrophobic core of aromatic residues (F66, F95, W97 and F226). Superimposition of the HCA II CO2 bound to the unbound structure revealed that F226 undergoes a 30° tilt with respect to the plane of the ring when the substrate is bound (Fig. 1B). Additionally, xenon has been reported (15) to be seen in this secondary binding site (Fig. 1C) in low occupancy (~0.3) which may suggest that this hydrophobic core is susceptible to solvent exposure through natural protein breathing, which is not uncommon for aromatic clusters (16). Several proteins isolated from thermophilic microorganisms contain aromatic clusters and could be a possible source for their stability (17-20).
Figure 1.
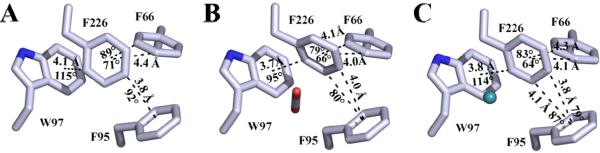
X-ray crystallographic structures of (A) native HCA II (PDB: 3KS3; (25)) (B) HCA II with CO2 bound (PDB: 3D92; (14)) and (C) HCA II with Xe bound (PDB: 3CYU; (15)). Models are shown as sticks with carbon colored light blue, oxygen red, nitrogen blue and xenon cyan. Potential C-H···π interactions between F226, F66, F95 and W97 are represented by dashed lines with distances and angles defined as in Table 4.
Sequence alignment for all of the HCA isoforms revealed that F226 is conserved within seven of the twelve catalytically active isoforms (HCA I, II, IV, VA, VB, VII, and XIII). Interestingly, HCA II, IV, VB and VII are the isoforms with the highest turnover rate (0.95 – 1.4 μs−1) and catalytic efficiencies (50 – 120 M−1 μs−1) (reviewed in (1)). The other five catalytically active isoforms (HCA III, VI, IX, XII and XIV) show a conserved hydrophobic leucine substituent at this position with overall lower turnover rates (0.01 – 0.42 μs−1) and catalytic efficiencies (0.3 – 55 M−1 μs−1). While some of these lowered catalytic rates can be explained via the structure of the active site (e.g., substitution of H64K in HCA III (21)), others are not so readily understood. It should be noted that F226 makes delicate interactions with F95 and W97, which are both just one position downstream from the zinc-coordinating residues H94 and H96, respectively. Distortion of the peptide backbone in this position could potentially lead to disruption of the catalytic efficiency of the enzyme.
This study investigates the affects from disruption of the lone aromatic cluster in HCA II via site-directed mutagenesis of F226 into the conserved variant F226L, as well as two nonconserved hydrophobic substitutions of F226I and F226W. X-ray crystallographic analysis of the variants revealed conservation of the hydrophobocity core centered on F226 allowed for proper folding of the enzymes with no large-scale conformational change in or around the active sites. Measurement of the catalytic rates via 18O mass spectrometry confirmed retention of HCAII's extreme catalytic efficiency in these variants. Differential scanning calorimetry (DSC) studies revealed, however, that the F226 variants displayed a ~3 K reduction in denaturing temperature compared to the native enzyme. Closer inspection of the X-ray crystallographic models suggest that the delicate geometry (i.e., distances and angles) of weak C-H···π bonds made between F226 and the surrounding aromatic residues are important in the thermostability of HCA II.
Materials and Methods
Enzyme Expression and Purification
Primers from Invitrogen and the Stratagene QuikChange II kit were used for site-directed mutagenesis in the preparation of HCAII cDNA with the individual F226I/F226L/F226W mutations from an expression vector consisting of the enzyme coding region (22). Transformation of the mutant cDNA occurred in E. coli XL1-Blue super-competent cells. The transformation was verified by sequencing the DNA of the whole coding region. The verified cDNA was transferred into E. coli BL21(DE3)pLysS cells. The cells were then grown at 37 °C to 0.6 turbidity at 600 nm in 1L of 2 × Luria broth medium, containing 0.1 mg/mL ampicillin (final concentration). Protein expression was induced by the addition of ~0.1 mg/mL isopropyl β-D-1-thiogalactopyranoside (IPTG) and ~1mM zinc sulfate (final concentrations). The cells were then incubated for an additional three hours and harvested by centrifugation.
