

Abstract
Here we introduce a series of thoroughly tested and well standardized research protocols adapted for use in remote marine environments. The sampling protocols include the assessment of resources available to the microbial community (dissolved organic carbon, particulate organic matter, inorganic nutrients), and a comprehensive description of the viral and bacterial communities (via direct viral and microbial counts, enumeration of autofluorescent microbes, and construction of viral and microbial metagenomes). We use a combination of methods, which represent a dispersed field of scientific disciplines comprising already established protocols and some of the most recent techniques developed. Especially metagenomic sequencing techniques used for viral and bacterial community characterization, have been established only in recent years, and are thus still subjected to constant improvement. This has led to a variety of sampling and sample processing procedures currently in use. The set of methods presented here provides an up to date approach to collect and process environmental samples. Parameters addressed with these protocols yield the minimum on information essential to characterize and understand the underlying mechanisms of viral and microbial community dynamics. It gives easy to follow guidelines to conduct comprehensive surveys and discusses critical steps and potential caveats pertinent to each technique.
Keywords: Environmental Sciences, Issue 93, dissolved organic carbon, particulate organic matter, nutrients, DAPI, SYBR, microbial metagenomics, viral metagenomics, marine environment
Introduction
Marine ecosystems are subjected to a wide range of perturbations that result in changes from nutrient availability to the biomass of apex predators. Over the last decade, multiple studies have demonstrated the importance of the microbial communities in marine ecosystems1-4. It is evident that changes in the bacterial and viral community are closely associated with the overall degradation of marine environments5. These changes may be facilitated by altered geochemistry in the water column such as oxygen availability or carbon remineralization 6,7. As a result of the interdependency between the macroorganisms, water chemistry and microbial activity feedback loops may accelerate the rate of ecosystem degradation8.
In the 1970s and 80s multiple attempts were made to unravel biogeochemical cycles in marine ecosystems9-11. One of the main challenges for these early studies was the lack of standardized approaches to measure the assessed biogeochemical parameters. Though there were protocols available, primarily on seawater nutrients12,13, the assessment of carbon dynamics and the microbial community structure were limited by the tools and methods available. With the JGOFS EqPac methods comparison, Sharp et al.14 suggested for the first time a reliable and standardized protocol for dissolved organic carbon (DOC) concentration assessments. By the early 2000s the analytical challenges to determine resources available to the microbial community had thus been largely resolved and approaches to characterize microbial communities in controlled culture environments had been established (see DeLong15). The traditional sequencing methods at that time largely relied on cultivated clonal cultures. Early environmental gene sequencing of natural samples revealed, however, that the majority of microbial biodiversity had been missed by cultivation-based methods16. In 2002, Breitbart et al.17 used shotgun sequencing for the first time to describe the viral community in environmental seawater samples establishing a method to sequence the entire genome of uncultured marine viral communities.
Within the existing microbial assessments, viruses still remain particularly under-studied, as most are difficult to culture and they lack a universally conserved marker such as the 16S ribosomal RNA (rRNA) genes typically used for assessing diversity and community profiling. The metagenomic sequencing approach provides an alternative to culture-dependent and gene-targeted methods for analyzing complex viral communities. While the first viral metagenomes generated from seawater were sequenced using Sanger sequencing17, the development of next-generation sequencing technologies and improved molecular techniques has led to a rapid increase in metagenomic studies18. The current laboratory workflow for viral metagenomics involves the separation, enrichment, and/or concentration of viral particles, followed by nucleic acid (DNA or RNA) extraction, library preparation, and sequencing19-21.
To further the existing knowledge on viral, microbial and biochemical functioning in aquatic ecosystems, comparing data sets in various systems around the world is essential. However, the established protocols have largely been developed for near-shore environments with readily accessible laboratory facilities. Thus, the lack of standardized field methods hinders these cross-ecosystem comparisons. Here we introduce thoroughly tested and well standardized adaptations from state of the art research protocols for use in remote field locations. These methods have been proven successful in multiple studies spanning the last decade5,22 and are currently used by various marine laboratories as well as on the NOAA Reef Assessment and Monitoring Program (RAMP) throughout the Pacific Ocean4. The sampling protocols include water chemistry parameters (inorganic and organic carbon concentrations, inorganic nutrient concentration), viral and microbial abundance (direct bacterial counts, direct viral counts, and flow cytometry to assess autotroph vs heterotroph ratios), and viral and microbial community structure and metabolic potential through metagenomic analysis (for an overview see Figure 1). The results obtained from the methods described here are required in order to elucidate key biogeochemical background parameters needed to comprehensively characterize aquatic ecosystems.
Protocol
1. Preparation of Instruments
- Preparation of Filtration Setup
- Prepare a 5% hydrochloric acid (HCl) solution (0.5-0.6 M; add 250 ml 32-36% HCl to 4.75 L distilled water to make 5 L of final solution). Store this solution in a high-density polyethylene container (HDPE) for up to 2 weeks.
- Leave all parts that will come in contact with the sample (except the filters) for 24 hr in the 5% HCl solution to leech out potential dissolved organic carbon (DOC) contaminants. After each sampling event rinse all parts and flush lines with about 30 ml 5% HCl solution to prevent carbon contamination (e.g., from gas fumes, oil, boats, mosquito spray).
- Keep all sampling items wrapped in pre-combusted aluminum foil or capped until to immediately prior to use.
- Precombust 25 mm GF/F filters wrapped in aluminum foil in a muffle furnace at 550 °C for 12 hr to volatilize off all carbon and store them in a clean, dry place until use. Use acid washed forceps and sterile, non-powdered gloves to handle the precombusted filters at all times.
- Preparation of Viral Metagenome Sampling Materials
- Thoroughly wash four 20 L collapsible low-density polyethylene carboys and four 20 L high density polyethylene buckets and lids with 10% bleach.
- Subsequently rinse all items 3x with distilled and then sample water and reseal carboys with spigot.
- Wash Tangential Flow Filters (TFF) after each use according to the following protocol, and clean preemptively if the filter has not been used in the past 5 days.
- Dissolve 50 g of NaOH pellets in 5 L of water in a wash bucket.
- Attach a wet TFF to the tubing and assemble according to Figure 3.
- Run peristaltic pump at ~30 rpm with no backpressure to move some of the NaOH wash solution through the system. Once wash flows from the return line, move transfer to wash bucket to complete the circulation of solution.
- Apply backpressure to the system by placing a clamp on the return line downstream of the TFF. This will force wash solution out the filtrate line. Excessive time (5 min) to flood the outside of the TFF and produce filtrate is indicative of a degraded TFF.
- Run the system until all of the NaOH solution is in the lines. Then remove backpressure, run solution out of the system and discard.
- Rinse the TFF by running water under backpressure until the pH of the water flowing from the return and filtrate lines is pH 7-8.
- Remove tubing from the peristaltic pump and TFF. Reseal TFF for storage. Ensure that the TFF remains wet until the next use.
2. Sample Collection
NOTE: During transport all collected samples should be stored cool (if possible 4 °C) and not exposed to direct sunlight until further processing.
- Sampling for Carbon, Nutrients, Microscopy, Flow Cytometry, and Microbial Metagenomics
- Take two Hatay Niskin units per sampling event (resulting in 4 L sample water). Open the sampling vessels just before submerging (otherwise trapped air will prevent descending).
- At the respective site, flush the inside of the sampling vessel with the sample water by opening both ends and moving the cylinder along the polar axis through the water.
- Take the sample and carefully close the sampling container. Make sure the sampling site is upstream from boat and divers to avoid contamination.
- Sampling for Viral Metagenomics
- On the target site mount bilge pump on 20 L carboy.
- Fill up carboy aiming bilge pump inlet at the each targeted area and seal carboy with the corresponding spigot.
- Collect 4 carboys of unfiltered seawater (all together 80 L) from each sampling site.
3. Sample preparation
NOTE: Process the samples in the order presented here, starting with DOC to minimize the chance of contamination. Use powder free gloves for the entire procedure.
- Dissolved Organic Carbon (DOC)
- Mount one of the two aliquot Hatay Niskin units in respective slot of the filtration set. Connect the outlet tubing which leads to the respective filter holder. Connect the pressurized air (0.2 bar) tubing to the Hatay Niskin unit (See Figure 2).
- Flush all lines with 100 ml of sample water before inserting filters in the filter holders. Place 25 mm pre-combusted GF/F filter in filter holder using the acid washed forceps. Rinse each DOC bottle and cap 3 times with about 20 ml filtered sampling water.
- Fill bottle with about 40 ml (~2/3 full) of sampling water. Collect at least duplicates of DOC samples to have a backup in case of potential contamination or loss during transport.
- Freeze sampling bottles standing upright at -20 °C until analysis.
- Particulate Organic Matter (POM)
- After DOC samples have been collected continue filtering until a total of 500 ml have passed the GF/F filter, including what passed through while collecting DOC samples.
- Unscrew inline filter holder.
- Remove the filter for POM analysis using forceps and place filter inside a square of pre-combusted aluminum foil, fold top-side turned in on itself, and wrap.
- Freeze filters at -20 °C for storage until subjected to elemental and isotopic analysis.
- Inorganic Nutrients
- Place a 0.2 µm Track-Etched filter into each filter holder. Re-attach the filter cassettes.
- Rinse each 20 ml plastic scintillation bottle three times with sampling water. Fill each bottle to the shoulder (~18 ml).
- Freeze at -20 °C until later analysis.
- Microscopy (SYBR Gold and DAPI)
- Take off filter holder.
- Collect 1 ml of water in each of two microcentrifuge tubes.
- Add 66 µl 32% paraformaldehyde to one of the aliquots (for later SYBR Gold staining). Add 12 µl 25% glutaraldehyde to the corresponding other (for later DAPI staining).
- Gently mix, and allow samples to “fix” for at least 15 min at RT in the dark.
- Flow Cytometry
- Place an 8.0 µm polycarbonate filter in one of the filter holders to exclude debris and large eukaryotic cells.
- Pass sampling water through to fill 2 cryovials from each sampling site with 1 ml sample.
- Add 5 µl of 25% glutaraldehyde to each cryovial (final concentration = 0.125%).
- Invert vials to mix.
- Allow the sample to fix for 15-30 min at RT. Do not exceed 30 min.
- Flash freeze samples in liquid nitrogen.
- Store samples at -80 °C or in liquid nitrogen dry shipper until analysis on a flow cytometer.
- Microbial Metagenomic Sample
- Remove in line filter holders.
- Directly mount a 0.22 µm cylindrical filter onto the respective line.
- Filter remaining sample water of both Hatay Niskin units from each site (totaling 3-4 L per filter) through one filter.
- After the filtration push the remaining water out of each filter by using a clean 10 ml syringe filled with air.
- Place filter back into original packaging and seal the package with lab tape.
- Store individually packed filters at -20 °C.
- Viral Metagenomic Sample
- Transfer viral metagenome samples immediately into the washed buckets to ensure no sample is lost or contaminated by the environment.
- Pre-filter using large pore size nylon mesh (25-100 µm) to remove debris and cellular material prior to concentration19.
- Set up the TFF as shown in Figure 3, placing the delivery line in a sample bucket, and leaving the return and filtrate lines running into a sink.
- Turn on peristaltic pump and flush lines with 1-2 L of sample water.
- Place return line in the sample bucket to complete cycle, add 0.7 bar of backpressure.
- While concentrating the seawater, top up sample reservoir as the level drops. When water level drops below the intake line in the bucket, transfer concentrate into a bleach-washed, triple rinsed tripour beaker and continue concentrating.
- If reservoir beaker is empty, remove backpressure, increase the pump rate and push the entire sample through the lines, recovering it in the tripour.
- Pass the concentrate through 0.45 µm cylindrical filters to remove most Bacteria while not discriminating against any viral lineages.
- Change 0.45 µm cylindrical filters after every 150 ml. Collect the 0.45 µm-filtered viral concentrate in 50 ml tubes.
- Add 250 µl of chloroform to each 50 ml aliquot of filtered viral concentrate to eliminate residual Bacteria. Invert to mix, and store, upright at 4 °C until subsequent processing.
- Dry the 0.45 µm filters and store them as described in Steps 3.6.5 and 3.6.6.
4. Processing of Microscopy Samples
NOTE: Rinse filter towers with 10% bleach followed by 95% ethanol to prevent potential stain or biological residues between runs. Process microscope samples within 1 hr and avoid exposing stains and stained filters to light if possible.
- Mount
- Add 100 µl of 10% ascorbic acid to 4.9 ml of 1x PBS, and mix thoroughly.
- Add 5 ml of 100% glycerol, and mix thoroughly.
- Filter mount using a 0.02 µm alumina matrix disposable syringe filter, aliquot and store at -20 °C.
- SYBR Gold Staining
- Pipette a 500 µl aliquot of each sample fixed with 2% final concentration paraformaldehyde in a microcentrifuge tube.
- Add 0.5 µl of 1,000x SYBR Gold solution to each of the aliquots.
- Mix sample gently and put it in the dark for 10 min.
- Place a 0.02 µm alumina matrix filter with annular polypropylene support ring on each filter stand of the vacuum pump system and attach the filter towers (Figure 4). Make sure the 0.02 µm filter is placed on the filter stand plastic-side up, as these filters have a dual porosity.
- With the vacuum pump turned off, add the 500 µl of SYBR stained sample, along with 2 ml of 0.02 µm filtered virus free water to ensure the sample is spread evenly across the filter.
- Turn on the vacuum pump to create a negative pressure of no more than 1 bar to avoid damaging viruses and microbes being filtered.
- Allow the entire sample to go through.
- Using forceps (not the same pair as used for DOC), carefully remove each filter from filtering tower, and pass over a delicate task wipe to ensure the filter is dry.
- Pipette 10 µl of mount on a microscope slide and place the dry filter containing the stained sample on the drop of mount.
- Add 10 µl mount to the top of each filter before adding a coverslip.
- Gently press coverslip down. Try to eliminate air-bubbles if possible and avoid sideways motion of the coverslip.
- Place slide in a slidebox and store at -20 °C.
- DAPI Staining
- Place a 0.2 µm alumina matrix filter with annular polypropylene support ring on each filter stand and attach the filter towers.
- With the vacuum pump turned off, add 2 ml of 0.02 µm filtered virus free water to each tower used.
- Pipette 1 ml of the glutaraldehyde-fixed sample into each tower, resulting in a total volume of 3 ml in each filter tower.
- Turn on vacuum pump to create a negative pressure of no more than 1 bar.
- Filter entire sample onto the 0.2 µm alumina matrix filter.
- Carefully remove filter from filtering tower using forceps (not the same pair as used for DOC).
- Place the filter on a 100 µl drop DAPI solution [25 µg/ml] in a Petri dish keeping the filter right side up and let the sample stain for 15 min. Avoid light during the staining process.
- After the 15 min, remove the filter from the stain, pass it over a delicate task wipe to dry and transfer to a 100 µl drop of 0.02 µm filtered virus free water, again keeping the filter right side up, to wash off remaining DAPI stain for 1 min. Repeat this wash once.
- Mount filter on microscope slide the identical way as described for the SYBR Gold samples.
5. DNA Extraction
- Microbial Metagenomic DNA
- Thaw the cylindrical filters for at least 20 min at RT.
- Remove any remaining seawater in the filter using a 10 ml syringe. Cap the bottom end of the cylindrical filter using a bleached and autoclaved cap.