Hen egg white lysozyme and DNaseI was added to the cells suspended in 0.2 M sodium sulfate, 0.1 M Tris-HCl, pH 9.0 followed by removal of cellular debris via centrifugation. An affinity column with p-(aminomethyl)-benzene-sulfonamide, a HCA II tight-binding inhibitor (23),coupled to an agarose resin was used for purification. In the purification process, the bound HCA II variants were eluted with 0.4 M sodium azide, 0.1 M Tris-HCl, pH 7.0 followed by extensive dialysis in 50 mM Tris-HCl, pH 7.8 to remove the azide. The purity of each sample was checked via SDS-PAGE and was found to be >95% (data not shown).
Crystallization and Diffraction Data Collection
Crystal trays were prepared for each HCA II variant with a 1:1 ratio of the reservoir solution and the protein sample (10 mg/mL). The reservoir solutions contained between 1.4 – 1.6 M sodium citrate, 50 mM Tris, pH 8.0 – 9.0. The crystals formed in approximately one week via the hanging drop diffusion method. Diffraction sets were collected on an in-house Rigaku R-Axis IV++ image plate detector with a RU-H3R rotating Cu anode (Kα = 1.5418 Å) operating at 50 kV and 22 mA. The X-rays were focused using Osmic optics, followed by a helium-purged beam path. The crystal-to-detector distance was 80 mm. Each image was collected for five minutes with 1° oscillations. HKL2000 (24) was used to integrate, merge and scale the data sets. The variants crystallized in the monoclinic space group P21 and diffracted to resolutions of 2.05, 1.63 and 1.70 for F226I, F226L and F226W, respectively. The diffraction and refinement statistics for all of the variants are summarized in Table 1.
Table 1.
Refinement and Final Model Statistics for F226 HCA II Variants.
| F226I | F226L | F226W | |
|---|---|---|---|
| PDB Accession Number | 4L5U | 4L5V | 4L5W |
| Wavelength (Å) | 1.5418 | ||
| Spacegroup | P21 | ||
| Unit-cell parameters (Å;°) | a = 42.3, b = 41.4, c = 71.5; β = 104.3 | a = 42.4, b = 41.4, c = 72.2; β = 104.4 | a = 42.1, b = 41.3, c = 72.0; β = 104.3 |
| Total number of measured reflections | 103017 | 107591 | 77729 |
| Total number of unique reflections | 15372 | 28174 | 24828 |
| Resolution (Å) | 20.0 – 2.05 | 20.0 – 1.63 | 20.0 – 1.70 |
| Rsyma (%) | 11.1 (42.7)e | 6.8 (35.2) | 4.1 (44.2) |
| I / σ (I) | 13.5 (3.9) | 37.2 (5.1) | 23.3 (2.6) |
| Completeness (%) | 99.0 (98.8) | 92.2 (85.0) | 92.8 (98.8) |
| Redundancy | 6.7 (6.5) | 3.8 (3.8) | 3.1 (3.0) |
| Rcrystb (%) | 15.4 | 13.0 | 14.2 |
| Rfreec (%) | 20.1 | 15.3 | 18.2 |
| Number of residues | 257 | 257 | 257 |
| Number of protein atoms (including alternate conformations) | 2300 | 2298 | 2293 |
| Number of water molecules | 209 | 338 | 290 |
| r.m.s.d.: Bond lengths (Å), angles (°) | 1.297, 0.011 | 1.333, 0.010 | 1.313, 0.010 |
| Ramachandran statistics (%): Most favored, allowed, outliers | 97.2, 2.8, 0.0 | 96.9, 3.1, 0.0 | 96.9, 3.1, 0.0 |
| Average B factors (Å2): All, main chain, side chain, solvent | 9.9, 5.9, 10.3, 27.4 | 21.2, 16.2, 21.7, 34.2 | 29.0, 24.3, 30.3, 39.8 |
| r.m.s.d.: Cα (Å)d | 0.10 | 0.08 | 0.14 |
Rsym = (∑|I - <I>|/∑ <I>) × 100
Rcryst = (∑|Fo - Fc|/∑ |Fo|) × 100
Rfree is calculated in the same way as Rcryst except it is for data omitted from refinement (5% of reflections for all data sets).
Root-mean-square deviation of the Cα backbone compared to HCA II (PDB: 3KS3 (25)).
Values in parentheses represent highest resolution bin.
Structure Refinement
Initial phases of the variants were calculated using molecular replacement using the wild-type (wt) HCA II structure (PDB: 3KS3; (25)) with 5% of the reflections set aside for Rfree calculations. PHENIX.REFINE (26) was used in cycles of restrained refinement of the molecular model, alternating with manual building using COOT (27). Initial Fo - Fc difference maps revealed negative density around the phenyl ring (present in the molecular replacement structure) at position 226 for the Leu and Ile variants, whereas F226W showed positive density that extended further away from the phenyl ring. Subsequent refinements showed excellent electron density in the 2Fo - Fc maps for the respective variants at this position (Fig. 2). The final Rcrys and Rfree values are shown in Table 1. MOLPROBITY (28) was used to assess the quality of the final model. All structural figures were created in PyMOL (29). Experimental data and structural coordination have been deposited with the Protein Data Bank under the accession numbers 4L5U, 4L5V and 4L5W for F226I, F226L and F226W, respectively.
Figure 2.

Final 2Fo – Fc electron density maps for (A) F226I (B) F226L and (C) F226W. The map is displayed as a light gray mesh, contoured at 1.6σ, around the corresponding mutation, represented in stick model. The carbon atoms are colored orange, pink and yellow for F226I, F226L and F226W, respectively.
Kinetics
18O-exchange mass spectrometry was used to measure the catalytic rate of the variants at chemical equilibrium (30). As the carbon dioxide in solution goes in an Extrel EXM-200 mass spectrometer via a semi-permeable membrane, the 18O-labeled isotopic content of CO2 is measured. There are two steps involved in the catalytic process that produce two catalytic rates, R1 and RH2O:
| [1] |
| [2] |
In the first step, there is a random probability of labeling the zinc-bound hydroxide with 18O (Eq. 1). In the next step, the protonation of 18OH−, via the proton shuttle residue His 64 (11), promotes the release of H 182O into the bulk solvent where it is infinitely diluted by unlabeled water (Eq. 2).
The rate of catalysis of the interconversion of CO2 and bicarbonate is indicated by R1 (see Eq. 3). The rate constant, kcatex, is a measure of the maximum conversion between the substrate and the product. The effective binding constant of the substrate is KeffS. Assuming steady state conditions, the ratio kcatex/KeffS in Eq. 4 is, in principle, the same as kcat/KM and is reported as such.
| [3] |
| [4] |
In Eq. 4, RH2O is the rate of release of H 182O into the bulk solvent. The proton transfer from His64 to the isotopically labeled zinc-bound hydroxide determines this rate (Eq. 2). The relationship between kB, which is the rate constant from the zinc-bound hydroxide's proton transfer and the ionization constants for the proton donor, (Ka)donor, and the acceptor, (Ka)ZnH2O, is shown in Eq. 4.
Using an enzyme concentration of approximately 1 mg/mL and all possible isotope-labeled species of carbon dioxide of 25 mM, the rates R1 and RH2O were measured in the absence of buffer at 25 °C. The kinetic and ionizations constants shown in Eqs. 3 and 4 were determined via a nonlinear least squares method (Enzfitter, Biosoft).
DSC Thermostability Studies
A VP-DSC calorimeter (Microcal, Inc., North Hampton, MA) was used to assess the thermal stability of the F226 variants. Samples of the F226 variants were dialyzed into 50 mM Tris-HCl, pH 7.8 at protein concentrations of approximately 1 mg/mL (30 μM). The samples and buffers were degassed under vacuum at 16 °C for 20 minutes, with stirring, prior to sample loading. The DSC scan rate was 60 °C/hour, with data being collected from 20 to 100 °C.
The melting temperature (TM) values of the F226 variants and wt HCAII were obtained from the midpoints of the DSC curves, indicating a two-state transition. The difference in Gibbs free energy (ΔG°) at a given temperature, T, was calculated via (31):
| [5] |
Where ΔH°M is the calorimetric enthalpy at TM and ΔCP is the observed change in heat capacity between the folded and unfolded states. The denaturation enthalpies (ΔH°) and entropies (ΔS°) were calculated at a given temperature using the Kirchoff's law equations (32):
| [6] |
| [7] |
Thermograms for all of the variants were obtained in triplicates and then averaged to obtain the final denaturation profile, followed by reference subtraction and data analysis. The pre- and post-translation baselines of the thermograms were adjusted via linear regression and the area under the peaks was determined through integration to calculate the enthalpy of unfolding (ΔH).