- For each filter, mix 360 μl T1 “pre-lysis” buffer and 50 μl Proteinase K, and then pipette the mixture into the filter. Cap the open end.
- Incubate the lysis reaction in a rotating oven O/N (or at least 2 hr) at 55 °C.
- Remove one of the cap and add 200 µl B3 “lysis” buffer into the filter to the pre-lyse sample. Re-cap filter and incubate the mixture in a rotating oven for 10-20 min at 70 °C.
- Extract the entire volume from the cylindrical filter using a 3 ml syringe by sucking the liquid out with the filter upside down.
- Expel the lysed sample into a new microfuge tube, and add 200 μl of 100% ethanol.
- Mix well, load the entire sample onto the spin column, and proceed as instructed by the manufacturer's protocol.
- Quantify DNA using a fluorescent-based assay.
- Marine Viral Metagenomics
- Purifying and Concentrating Phage Particles (modified from Thurber et al.19)
- Dissolve CsCl in seawater to prepare solutions of 1.7 g/ml, 1.5 g/ml, 1.35 g/ml, 1.2 g/ml (calibrated by weighing out 1 ml of solution). Filter each fraction with 0.02 µm filter prior to use. NOTE: Ensure that the density of each fraction is accurate to three significant figures before using the solutions and filter all solutions with a 0.02 µm filter before use. Use seawater or seawater saturated with CsCl to lower or increase density, respectively.
- Make a microscope slide from 1 ml of combined reagents as described in section 4.2 to ensure that all reagents are devoid of viruses before proceeding.
- Add 9 g CsCl to 45 ml of viral TFF concentrate to a final density of 1.12 g/ml. Refrigerate while setting up the CsCl step gradient.
- Starting with the 1.7 g/ml solution successively add 1 ml of each sequentially less dense fraction slowly into each tube using serological pipettes. Mark the level of each individual meniscus and ensure that the pycnoclines between fractions are not disturbed. For more detail see the companion paper (Lim et al. 2014).
- With a serological pipette load 7.5 ml of sample into each of 6 tubes and centrifuge at ~83,000 x g, 4 °C for 2 hr.
- Without disrupting the density gradients unload the rotor and pierce the tube just below the previously marked 1.5 g/ml density layer (Figure 5) with an 18 G needle on a 3 ml syringe. Draw off 1.5 ml of purified VLPs and transfer to a new microcentrifuge tube. Repeat for all samples.
- Add 0.2 volume of chloroform to the purified VLPs, mix vigorously, incubate at RT for 10 min.
- Add 150 µl of 10x DNAse buffer to each microcentrifuge tube.
- Spin at 16,000 x g for 5 min, and collect the aqueous phase. If the chloroform does not aggregate in the bottom of the microcentrifuge tube, add more DNAse buffer, mix and repeat.
- Add DNase I (final concentration = 2.5 unit/µl) and incubate at 37 °C for 2 hr, then inactivate DNase activity by incubating at 65 °C for 15 min.
- DNA Extraction from Viral-like Particles (VLPs)
- Evenly pool the purified VLPs from each sample into two cleaned and autoclaved Oak Ridge tubes.
- Add 0.1 volume 2 M Tris-HCl (pH 8.5) with 0.2 M EDTA, 0.01 volume 0.5 M EDTA, 1 volume of formamide, 10 μl glycogen (10 mg/ml). Mix well and incubate at RT for 30 min.
- Referring to new volume, add 2 volumes RT 100% ethanol. Mix and incubate at 4 °C for more than 30 min.
- Pellet DNA by spinning the Oak Ridge tubes at 17,200 x g for 60 min, at 4 °C.
- Remove the supernatant. Wash the DNA pellet twice with ice-cold 70% ethanol using serological pipettes.
- Remove all ethanol (re-spinning briefly if necessary), cover, and allow pellet to air-dry at RT for >15 min.
- Resuspend the DNA pellet for >15 min in 567 μl of 1X TE buffer (pH 8.0). Transfer the 567 μl of resuspended DNA into a new 1.5 ml microcentrifuge tube.
- Add 30 μl of 10% SDS (pre-warm at 65 °C prior to use) and 3 μl of proteinase K (20 μg/ml), mix thoroughly and incubate for 1 hr at 56 °C.
- Add 100 μl of 5 M NaCl and mix thoroughly. Add 80 μl of pre-warmed CTAB NaCl solution, vortex, and incubate for 10 min at 65 °C.
- Add 0.2 parts of volume of chloroform, vortex, and spin at 16,000 x g for 2 min. Transfer supernatant to a new 1.5 ml microcentrifuge tube.
- Add equal volume of phenol chloroform isoamyl 25:24:1 solution, vortex to mix, and spin at 16,000 x g for 2 min. Transfer the supernatant to a new 1.5 ml microcentrifuge tube.
- Add equal volume of chloroform, vortex, and spin at 16,000 x g for 2 min. Transfer the supernatant to a new 1.5 ml microcentrifuge tube.
- Add 0.7 volume of isopropanol to the supernatant fraction and incubate at -20 °C for at least 30 min to precipitate DNA.
- Pellet the DNA by spinning at 16,000 x g for 20 min at 4 °C. Pipette off the supernatant and wash the pellet twice with 500 µl of ice-cold 70% ethanol.
- Spin at 16,000 x g and remove the remaining ethanol from the tube. Air-dry the pellet for 15 min.
- Resuspend DNA pellet with 50 μl of elution buffer (5 mM Tris, pH 7.5) or Tris-EDTA pH 8.0 for at least 5 min at RT.
- Quantify the DNA using high-sensitivity fluorescence-based assay.
Representative Results
Microscopy
Microscopy samples can and should be analyzed immediately to ensure they are of the desired quality. To measure abundance and size distribution of the bacterial community, DAPI slides will be examined by epifluorescence microscopy (excitation/emission: 358/461 nm, see McDole et al. 2012) (Figure 6A). Cell counts and dimensions can be collected using imaging software (e.g., ImagePro or ImageJ). From length and width measurements cell volumes (V) are derived by making the assumption that all cells have the shape of cylinders with hemispherical caps using the following equation:
V = π /4 × w2 (L –w/3)
where L is length and w is width of each cell23,4. Microbial biomass can then be estimated using previously established size-dependent relationships for marine microbial communities24. Generally marine bacteria range in length from 0.1-4 µm, but go up to ~ 8 µm in some locations.
While the filters (0.2 µm) stained with DAPI will only show bacteria, the filters used for the SYBR Gold stain (0.02 µm) contain bacteria and viruses. Measuring the abundance of viruses follows the same protocol as for microbes, however an excitation of 325-375 nm will be used and the emission maximum is at 537 nm (Figure 6B).
In order to generate quantitative data the sampling volume may need to be adjusted depending on the viral abundance in the original sample. The correct volume to filter is best determined empirically for a given body of water. Examples of micrographs containing samples with varying viral concentrations are illustrated in Figure 7.
Results from previous studies suggest that Virus to Microbe ratios (VMR) generally range from 1 to 50 in aquatic systems25-29, and in between 3 and 20, with an average of approximately 6 in coral reef systems (Knowles unpublished data).
Flow Cytometry
In addition to the direct counts and size estimation of the microbial community, assessments of the ratio of autotrophic to heterotrophic microbes via flow cytometry can further be extracted from the collected samples (exemplary flow cytometry output given in Figure 8). To determine the number of total Bacteria cells, samples are stained with SYBR Green I and a photomultiplier tube with a 530/30 bandpass filter is used for detection. A channel for chlorophyll (back to back LP mirrors resulting in a range of 675-735 nm) and phycoerytherin (585/42 bandpass filter) is used to count the abundance of autotrophs in unstained samples. To determine the abundance of heterotrophic microbes, the autotrophic counts from the non-stained portion are then subtracted from the SYBR-stained total count (McDole et al. submitted).