Results
X-ray Crystallographic Structure
The F226 HCA II variants crystallized in the monoclinic P21 space group with approximate unit cell dimensions of a = 42, b = 41 and c = 72 Å, β = 104° and diffracted to medium resolution (1.63, 1.70 and 2.05 for F226L, F226W and F226I, respectively). The final Rcryst/Rfree values were 13.0/15.3, 14.2/18.2 and 15.4/20.1 for F226L, F226W and F226I, respectively. A summary of the collected diffraction and final refinement statistics is shown in Table 1. The 2Fo – Fc maps for each variant are shown in Fig. 2 and confirm the successful amino acid substitutions at residue 226 in HCA II.
A linear least-square fit of the Cα atoms in the F226 variants to wt HCA II structure (PDB: 3KS3; (25)) revealed little structural variation (r.m.s.d. ~0.10 Å; Table 1). There were, however, minor local rearrangements (~0.2 Å shift in Cα positioning) of F95 and W97 within the secondary CO2 binding pocket of the F226W variant. These structural conformational changes could be contributed to error within the positioning of the atomic coordinates (~10% of the high resolutions) and are not considered to be of significance.
Interestingly, the active site residues and the proton-shuttle water network of the F226 variants were essentially identical to that of the wt HCA II structure (< 0.1 r.m.s.d.), indirectly indicating that these variants should have comparable catalytic rates to one another and to wt HCA II. The proton-shuttle residue H64 (11) was seen to be in a dual conformation, ‘in’ and ‘out’, for the F226L and F226W variants, but was found only in the ‘in’ conformation (i.e., towards the active site) in the F226I variant. These variable conformations among crystallographic structures of HCA II are not an uncommon occurrence and are dependent on the crystallization conditions (1, 9, 10, 33, 34).
Catalytic Rates
The pH profiles for the two rate constants, kcat/KM and RH2O, were determined by measuring the exchange of 18O-label between CO2 and water via mass spectrometry. The rates of catalyzed interconversion between CO2 and bicarbonate as measured by kcat/KM (Eq. 3; Fig. 3A; Table 2) for the F226 variants were comparable to that of wt HCA II. F226I showed the most similarity in catalytic efficiency to wt HCA II (~120 M−1 μs−1) whereas F226L was slightly higher at 140 M−1 μs−1 and F226W slightly lower (87 M−1 μs−1). These less than two-fold fluctuations, however, are not considered to be statistically significant. Similar catalytic rates are to be expected, however, as a Cα atom r.m.s.d. linear least-square fit of the F226 variants onto that of the wt HCA II structure (25) yielded a deviation of ~0.1 Å (Table 1).
Figure 3.

Catalytic constants of F226 variants via 18O exchange. Data for each variant are represented throughout as follows: HCA II, solid black square (■); F226I, open triangle (Δ); F226L, cross (X); F226W, open circle (○). (A) Calculation of kcat/KM (M−1 μs−1) versus pH from Eq. 3. (B) The pH profile for the rate constant RH2O/[E] (μs−1) used in calculation of the proton transfer constant, kB, and pKa values via Eq. 4.
Table 2.
Apparent Values of pKa and Maximal Rate Constants for F226 HCA II Variants.
The measured rate constant for proton transfer through the water network in the active site, kB, from Eq. 4 reveals a similar pattern as that seen in the kcat/KM measurements (Fig. 3A; Table 2). Both the F226I and F226L variants show comparable kB values to that of wt HCA II (1.4 μs−1) whereas F226W had a slight reduction in proton transfer at 1.0 μs−1. As discussed with the catalytic efficiency measurements, these variations in rates are not considered to be of significance. Additionally, the overall microenvironment of the active site is not significantly different between the variants as indicated by the comparable pKa values for H64 (~6.8) and ZnOH− (~7.0) as calculated by Eq. 3 and 4 fitted to the data in Figs. 3A and 3B, respectively. These results correlate well to the structural similarity between the active site residues and water network of the F226 variants to that of wt HCA II.
Thermostability
The thermal unfolding transitions of the F226 variants and wt HCA II at pH 7.8 were studied in triplicate utilizing differential scanning calorimetry. The peaks were fitted assuming a simple two-state transition, with further complexity (e.g., multiple transition peaks) added if necessary (thermograms not shown). These peaks were calculated to be endothermic and were centered at the denaturing temperature (TM), with the results summarized in Table 3.
Table 3.