Viral Metagenomics
Viral metagenomics utilizes a culture-independent molecular approach to viral ecology, using total genomic sequence information to determine community structure and function. Metagenomics has been advancing our understanding of the complexity and diversity of viral communities at local17 to global scales30. The recent development of a wide array of bioinformatic tools for analysis of viral metagenomic data has decreased computational bottlenecks, allowing for a more comprehensive view the of the virosphere through the lens of metagenomics.
The methods presented here highlight the isolation and enrichment of viral particles from seawater using a combination of pre-filtration with large pore size nylon mesh to remove debris and cellular material, concentration of samples with TFF (Figure 3) and a subsequent 0.45 μm filtration to remove larger cells. This method produces roughly a 100x concentrated sample (Figures 5B-5E) which allows us to perform downstream processes in smaller volumes. In order to prevent growth of any remaining microbial cells in the viral lysate and therefore changes in the viral abundance of the sample during the long transit time between field station and laboratory, chloroform is often added to a final concentration of 2% for storage. Following isolation and purification of VLPs, epifluorescence microscopy with nucleic acid dyes such as SYBR Gold are used to verify the presence and purity of the viral particles (Figures 5B-5E).
Here, we present two viral metagenomes from the Southern Line Island coral reef, specifically, the Starbuck (Star7) and Millennium (CAR9; previously known as Caroline) islands. A total volume of 120 L sample water was collected from each site at a depth of 10 meters and processed as described in section 3.7 and 5.2. The VLPs purified from the cesium chloride gradient centrifugation were 3.3 x 108 particles/ml for CAR9 (Figure 5D) and 2.9 x 109 particles per ml for Star7 as verified according to the method described in paragraph 4 (Figure 5B). The total amount of DNA isolated from these samples were around 400 ng based on the Nanodrop measurement. DNA was amplified using Phi29 polymerase and sequenced by the Environmental Genomics Core facility in 2011. The characteristics of the sequence data is presented in Table 1. Based on similarity searches using existing databases, the majority (>70%) of the sequences often ended up uncharacterized, i.e., unknown origins and functions. Similarly, the two viromes presented here presented more than 97% of unknown sequences. As a result, non-database dependent analysis was used for analysis and opened up a new arm of research opportunity on the “dark matter” in viral metagenomics (Seguritan et al.in press).
Microbial Metagenomics
The analysis of microbial metagenomes allows for the characterization of the microbial community present and the functional description of these communities. Examples include estimates of the presence of pathogens and virulence5,22 or comparisons between changes in macrobial community structure or available nutrients and abundance of species and their predominant metabolic pathways (Figure 9; Kelly et al. 2014).
Water Chemistry
Water chemistry parameters generally take extensive analytical processing to generate the desired data; samples for DOC analysis will be measured via high temperature catalytic oxidation31,32, particulate organic matter (POM) via isotope ratio mass spectrometry coupled to an elemental analyzer 33-35, and inorganic nutrients by Flow Injection Analysis36,12,37. After a successful completion of all the analyses, information as shown in Table 1 will be available to complement the characterization of the microbial and viral communities. This information can be complimented with stable isotope ratios of organic carbon and nitrogen samples (See Haas et al.35) and ratios between autotrophs and heterotrophs (McDole et al. submitted).
 Figure 1. Overview of methods described in the protocol section.
Please click here to view a larger version of this figure.
Figure 1. Overview of methods described in the protocol section.
Please click here to view a larger version of this figure.
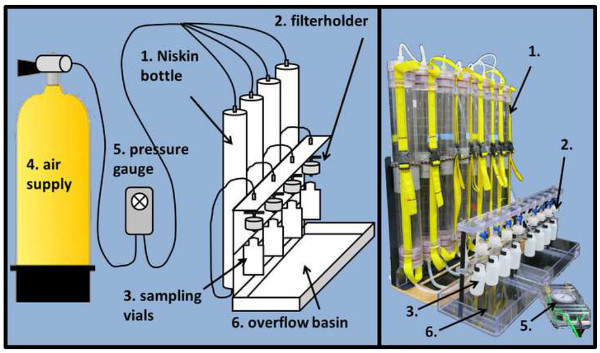 Figure 2. Sample processing rig setup. The filtration setup introduced here (schematic drawing left, actual picture right panel) is not necessarily required to generate samples but will expedite the process significantly. The setup consists of a backplate, on which the Hatay Niskin units (1) are mounted. On the outlet facing downwards, Hatay Niskin units are connected with silicone tubing to the in-line filter holders (2). These let the sampled water pass through the respective filter into the HDPE sampling vials (3) mounted right underneath. The filtration process is accelerated through pressurized air (4), which and regulated by a pressure gage (5). An overflow basin prevents spilling water during the changing of the HDPE vials or POM filtration. Please click here to view a larger version of this figure.
Figure 2. Sample processing rig setup. The filtration setup introduced here (schematic drawing left, actual picture right panel) is not necessarily required to generate samples but will expedite the process significantly. The setup consists of a backplate, on which the Hatay Niskin units (1) are mounted. On the outlet facing downwards, Hatay Niskin units are connected with silicone tubing to the in-line filter holders (2). These let the sampled water pass through the respective filter into the HDPE sampling vials (3) mounted right underneath. The filtration process is accelerated through pressurized air (4), which and regulated by a pressure gage (5). An overflow basin prevents spilling water during the changing of the HDPE vials or POM filtration. Please click here to view a larger version of this figure.
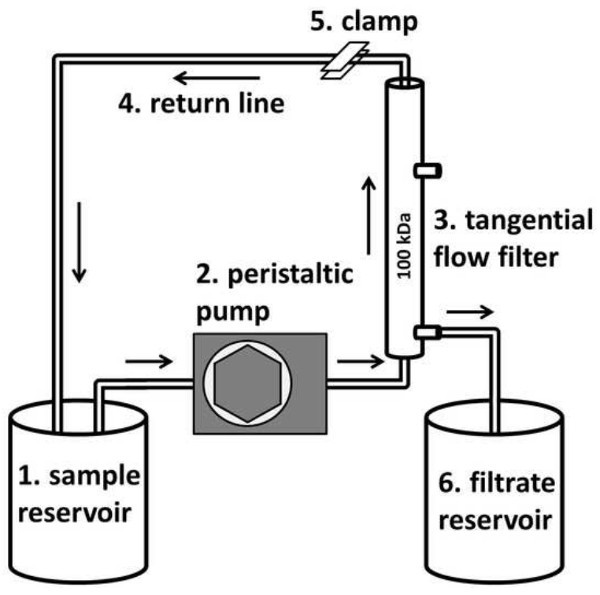 Figure 3. Tangential flow filter lineup (modified from Thurber et al.19). Nylon mesh filtered sample water is pumped from the reservoir (1) via a peristaltic pump (2) to a tangential flow filter (3). The sample water returns to the reservoir (1) along the return line (4) in the absence of backpressure. When backpressure is applied through a hose clamp (5) on the return line, water passes through the filter and is discarded or delivered to the filtrate reservoir (6). Sample water is replaced as it is concentrated out the filtrate line until all the sample water is concentrated in the tubing lines.
Figure 3. Tangential flow filter lineup (modified from Thurber et al.19). Nylon mesh filtered sample water is pumped from the reservoir (1) via a peristaltic pump (2) to a tangential flow filter (3). The sample water returns to the reservoir (1) along the return line (4) in the absence of backpressure. When backpressure is applied through a hose clamp (5) on the return line, water passes through the filter and is discarded or delivered to the filtrate reservoir (6). Sample water is replaced as it is concentrated out the filtrate line until all the sample water is concentrated in the tubing lines.