Thermodynamic properties of F226 HCA II Variants.
| Property | HCA II | F226I | F226L | F226W |
|---|---|---|---|---|
| TM (K)a | 330.2 ± 0.1 | 326.1 ± 0.1 | 326.3 ± 0.1 | 324.1± 0.2 327.2± 0.7 329.2 ± 0.2 |
| ΔH°m(kcal mol−1)a | 223 ± 5 | 104 ± 4 | 208 ± 9 | 110 ± 9 71 ± 11 25 ± 3 |
| ΔHvH(kcal mol−1)b | 195 ± 6 | 113 ± 10 | 114 ± 6 | 140 ± 3 240 ± 20 480 ± 40 |
| ΔCp (kcal mol−1 K−1)c | 0.69 | 0.32 | 0.65 | 0.32 0.22 0.67 |
| ΔGT (kcal mol−1)d | 1.98 | −0.63 | −1.21 | −1.31 −0.21 0.081 |
| ΔHT (kcal mol−1)e | 311 | 103 | 212 | 107 25 71 |
| ΔST (kcal mol−1 K−1)f | 0.93 | 0.32 | 0.65 | 0.33 0.67 0.076 |
Calorimetric parameters determined by DSC.
van't Hoff enthalpy (ΔHvH) was determined by fitting thermograms to a two-state reversible unfolding model.
Heat capacity (ΔCp) of protein unfolding obtained by plotting calorimetric enthalpy (ΔH°m) versus melting temperature (TM).
Thermodynamic parameter extrapolated to reference temperature of 328.2 K using Eq. 5.
Thermodynamic parameter extrapolated to reference temperature of 328.2 K using Eq. 6.
Thermodynamic parameter extrapolated to reference temperature of 328.2 K using Eq. 7.
Interestingly, the TM for the F226I/L variants was 3-4 K lower than that of the native enzyme (~326 versus 330 K, respectively; Table 3). However, only the F226I variant displayed unfolding characteristics similar to that of the wt enzyme, that of a single domain and intermediate transition (i.e., the ΔH°M / ΔHvH ratios for F226I and wt HCA II are ~1; Table 3). Comparison of the ΔH°M / ΔHvH ratio for the F226L variant (~2) shows a non-2 state phase transition. The change in hydration of side-chain residues, as measured by ΔCP (Table 3), shows that the F226L variant, however, unfolds in similar manner to that of the wt enzyme whereas F226I displays less hydrophobic exposure (35, 36). The thermogram for the F226W variant displayed a broad dissociation peak consistent with three intermediate states: one that has a TM similar to that of F226I/L at 327 K, one similar to that of wt HCA II at 329 K, and one at a lower temperature than observe for the other variants at 324 K (Table 3). However, the ΔH°M / ΔHvH ratios for the F226W variant at the higher denaturation temperature are indicative of oligomeric aggregation. Comparison of the calculated ΔG (Eq. 5) at 328.2 K for the variants and wt HCA II shows the energy difference between these variants are ~2-3 kcal mol−1 (Table 3), an energy dissociation that can be correlated to C-H···π bonds (37). The absence of local reconfiguration (Fig. 2) and decreased thermostability of the F226 variants compared to that of wt HCA II suggests that a phenyl ring at position 226 makes important enthalpic contributions to the overall stability of the enzyme.
Discussion
This study investigated the significance of the proximity of the lone aromatic cluster in HCA II to the active site (~12 Å from the zinc metal) in the catalytic efficiency and thermostability of the enzyme. X-ray crystallographic analysis revealed that site-directed mutagenesis of position 226 to a conserved human variant (F226L) and other non-conserved residues (F226I/W) did not significantly alter the secondary CO2 binding site (Fig. 2) or the active site of the enzymes. Kinetic measurements via 18O-mass spectrometry confirmed the overall catalytic rate and proton transfer of the F226 variants did not significantly change compared to that of wt HCA II (Fig. 3; Table 2). DSC studies revealed, however, some reduction in the denaturing temperature (ΔTM ≅ 4 K) of the variants as compared to wt HCA II (Table 3).