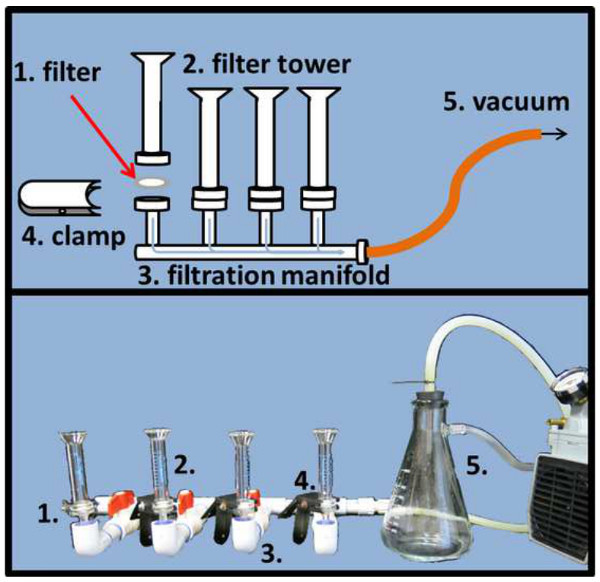 Figure 4. Microscope slide filtration setup. To evenly distribute DAPI and SYBR Gold stained samples on the respective alumina matrix filter (1), filters need to be placed in between the filter tower (2) and stem of the filtration manifold and fixed with a clamp (4). A pressure regulated vacuum (5) will expedite the filtration process (schematic drawing top, actual picture bottom panel). Please click here to view a larger version of this figure.
Figure 4. Microscope slide filtration setup. To evenly distribute DAPI and SYBR Gold stained samples on the respective alumina matrix filter (1), filters need to be placed in between the filter tower (2) and stem of the filtration manifold and fixed with a clamp (4). A pressure regulated vacuum (5) will expedite the filtration process (schematic drawing top, actual picture bottom panel). Please click here to view a larger version of this figure.
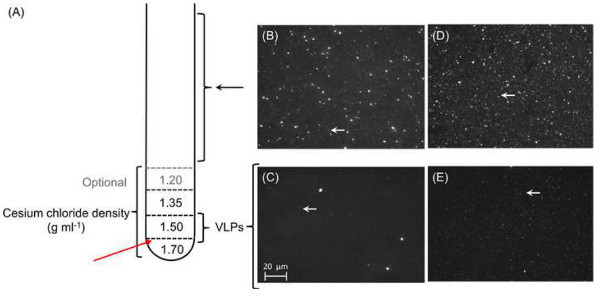 Figure 5. Purification and concentration of phage particles. A total of 1 ml of each cesium chloride density gradient is layered on top of each other prior to leading the pre-treated sample (A). Each layer should be marked outside the tube to facilitate the extraction after ultracentrifugation. Red arrow marks where the tube will be pierced to extract the VLP fraction. (B-E) Micrographs of viral concentrates: Epifluorescence micrographs of 0.45 μm-filtered representative viral concentrates of Star7 (B) and CAR9 (D). Large particles including bacterial cells were visible from the 0.45 μm-filtrates from both Star7 (B) and CAR9 (D) samples, whereas only VLPs (white arrows) remain after cesium chloride gradient ultracentrifugation; Star 7 (C) and CAR9 (E). Please click here to view a larger version of this figure.
Figure 5. Purification and concentration of phage particles. A total of 1 ml of each cesium chloride density gradient is layered on top of each other prior to leading the pre-treated sample (A). Each layer should be marked outside the tube to facilitate the extraction after ultracentrifugation. Red arrow marks where the tube will be pierced to extract the VLP fraction. (B-E) Micrographs of viral concentrates: Epifluorescence micrographs of 0.45 μm-filtered representative viral concentrates of Star7 (B) and CAR9 (D). Large particles including bacterial cells were visible from the 0.45 μm-filtrates from both Star7 (B) and CAR9 (D) samples, whereas only VLPs (white arrows) remain after cesium chloride gradient ultracentrifugation; Star 7 (C) and CAR9 (E). Please click here to view a larger version of this figure.
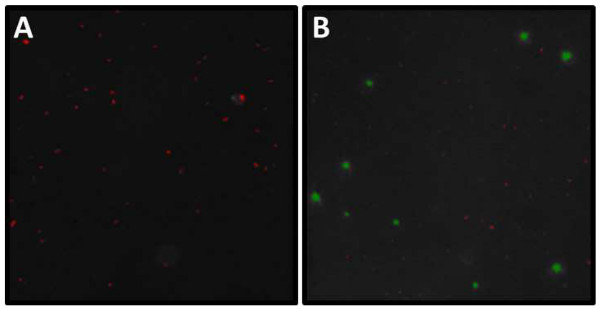 Figure 6. Example of DAPI and SYBR Gold micrograph analysis. Screenshot from the analysis of (A) DAPI and (B) SYBR Gold stained slide example. (A) Length and width (µm) of all highlighted particles (= microbes) can be assessed with the help of an imaging program. Note the aggregation in the center of the image. These clusters will need to be excluded from subsequent analysis. (B) During analysis viruses and bacteria need to be binned by size thresholds into VLP and cellular size ranges (VLP red, cellular green). Note that there are usually some faint VLPs uncategorized by the respective imaging program. These VLPs appearing as faint white dots must be manually counted and added to the automated viral counts. Please click here to view a larger version of this figure.
Figure 6. Example of DAPI and SYBR Gold micrograph analysis. Screenshot from the analysis of (A) DAPI and (B) SYBR Gold stained slide example. (A) Length and width (µm) of all highlighted particles (= microbes) can be assessed with the help of an imaging program. Note the aggregation in the center of the image. These clusters will need to be excluded from subsequent analysis. (B) During analysis viruses and bacteria need to be binned by size thresholds into VLP and cellular size ranges (VLP red, cellular green). Note that there are usually some faint VLPs uncategorized by the respective imaging program. These VLPs appearing as faint white dots must be manually counted and added to the automated viral counts. Please click here to view a larger version of this figure.
 Figure 7. Concentrations of SYBR Gold stained samples. Micrographs of a 0.02 alumina matrix filter containing various numbers of SYBR stained virus samples. Panel A shows a filter containing a suitable amount of seawater filtered, whereas the concentrations on the filter shown in B are too high and in C too low for reliable counts. Please click here to view a larger version of this figure.
Figure 7. Concentrations of SYBR Gold stained samples. Micrographs of a 0.02 alumina matrix filter containing various numbers of SYBR stained virus samples. Panel A shows a filter containing a suitable amount of seawater filtered, whereas the concentrations on the filter shown in B are too high and in C too low for reliable counts. Please click here to view a larger version of this figure.
 Figure 8. Results of flow cytometry analysis output of a reef-water sample. Panel (A) shows molecular grade water only with yellow-green fluorescent microspheres (0.75 μm), used to verify minimal background with the instrument settings. Panel (B) depicts the output of the reef water sample run with identical settings and layout as in A. Note that the beads can still be seen in the same location, along with several populations of autotrophs. (C) The same sample as in (B), used to back-gate the flow cytometer, targeting SYBR positive events (autotrophic + heterotrophic count) and minimizing background. (D) The representative reef-water sample stained with SYBR Green I. Using the gating established in (C), total microbe counts were generated, and heterotrophic and autotrophic microbes partitioned by chlorophyll autofluorescence (see McDole et al.4). Please click here to view a larger version of this figure.
Figure 8. Results of flow cytometry analysis output of a reef-water sample. Panel (A) shows molecular grade water only with yellow-green fluorescent microspheres (0.75 μm), used to verify minimal background with the instrument settings. Panel (B) depicts the output of the reef water sample run with identical settings and layout as in A. Note that the beads can still be seen in the same location, along with several populations of autotrophs. (C) The same sample as in (B), used to back-gate the flow cytometer, targeting SYBR positive events (autotrophic + heterotrophic count) and minimizing background. (D) The representative reef-water sample stained with SYBR Green I. Using the gating established in (C), total microbe counts were generated, and heterotrophic and autotrophic microbes partitioned by chlorophyll autofluorescence (see McDole et al.4). Please click here to view a larger version of this figure.
 Figure 9. Example of microbial metagenome data. Patterns between the microbial metagenomic sequence data and site dependent variables. Here, the relative abundance of Synechococcus spp. was positively correlated with the percent cover of coral, whereas Pelagibacter spp. was not (left panels). The metabolic pathway for conjugative transfer was positively correlated with nitrate concentrations, while the metabolic pathway for DNA repair was consistent between all metagenomes (right panels). Please click here to view a larger version of this figure.
Figure 9. Example of microbial metagenome data. Patterns between the microbial metagenomic sequence data and site dependent variables. Here, the relative abundance of Synechococcus spp. was positively correlated with the percent cover of coral, whereas Pelagibacter spp. was not (left panels). The metabolic pathway for conjugative transfer was positively correlated with nitrate concentrations, while the metabolic pathway for DNA repair was consistent between all metagenomes (right panels). Please click here to view a larger version of this figure.
| Star 7 | CAR9 | |
| Total Number of Reads | 939,311 | 591,600 |
| Number of Preprocessed Reads | 579,664 | 360,246 |
| Mean Sequence Length (bp) | 395 ± 131 | 451 ± 159 |
| Annotated Sequences (%) | 42,218 (7.3%) | 9,117 (2.5%) |
| Unknown (%) | 537,446 (92.7%) | 351,129 (97.5%) |
Table 1. Data characteristics of two viromes generated from the Southern Line Islands, Starbuck and Millennium. The degree of annotation is based on BLAT using the MG-RAST server.
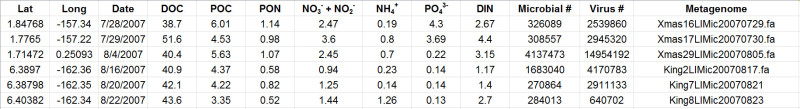 Table 2. Representative results showing data from various sites during one expedition. The here assessed parameters include water chemistry measurements paired with microbial and viral abundances. The table further contains file names referring to the respective metagenomic data for each sampling location. Carbon and inorganic nutrient concentrations are given in μmol/L. Please click here to view a larger version of this table.
Table 2. Representative results showing data from various sites during one expedition. The here assessed parameters include water chemistry measurements paired with microbial and viral abundances. The table further contains file names referring to the respective metagenomic data for each sampling location. Carbon and inorganic nutrient concentrations are given in μmol/L. Please click here to view a larger version of this table.
Discussion
The methods presented here will provide a tool to generate a comprehensive assessment of viral and microbial dynamics in aquatic ecosystems. They are targeted to quantify the standing stock of organic and inorganic resources available to microbial communities and to characterize the makeup of the viral and microbial community present. Next to quantifying viral abundance and microbial abundance and biomass, genomic sequence information reveals their community structure and function. All methods have been developed or modified to allow for application in remote field locations. However the lack of controlled laboratory settings inherently creates some potential for errors. Here we discuss some caveats associated with each of the parameters assessed. Next to potential for contamination during sample handling some of the parameters also yield potential for error in the analytical process.
Dissolved Organic Carbon
Carbon contamination can occur during the sampling procedures (sampling downstream, or in the vicinity of a boat or diver), during the sample preparation (contamination of equipment or inadvertent handling), and even during storage of the sample (other samples in freezer containing organic solvents/volatiles). To avoid the various sources of contamination conduct as few handling steps as possible, as each sample relocation poses an additional source of contamination. The sample should not get in contact with any item not acid washed or combusted. Finally, designating a storage freezer exclusively for samples not containing volatile organic substances will ensure the integrity of the samples during longer storage periods. If samples cannot be kept frozen during an extended travel period acidification of DOC samples with concentrated HCl (as described38-41) may be an applicable alternative. To verify the measurement accuracy DOC consensus reference materials should be used regularly during the DOC measurement runs. Especially deep sea water (>2,000 m) references are valuable in assessing the performance of the analytical machine as it is very stable in its DOC content42.
Particulate Organic Matter
In addition to avoiding organic carbon contamination, POM sampling requires special attention to ensure the accuracy of the filtered volume. If the targeted volume of 500 ml should deviate for any potential reason this needs to be noted to relate the amount of POM captured on the filter to the filtrate of which it was derived.
Inorganic Nutrients
As with organic carbon sampling, attention must be paid to possible nutrient sample contamination generated by various sources like boats or wastewater discharges12. Filters used for organic carbon sampling with a nominal pore sizes of 0.8 µm, may not exclude all microbial biomass and should therefore be changed to 0.2 µm filters for this filtration step. Further, detection limits pose a problem with nutrient samples; especially in the oligotrophic surface water around many coral reef locations (e.g., Cotner et al.43). Detection limits are roughly around 0.1, 0.2, and 0.1 μmol/L for ammonium, nitrate and nitrite, and ortho-phosphate respectively (see 36,12,37)
Microscopy
Beside variations in the concentration of samples image analysis from microscopy slides further yields potential for errors. For instance, images may be out of focus in some areas of the field of view, and in focus in others. As a high-purity alumina matrix filter is a very rigid filter type, any debris under the filter may cause the whole filter to sit on an angle to the slide. As a result, images can be consistently out of focus in various parts of the field of view for a given filter, regardless of microscope alignment. Remounting filters, ensuring the underside of the filter is clean, can ameliorate this if it is observed. Also, in some cases there may be two focal planes in which viruses and microbes can be found. This is the result of stained objects becoming detached from the filter upon mounting, and floating up to the underside of the cover slip which can make counts unreliable. Therefore it is always recommended to check the quality of the samples during the process.
Flow Cytometry
As with the microscopy samples, there may be naturally occurring differences between flow cytometry samples which require adjustments of the analytical process. For instance, depending on the sensitivity of the instrument, dimly fluorescing Prochlorococcus cells may be below the noise level and will not be quantified. Beads serve as an internal sample control (e.g., to confirm that the instrument’s response to fluorescent signals is consistent from day to day (and remains stable during the run itself). If added to the sample at known concentrations, they can also serve as an internal control to verify the sample volume run. However, it is recommended to always run an aliquot of a previously frozen “standard” seawater sample that is thawed and stained along with the samples of interest to provide an additional biological control for sample handling between 96-well plate runs. Depending on the location, seawater samples may look very different from each other. Sorting “representative” populations and then viewing under an epifluorescent microscope (EM) can be a good way to quickly verify phytoplankton populations as either Synechococcussp. or photosynthetic eukaryotes. Prochlorococcus cells are typically not visible under EM because they fade too fast. In addition to chlorophyll a, Synechococcussp. also contain the photopigment phycoeurythrin (PE), which emits light at shorter wavelengths (540-630 nm) than chlorophyll A (660-700 nm). When Synechococcus cells are excited with blue/green light (470-490 nm), they will appear golden if viewed under an emission filter that removes all wavelengths of light below 510 nm. By comparison, the eukaryotic fraction will look red, when excited with the same wavelength of light.