The source for this destabilization in the F226 variants may be explained via disruption of weak C-H···π interactions among the cluster residues F66, F95 and W97 (Fig. 4; Table 4). These weak hydrogen bonds between soft acids and bases are highly orientation and spatially dependent (ideally perpendicular and 3.2 – 3.8 Å distance) and contribute ~2 kcal mol−1 enthalpically (16, 20, 38-40). The structure of HCA II without CO2 bound (25) reveals that F226 has favorable interaction geometry with F95 with a bond length of 3.8 Å and 92° bond angle along with a slightly distorted C-H···π bonding association with W97 at a distance and torsion of 4.1 Å and 115°, as well as elongated bonding distances with F66 (Fig. 1A; Table 4). Substitution of F226 with either I/L results in complete loss of the meta- and para-interactions with F66 and F95 (Fig. 4 A, B). The methyl group of F226I, however, is seen to be within 3.3 Å of the center of the indole group of W97 at an angle of 112° (Fig. 4A), suggesting a potential weak C-H···π interaction. Similarly, in F226L, two long-distance relationships (4.5 and 4.7 Å) with W97 can be found between the terminal carbons, the closer of the two being nearly perpendicular (Fig. 4B). The interaction between F95 is restored in the F226W mutant (Fig. 4C) at a closer distance (3.5 Å), but a larger angle from ideal (115°), due to molecular crowding of the introduced indole ring. The interaction of F226W with F66 and W97 is also weakened with increased distances (>4.4 Å) and angles (~120°) for C-H···π interactions (Fig. 4C). A summary of the geometric properties of the F226 variants is shown in Table 4.
Figure 4.
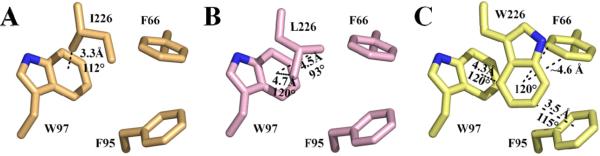
X-ray crystallographic models of (A) F226I (B) F226L and (C) F226W with the potential C-H···π interactions between F226, F66, F95 and W97 represented as dashed lines with distances and angles defined as in Table 4. Colors are same as in Figure 2.
Table 4.
Geometric Properties of F226 Variant C-H···π Interactions.
| Residue | Property | F226I | F226L | F226W | HCA IIa | HCA II-CO2b | HCA II-Xec |
|---|---|---|---|---|---|---|---|
| F66 | Distance (Å)d | N/A | N/A | 4.5, 4.8 | 4.4, 4.4 | 4.0, 4.1 | 4.1, 4.3 |
| Anglee | N/A | N/A | 120, 120 | 71, 89 | 66, 79 | 83, 64 | |
| F95 | Distance (Å) | N/A | N/A | 3.5 | 3.8 | 4.0, 4.0 | 3.8, 4.1 |
| Angle (°) | N/A | N/A | 115 | 92 | 82, 88 | 79, 87 | |
| W97 | Distance (Å) | 3.3 | 4.5, 4.7 | 4.3 | 4.1 | 3.7 | 3.8 |
| Angle (°) | 112 | 93, 120 | 120 | 115 | 95 | 114 |
The structural identity of the F226 variants compared to wt HCA II (Fig. 2) coupled with their decreased thermostability (Table 3) suggests that the loss of C-H···π bonds with F66 and F95 (for F226I/L) decreases the denaturation temperature by ~4 K. The broader transition phase seen with F226W that restores stability up to near wt levels emphasizes the apparent importance of maintaining these weak interactions among these aromatic cluster residues. It is noteworthy that the F226L variant is accompanied by F66L and/or F95L replacements in HCA III, IX, XII and XIV as well as in the HCA-related proteins (HCA-RPs) X and XI (~85% conservation). Additionally, at least three of these phenyl rings at residues 66, 95 and 226 in HCA II are conserved in I, VA, VB, VII and HCA-RP VIII (80% conservation). Taken together, the absence or conservation of a phenyl ring at position 226 (HCA II numbering) suggests an important evolutionary branching point among the human CAs.
Conclusions
This study suggests that while the aromatic cluster centered on F226 in HCA II does not contribute to enzymatic activity (Table 2; Fig. 3), it does play an important role in protein stability. The F226 variants all showed denaturation profiles that were ~4 K lower in thermostability compared to native HCA II (Table 3). This could be due to distorted or absent CH···π bonds between residue 226 and F66, F95 and W97 (Table 4; Fig. 4), illustrating the importance of these weak interactions in the global stability of HCA II.