Viral Metagenomics
It is important to note that the process of VLP concentration and storage influences the community recovered. Viral particle loss can occur at every step in the process, and thus, the selection of viral purification and enrichment protocols can ultimately affect the observed taxonomic composition and diversity of the resulting viral metagenomes44,45,18. Concentration of samples can be done using TFF19 or chemical-based flocculation20. In the chemical-based approach, positively charged iron ions bind the naturally negatively charged viral particles forming large (>8 µm) iron-viral complexes that flocculate out of solution and can be recovered using 8 µm filters. Subsequently the iron is chelated off the viral particles and re-dissolved using magnesium, ascorbic acid, and EDTA, leaving concentrated viral particles for nucleic acid extraction20. After comparing these two current methods, we concluded that the iron chloride method is less time consuming than TFF viral concentration, but may yield some caveats. We found that dissociation and dissolution of the iron-viral requires a two-fold more concentrated magnesium-ascorbic acid-EDTA solution than reported20 in order for dissolution to proceed before the ascorbic acid degrades. Aside from this issue, the protocol purifies viral particles by size-fractionation alone, removing microbial contaminants using 0.2 µm filtration. Large viruses do not pass through the filter (e.g., Yang et al.46) and will thus not be detected in the metagenome, while some Bacteria do (McDole, unpublished data). This results in bacterial contamination while discriminating against large viruses.
That specific issue however is also relevant for the removal of microbes in connection with TFF concentration. As the chloroform treatment has been shown to remove chloroform-sensitive viruses with external lipid membranes47 some studies suggest that 0.22 µm filtration can be used to remove the majority of bacteria. It should also be noted that the presented method primarily targets bacteriophage, the most abundant carrier of genetic material in marine environments. Because phage are generally thought not to commonly contain lipids 48 they are more resistant to chloroform treatment. To our knowledge a “catch-all” method does not exist to date. The method presented here is adapted from our field experience over the last 10 years in various marine environments spanning tropical to arctic systems.
Microbial Metagenomics
The construction of metagenomic libraries for microbes (i.e., bacteria and archaea) is more straightforward than viral metagenomes as it is easier to generate the minimal DNA concentrations required for library preparation. Linker amplification shotgun libraries (LASLs)17,49,50 and whole genome amplification based on multiple displacement amplification (MDA) are the two most commonly used methods to generate sufficient DNA for sequencing. The use of MDA to obtain higher DNA concentrations in microbial metagenomes is sometimes necessary. This however can cause artifacts in the sequence data, such as the overamplification of dinoflagellate minicircles. MDA methods are known to preferentially amplify single-stranded and circular DNA, resulting in non-quantitative results51,52. The optimized LASLs approach50 may thus serves as a better alternative in amplifying both microbial and viral metagenomic DNA for sequencing. It is important to note that the LASLs approach has multiple steps, requires sophisticated equipment, and is limited to dsDNA templates. Further, the tissue DNA extraction kit has been shown to extract DNA from both Gram positive and Gram negative phyla, but it has not been verified to effectively lyse recalcitrant microbial taxa or their associated structures (e.g., certain archaea, endospores, or fungal spores). Therefore a bead beating step may be necessary to obtain DNA from certain microbes.
These comprehensive assessments will result in a better understanding of viral and microbial ecosystem functioning. Although few studies have undertaken the effort to assess all proposed parameters, many have been investigating selected ones. Nelson et al.53 suggested a connection between specific reef habitats and depletions in both DOC and bacterioplankton concentrations relative to offshore waters. These concentration changes were accompanied by distinct differentiations of bacterioplankton communities. Dinsdale et al.5 showed that increased anthropogenic impact was accompanied by 10x higher abundances of microbial cells and virus-like particles. Microbial communities in marine waters surrounding inhabited islands also had larger fractions of heterotrophs and potential pathogens5. Finally McDole et al.4 showed, by introducing the microbialization score, that human activities are shifting energy to the microbes, at the expense of the macrobes. These studies, focused on specific microbial and water chemistry parameters, provide novel insights into the biochemical structure of marine ecosystems. Combining traditional data of ecosystem monitoring efforts - like temperature and pH values, fish biomass and diversity, benthic cover, or exposure to human impact - with water chemistry and microbial assessments, will likely result in more sensitive environmental monitoring efforts, which may lead to more specifically targeted conservation efforts.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Elisha Wood-Charlson, Jackie Mueller, Karen Weynberg, and Kathy Morrow for their help and constructive input on this manuscript. We also thank the crew and captain of the Hanse explorer which provided us with a perfect working environment to conduct large parts of this research. Further we would like to thank the three anonymous reviewers for their time and helpful comments to improve the manuscript. This work was funded by the NSF Dimensions of Biodiversity and PIRE awards DEB-1046413 and OISE/IIA-1243541, respectively, the Gordon and Betty Moore Foundation, Investigator Award 3781, and the CIFAR Integrated Microbial Diversity Fellowship IMB-ROHW-141679, all to FR.
References
- Fuhrman JA. Marine viruses and their biogeochemical and ecological effects. Nature. 1999;399(6736):541–548. doi: 10.1038/21119. [DOI] [PubMed] [Google Scholar]
- Azam F, Malfatti F. Microbial structuring of marine ecosystems. Nature Reviews Microbiology. 2007;5(10):782–791. doi: 10.1038/nrmicro1747. [DOI] [PubMed] [Google Scholar]
- Huber JA, et al. Microbial population structures in the deep marine biosphere. Science. 2007;318(5847):97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- McDole T, et al. Assessing coral reefs on a Pacific-wide scale using the microbialization score. PloS ONE. 2012;7(9) doi: 10.1371/journal.pone.0043233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinsdale EA, et al. Microbial ecology of four coral atolls in the Northern Line Islands. PloS ONE. 2008;3(2) doi: 10.1371/journal.pone.0001584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CE, et al. Coral and macroalgal exudates vary in neutral sugar composition and differentially enrich reef bacterioplankton lineages. The ISME Journal. 2013;7(5):962–979. doi: 10.1038/ismej.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AF, et al. Influence of coral and algal exudates on microbially mediated reef metabolism. PeerJ. 2013;1 doi: 10.7717/peerj.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, et al. Indirect effects of algae on coral: algae‐mediated, microbe‐induced coral mortality. Ecology Letters. 2006;9(7):835–845. doi: 10.1111/j.1461-0248.2006.00937.x. [DOI] [PubMed] [Google Scholar]
- Burdige DJ, Martens CS. Biogeochemical cycling in an organic-rich coastal marine basin: 10. The role of amino acids in sedimentary carbon and nitrogen cycling. Geochimica et Cosmochimica Acta. 1988;52(6):1571–1584. [Google Scholar]
- Capone DG, Kiene RP. Comparison of microbial dynamics in marine and freshwater sediments: Contrasts in anaerobic carbon catabolism. Limnology and Oceanography. 1988;33(4):725–749. [Google Scholar]
- Krom MD, Sholkovitz ER. Nature and reactions of dissolved organic matter in the interstitial waters of marine sediments. Geochimica et Cosmochimica Acta. 1977;41(11):1565–1574. [Google Scholar]
- Grasshoff K. Verlag Chemie. Weinheim and New York: 1976. Methods of sea-water analysis. [Google Scholar]
- Strickland JD. Practical handbook of seawater analysis. Bulletin Fisheries Research Board of Canada. 1968;167 [Google Scholar]
- Sharp JH, et al. Analyses of dissolved organic carbon in seawater: the JGOFS EqPac methods comparison. Marine Chemistry. 1995;48(2):91–108. [Google Scholar]
- DeLong EF. Microbial population genomics and ecology. Current Opinion in Microbiology. 2002;5(5):520–524. doi: 10.1016/s1369-5274(02)00353-3. [DOI] [PubMed] [Google Scholar]
- Hugenholz P, Goebel BM, Pace NR. Impact of Culture-Independent Studies on the Emerging Phylogenetic View of Bacterial Diversity. Journal of Bacteriology. 1998;180(18):4765–4774. doi: 10.1128/jb.180.18.4765-4774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, et al. Genomic analysis of uncultured marine viral communities. Proceedings of the National Academy of Sciences. 2002;99(22):14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willner D, Hugenholtz P. From deep sequencing to viral tagging: Recent advances in viral metagenomics. BioEssays. 2013;35:436–442. doi: 10.1002/bies.201200174. [DOI] [PubMed] [Google Scholar]
- Thurber RV, Haynes M, Breitbart M, Wegley L, Rohwer F. Laboratory procedures to generate viral metagenomes. Nature Protocols. 2009;4:470–483. doi: 10.1038/nprot.2009.10. [DOI] [PubMed] [Google Scholar]
- John SG, et al. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environmental Microbiology Reports. 2011;3:195–202. doi: 10.1111/j.1758-2229.2010.00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, et al. The Microbiome and Virome of CF. Pediatric Pulmonology. 2012;47:321–322. [Google Scholar]
- Kelly LW, et al. Black reefs: iron-induced phase shifts on coral reefs. The ISME Journal. 2012;6(3):638–649. doi: 10.1038/ismej.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsen PK. Automatic determination of bacterioplankton biomass by image analysis. Applied Environmental Microbiology. 1986;51 doi: 10.1128/aem.51.6.1199-1204.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Azam F. Protein content and protein synthesis rates of planktonic marine bacteria. Marine Ecology Progress Series. 1989;51(3):201–213. [Google Scholar]
- Hara S, Koike I, Terauchi K, Kamiya H, Tanoue E. Abundance of viruses in deep oceanic waters. Marine Ecology Progress Series. 1996;145(1):269–277. [Google Scholar]
- Wommack KE, Colwell RR. Virioplankton: viruses in aquatic ecosystems. Microbiology and Molecular Biology Reviews. 2000;64(1):69–114. doi: 10.1128/mmbr.64.1.69-114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbauer MG, Brettar I, Hofle MG. Lysogeny and virus-induced mortality of bacterioplankton in surface, deep, and anoxic marine waters. Limnology and Oceanography. 2003;48(4):1457–1465. [Google Scholar]
- Clasen JL, Brigden SM, Payet JP, Suttle CA. Evidence that viral abundance across oceans and lakes is driven by different biological factors. Freshwater Biology. 2008;53(6):1090–1100. [Google Scholar]
- Parada V, Baudoux AC, Sintes E, Weinbauer MG, Herndl GJ. Dynamics and diversity of newly produced virioplankton in the North Sea. The ISME Journal. 2008;2(9):924–936. doi: 10.1038/ismej.2008.57. [DOI] [PubMed] [Google Scholar]
- Angly FE, et al. The marine viromes of four oceanic regions. PLoS Biology. 2006;4(11) doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansell DA, Carlson CA. Deep ocean gradients in dissolved organic carbon concentrations. Nature. 1998;263(266) [Google Scholar]
- Carlson CA, et al. Dissolved organic carbon export and subsequent remineralization in the mesopelagic and bathypelagic realms of the North Atlantic basin. Deep Sea Research Part II: Topical Studies in Oceanography. 2010;57(16):1433–1445. [Google Scholar]
- Sharp JH. Improved analysis for particulate organic carbon and nitrogen from seawater. Limnology and Oceanography. 1974;19(6):984–989. [Google Scholar]
- Ehrhardt M, Koeve W. Determination of particulate organic carbon and nitrogen. Methods of Seawater Analysis. 2007;Third Edition:437–444. [Google Scholar]
- Haas AF, et al. Organic matter release by coral reef associated benthic algae in the Northern Red Sea. Journal of Experimental Marine Biology and Ecology. 2010;389(1):53–60. [Google Scholar]
- Guildford SJ, Hecky RE. Total nitrogen, total phosphorus, and nutrient limitation in lakes and oceans: Is there a common relationship. Limnology and Oceanography. 2000;45(6):1213–1223. [Google Scholar]
- Allison SD, Chao Y, Farrara JD, Hatosy S, Martiny AC. Fine-scale temporal variation in marine extracellular enzymes of coastal southern California. Frontiers in Microbiology. 2012;3 doi: 10.3389/fmicb.2012.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyl FC, Gast GJ. Linkage of small-scale spatial variations in DOC, inorganic nutrients and bacterioplankton growth with different coral reef water types. Aquatic Microbial Ecology. 2001;24(1):17–26. [Google Scholar]
- De Goeij JM, Van Duyl FC. Coral cavities are sinks of dissolved organic carbon (DOC) Limnology and Oceanography. 2007;52(6):2608–2617. [Google Scholar]
- De Goeij JM, et al. Major bulk dissolved organic carbon (DOC) removal by encrusting coral reef cavity sponges. Marine Ecology Progress Series. 2008;357:139–151. [Google Scholar]
- Mueller B, et al. Natural diet of coral-excavating sponges consists mainly of dissolved organic carbon (DOC) PLoS ONE. 2014;9(2) doi: 10.1371/journal.pone.0090152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CA, Ducklow HW, Hansel DA. Organic carbon partitioning during spring phytoplankton blooms in the Ross Sea polynya and the Sargasso Sea. Oceanography. 1998;43(3):375–386. [Google Scholar]
- Cotner JB, et al. Nutrient limitation of heterotrophic bacteria in Florida Bay. Estuaries. 2000;23(5):611–620. [Google Scholar]
- Willner D, et al. Metagenomic detection of phage-encoded platelet-binding factors in the human oral cavity. Proceedings of the National Academy of Science USA. 2011;108(1):4547–4553. doi: 10.1073/pnas.1000089107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RJ, et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. Journal of Virological Methods. 2013;195:194–204. doi: 10.1016/j.jviromet.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, et al. Nanoporous membranes with ultrahigh selectivity and flux for the filtration of viruses. Advanced Materials. 2006;18(6):709–712. [Google Scholar]
- Feldman HA, Wang SS. Sensitivity of various viruses to chloroform. Proceedings of the Society for Experimental Biology and Medicine. 1961;106(4):736–738. doi: 10.3181/00379727-106-26459. [DOI] [PubMed] [Google Scholar]
- Ackermann HW. In: The Bacteriophages., Second Edition. Calendar R, editor. New York: Oxford University Press; 2006. pp. 8–16. [Google Scholar]
- Henn MR, et al. Analysis of High-Throughput Sequencing and Annotation Strategies for Phage Genomes. PLoS ONE. 2010;5(2) doi: 10.1371/journal.pone.0009083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime MB, Deng L, Poulos BT, Sullivan MB. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: a rigorous assessment and optimization of the linker amplification method. Environmental Microbiology. 2012;14(9):2526–2537. doi: 10.1111/j.1462-2920.2012.02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz S, Allgaier M, Hugenholtz P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nature Methods. 2010;7(12):943–944. doi: 10.1038/nmeth1210-943. [DOI] [PubMed] [Google Scholar]
- Kim KH, Bae JW. Amplification Methods Bias Metagenomic Libraries of Uncultured Single-Stranded and Double-Stranded DNA Viruses. Applied Environmental Microbiology. 2011;77(21):7663–7668. doi: 10.1128/AEM.00289-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CE, Alldredge AL, McCliment EA, Amaral-Zettler LA, Carlson CA. Depleted dissolved organic carbon and distinct bacterial communities in the water column of a rapid-flushing coral reef ecosystem. The ISME Journal. 2011;5(8):1374–1387. doi: 10.1038/ismej.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly LW, et al. Local genomic adaptation of coral reef-associated microbiomes to gradients of natural variability and anthropogenic stressors. Proceedings of the National Academy of Sciences. 2014;111(28):10227–10232. doi: 10.1073/pnas.1403319111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Haynes M, Furlan M, Robertson CE, Harris JK, Rohwer F. Purifying the Impure: Sequencing Metagenomes and Metatranscriptomes from Complex Animal-associated Samples. J. Vis. Exp. 2014. [DOI] [PMC free article] [PubMed]