Closer inspection of the previously determined CO2 bound structure (14) revealed that the 30° shift of F226 upon binding the ligand (Fig. 1B) promotes favorable conditions for C-H···π interactions (Table 4) between F66, F95 and W97, three highly conserved residues in HCA isoforms. These conditions are satisfied well with the ortho-position carbon on the phenyl ring of F226 being 3.7 Å and 95° from that of the aromatic ring in the indole group of W97. Additionally, the meta- and para-position carbons could make two potential weak C-H···π interactions with F95 at 4.0 Å distance and an 82 and 88° tilt. There are similar bond sites in F66 as it could make two potential interactions at 4.0 and 4.1 Å distance and angles of 66 and 79° (Fig. 1B; Table 4). The promotion of more favorable C-H···π interactions among the F226 aromatic cluster upon CO2 binding may contribute significantly to thermostability as has been seen in previous ligand-HCA II complexes (1, 34) and among other proteins (41-43). The extra stability inferred from reorientation of F226 upon ligation of CO2 may provide additional protection against oxidative (44, 45) and acidic (46, 47) microenvironments encountered within the cell. If this were the case, the F226 aromatic cluster in HCA II not only acts as an innate stabilizing element during ambient conditions, but may also provide a fail-safe stabilizing mechanism during cellular stress. Future stability studies in the presence of CO2 are needed for further assessment.
The importance of aromatic clustering in HCA II stability is investigated.
DSC studies revealed lowered TM for F226 variants relative to wtHCA II.
The variants have identical catalytic rates compared to wtHCA II.
Structures suggest that weak CH···π interactions are vital in protein stability.
Acknowledgements
This research has partially been funded by the NIH (GM25154) awarded to DS and RM. The authors would like to thank the Center of Structural Biology for support of the X-ray facility at UF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aggarwal M, Boone CD, Kondeti B, McKenna R. J Enzyme Inhib Med Chem. 2013;28:267–77. doi: 10.3109/14756366.2012.737323. [DOI] [PubMed] [Google Scholar]
- 2.Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, et al. Chem. Rev. 2008;108:946–1051. doi: 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindskog S. Pharmacol Ther. 1997;74:1–20. doi: 10.1016/s0163-7258(96)00198-2. [DOI] [PubMed] [Google Scholar]
- 4.Lindskog S, Coleman JE. Proceedings of the National Academy of Sciences of the United States of America. 1973;70:2505–8. doi: 10.1073/pnas.70.9.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindskog S, Silverman DN. In: The Carbonic Anhdyrases: New Horizons. Chegwidden WR, Carter ND, Edwards YH, editors. Birkhäuser Verlag; Boston, USA: 2000. pp. 175–95. [Google Scholar]
- 6.Silverman DN, Lindskog S. Acc Chem Res. 1988;21:30–6. [Google Scholar]
- 7.Khalifah RG. Journal of Biological Chemistry. 1971;246:2561–73. [PubMed] [Google Scholar]
- 8.Steiner H, Jonsson BH, Lindskog S. European Journal of Biochemistry. 1975;59:253–9. doi: 10.1111/j.1432-1033.1975.tb02449.x. [DOI] [PubMed] [Google Scholar]
- 9.Fisher SZ, Tu C, Bhatt D, Govindasamy L, Agbandje-McKenna M, et al. Biochemistry. 2007;46:3803–13. doi: 10.1021/bi602620k. [DOI] [PubMed] [Google Scholar]
- 10.Fisher Z, Kovalevsky AY, Mustyakimov M, Silverman DN, McKenna R, Langan P. Biochemistry. 2011;50:9421–3. doi: 10.1021/bi201487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tu CK, Silverman DN, Forsman C, Jonsson BH, Lindskog S. Biochemistry. 1989;28:7913–8. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]
- 12.Mikulski R, West D, Sippel KH, Avvaru BS, Aggarwal M, et al. Biochemistry. 2013;52:125–31. doi: 10.1021/bi301099k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mikulski RL, Silverman DN. Biochim Biophys Acta. 2010;1804:422–6. doi: 10.1016/j.bbapap.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Domsic JF, Avvaru BS, Kim CU, Gruner SM, Agbandje-McKenna M, et al. J Biol Chem. 2008;283:30766–71. doi: 10.1074/jbc.M805353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aaron JA, Chambers JM, Jude KM, Di Costanzo L, Dmochowski IJ, Christianson DW. J Am Chem Soc. 2008;130:6942–3. doi: 10.1021/ja802214x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burley SK, Petsko GA. Science. 1985;229:23–8. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 17.Dong G, Vieille C, Savchenko A, Zeikus JG. Appl Environ Microbiol. 1997;63:3569–76. doi: 10.1128/aem.63.9.3569-3576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teplyakov AV, Kuranova IP, Harutyunyan EH, Vainshtein BK, Frömmel C, et al. J. Mol. Biol. 1990;214:261–79. doi: 10.1016/0022-2836(90)90160-n. [DOI] [PubMed] [Google Scholar]
- 19.Ishikawa K, Okumura M, Katayanagi K, Kimura S, Kanaya S, et al. J Mol Biol. 1993;230:529–42. doi: 10.1006/jmbi.1993.1169. [DOI] [PubMed] [Google Scholar]
- 20.Serrano L, Bycroft M, Fersht AR. J Mol Biol. 1991;218:465–75. doi: 10.1016/0022-2836(91)90725-l. [DOI] [PubMed] [Google Scholar]
- 21.Duda DM, Tu C, Fisher SZ, An H, Yoshioka C, et al. Biochemistry. 2005;44:10046–53. doi: 10.1021/bi050610h. [DOI] [PubMed] [Google Scholar]
- 22.Forsman C, Behravan G, Osterman A, Jonsson BH. Acta Chem Scand B. 1988;42:314–8. doi: 10.3891/acta.chem.scand.42b-0314. [DOI] [PubMed] [Google Scholar]
- 23.Khalifah RG, Strader DJ, Bryant SH, Gibson SM. Biochemistry. 1977;16:2241–7. doi: 10.1021/bi00629a031. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Avvaru BS, Kim CU, Sippel KH, Gruner SM, Agbandje-McKenna M, et al. Biochemistry. 2010;49:249–51. doi: 10.1021/bi902007b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, et al. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, et al. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 30.Silverman DN. Methods Enzymol. 1982;87:732–52. doi: 10.1016/s0076-6879(82)87037-7. [DOI] [PubMed] [Google Scholar]
- 31.Becktel WJ, Schellman JA. Biopolymers. 1987;26:1859–77. doi: 10.1002/bip.360261104. [DOI] [PubMed] [Google Scholar]
- 32.Avvaru BS, Busby SA, Chalmers MJ, Griffin PR, Venkatakrishnan B, et al. Biochemistry. 2009;48:7365–72. doi: 10.1021/bi9007512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boone CD, Habibzadegan A, Tu C, Silverman DN, McKenna R. Acta Crystallogr D Biol Crystallogr. 2013;69:1414–22. doi: 10.1107/S0907444913008743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aggarwal M, Boone CD, Kondeti B, Tu C, Silverman DN, McKenna R. Acta Crystallogr D Biol Crystallogr. 2013;69:860–5. doi: 10.1107/S0907444913002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sturtevant JM. Ann. Rev. Phys. Chem. 1987;38:463–88. [Google Scholar]
- 36.Haynie DT, Freire E. Anal Biochem. 1994;216:33–41. doi: 10.1006/abio.1994.1004. [DOI] [PubMed] [Google Scholar]
- 37.Novoa JJ, Mota F. Chemical Physics Letters. 2000;318:345–54. [Google Scholar]
- 38.Dill KA. Biochemistry. 1990;29:7133–55. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 39.Jaenicke R. Eur J Biochem. 1991;202:715–28. doi: 10.1111/j.1432-1033.1991.tb16426.x. [DOI] [PubMed] [Google Scholar]
- 40.Jaenicke R, Bohm G. Curr Opin Struct Biol. 1998;8:738–48. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 41.Maes D, Zeelen JP, Thanki N, Beaucamp N, Alvarez M, et al. Proteins. 1999;37:441–53. [PubMed] [Google Scholar]
- 42.Samiotakis A, Homouz D, Cheung MS. J Chem Phys. 2010;132:175101. doi: 10.1063/1.3404401. [DOI] [PubMed] [Google Scholar]
- 43.Schall CA, Wiencek JM. Biotechnol Bioeng. 1997;53:41–8. doi: 10.1002/(SICI)1097-0290(19970105)53:1<41::AID-BIT7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 44.Schafer FQ, Buettner GR. Free Radical Biology and Medicine. 2001;30:1191–212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 45.Martindale JL, Holbrook NJ. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 46.Pintsch T, Satre M, Klein G, Martin J-B, Schuster SC. BMC Cell Biology. 2001;2:9. doi: 10.1186/1471-2121-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daugirdas JT, Arrieta J, Ye M, Flores G, Battle DC. J Clin Invest. 1995;95:1480–9. doi: 10.1172/JCI117819. [DOI] [PMC free article] [PubMed] [Google Scholar]